

Abstract
Autophagy genes' expression is upregulated in visceral fat in human obesity, associating with obesity-related cardio-metabolic risk. E2F1 (E2F transcription factor 1) was shown in cancer cells to transcriptionally regulate autophagy. We hypothesize that E2F1 regulates adipocyte autophagy in obesity, associating with endocrine/metabolic dysfunction, thereby, representing non-cell-cycle function of this transcription factor. E2F1 protein (N=69) and mRNA (N=437) were elevated in visceral fat of obese humans, correlating with increased expression of ATG5 (autophagy-related 5), MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 β), but not with proliferation/cell-cycle markers. Elevated E2F1 mainly characterized the adipocyte fraction, whereas MKI67 (marker of proliferation Ki-67) was elevated in the stromal-vascular fraction of adipose tissue. In human visceral fat explants, chromatin-immunoprecipitation revealed body mass index (BMI)-correlated increase in E2F1 binding to the promoter of MAP1LC3B, but not to the classical cell cycle E2F1 target, CCND1 (cyclin D1). Clinically, omental fat E2F1 expression correlated with insulin resistance, circulating free-fatty-acids (FFA), and with decreased circulating ADIPOQ/adiponectin, associations attenuated by adjustment for autophagy genes. Overexpression of E2F1 in HEK293 cells enhanced promoter activity of several autophagy genes and autophagic flux, and sensitized to further activation of autophagy by TNF. Conversely, mouse embryonic fibroblast (MEF)-derived adipocytes from e2f1 knockout mice (e2f1−/−) exhibited lower autophagy gene expression and flux, were more insulin sensitive, and secreted more ADIPOQ. Furthermore, e2f1−/− MEF-derived adipocytes, and autophagy-deficient (by Atg7 siRNA) adipocytes were resistant to cytokines-induced decrease in ADIPOQ secretion. Jointly, upregulated E2F1 sensitizes adipose tissue autophagy to inflammatory stimuli, linking visceral obesity to adipose and systemic metabolic-endocrine dysfunction.
Keywords: adipose tissue, autophagy, E2F1, obesity, stress response, transcriptional regulation
Abbreviations
- BafA1
bafilomycin A1
- BMI
body mass index
- ChIP
chromatin immunoprecipitation
- FFA
free fatty acids
- IBMX
3-isobutylmethylxanthine
- IL1B
interleukin 1, β
- KRBH
Krebs-Ringer buffer
- MEF
mouse embryonic fibroblast
- Om
omental
- Sc
subcutaneous
- TNF
tumor necrosis factor.
Introduction
The highly evolutionarily conserved process of (macro)autophagy is currently receiving attention as a putative pathway dysregulated in diverse human pathologies, including malignant, neurodegenerative, infectious, autoimmune, inflammatory, and metabolic diseases.1 As a homeostatic-housekeeping process, autophagy is mostly considered to potentially contribute to pathogenesis when it is impaired (downregulated), resulting in diminished ability to clear protein aggregates, dysfunctional organelles, and to limit inflammation. Paradoxically, current clinical trials seeking to manipulate autophagy include mainly attempts to inhibit the process by antimalarial agents as adjuvant anticancer chemotherapy: by inhibiting lysosomal acidification, it is hoped that these drugs will effectively interfere with autophagy as a major cancer cell survival mechanism.2-8 The lack of clinical studies aiming to activate autophagy undoubtedly partly reflects the paucity of therapeutic tools to selectively activate the process. Yet, it likely also highlights the possibility that overactivated, and not only impaired autophagy, may contribute to pathogenesis, as has been demonstrated in chronic obstructive pulmonary diseases (COPD)9 and in cancer.
Recently, there has been growing interest in the role of autophagy in adipose tissue biology.10-12 We and others have reported that, in adipose tissue in obesity, autophagy gene expression is increased, and the process is most likely activated (recently reviewed in ref. 16).13-16 Interestingly, this appears to be a tissue-type selective response to obesity,17 as the liver has been shown to exhibit impaired autophagy that contributes to obesity-induced hepatic insulin resistance.18 Although activated autophagy at the whole tissue level could reflect the altered cellular composition of adipose tissue in obesity, several studies demonstrate that adipocytes per se likely exhibit activated autophagic flux.13,14 The functional significance of this autophagic activation is unclear: ex-vivo pharmacological inhibition of autophagy in human adipose tissue explants by 3MA (3-methyladenine) increased secretion of IL6 (interleukin 6), TNF (tumor necrosis factor) and IL1B (interleukin 1, β) by the tissue, suggesting that autophagy may act to limit adipose tissue inflammation.15 Yet, autophagy activation is more pronounced in visceral than in subcutaneous fat, which is particularly evident in patients with intra-abdominal fat distribution, and in patients whose obesity is associated with insulin resistance—all linking activated autophagy to more severe obesity subphenotypes.13 An intriguing finding has been the increased expression of key autophagy genes, not only at the protein, but also at the mRNA level.13 Indeed, although as an autodegradative lysosomal process, autophagy was mostly considered to be regulated post-transcriptionally,1,19 recently there has been accumulating evidence for transcriptional level regulation of the process.20,21 Thus, the mechanisms and impact of transcriptional regulation of autophagy in human diseases is currently a significant gap of knowledge.
In searching for potential mechanisms for transcriptional activation of autophagy genes in adipose tissue in obesity, the possible involvement of the transcription factor E2F1 has been raised by studies in cancer cells.22 Indeed, E2F1 is a member of the E2F family of transcription factors most studied for their involvement in cell-cycle regulation.23-25 Yet, evidence for non-cell-cycle-related functions of E2F1 has been accumulating, suggesting that it may exert significant gene regulatory functions also in quiescent cells.26 Most relevant to adipocyte biology, such functions may include promotion of adipogenesis, regulation of apoptosis, immune functions, and, importantly, cellular fuel metabolism via a range of metabolic genes, in particular PDK4 (pyruvate dehydrogenase kinase, isoform 4) and other mitochondrial respiratory genes.27-32 The overall metabolic effect of E2F1 action seems to indicate inhibition of cellular nutrient oxidation capacity.33 Given the possibility that E2F1 regulates autophagy genes shown to be elevated in human adipose tissue in obesity and to associate with obesity-related cardio-metabolic morbidity risk,13 we set out to test a putative role of E2F1-associated autophagy in adipose tissue in obesity.
Results
E2F1 in human adipose tissue in obesity: Potential role in regulating autophagy gene expression
E2F1 was proposed in cancer cells to regulate autophagy gene expression and activity.22,34 Thus, we first assessed the expression of E2F1 in human adipose tissue and its possible association with obesity-related clinical characteristics, and with autophagy gene expression. For this, we utilized 2 human adipose tissue biobanks (Leipzig N=437, and Beer-Sheva N=69). These complementary cohorts were utilized jointly and are described in detail in previous publications.13,35-37 The clinical characteristics of participants included in the current analyses are presented in Table 1 and 2, respectively. Subcutaneous (Sc) and omental (Om) fat E2F1 mRNA levels were nearly similar in lean persons, but were elevated in Om in obese persons: In those with predominant subcutaneous adiposity (Sc accounting for >50% of total abdominal fat as assessed by magnetic resonance imaging) a ∼2-fold increase in E2F1 mRNA levels was observed in Om compared to Sc fat (p<0.001) (Fig. 1A). This depot-difference was even more pronounced in those with intra-abdominal (visceral) adiposity, a fat distribution associated with greater obesity-related cardio-metabolic risk. These patients exhibited a 6-fold difference in E2F1 mRNA between Om and Sc depots (P<0.001) (Fig. 1A). There were no significant differences between males and females in E2F1 mRNA levels (data not shown). To assess the relation of Om-E2F1 mRNA levels with clinical characteristics of the patients, with emphasis on obesity-related cardio-metabolic parameters, the study population was stratified into quintiles of Om-E2F1 mRNA. Om-E2F1 mRNA levels did not associate with age (Fig. 1B). Yet, highly significant associations (P< 0.001) were observed with abdominal adiposity and insulin resistance (as determined by hyperinsulinemic-euglycemic clamp). Moreover, high levels of IL6, free fatty acids (FFA), triglycerides, LEP/leptin and low levels of ADIPOQ—all associated with increased risk of cardiovascular disease—38,39 strongly associated with higher Om-E2F1 mRNA levels (P< 0.001 for all, Fig. 1C). Indeed, diagnosis of diabetes was present in ˜8% of those in the lower Om-E2F1 mRNA quintile, but was >80% in those in the top quintile. Importantly, the association of Om-E2F1 mRNA with increased circulating FFA, LEP and decreased ADIPOQ, suggests correspondence to adipocyte metabolic and endocrine dysfunction (Fig. 1B, 1C and 1D).
Table 1.
Clinical and Biochemical Characteristics: Leipzig Cohort (N=437)
| Lean | Overweight | Obese | |||
|---|---|---|---|---|---|
| BMI≤25 | 25<BMI<30 | BMI≥30 | Entire group | P ANOVA | |
| n | 102 | 67 | 268 | 437 | |
| Sex (male/female) | 45/57 | 38/29 | 99/169 | 182/255 | |
| Age (y) | 55.3 ± 1.6 | 59.7 ± 1.6 | 49.6 ± 0.8 | 52.5 ± 0.7 | <0.01 |
| BMI (kg/m2) | 23.0 ± 0.2 | 27.8 ± 0.2 | 44.4 ± 0.6 | 36.8 ± 0.6 | <0.01 |
| Waist circumference (cm) | 78.9 ± 1.5 | 97.3 ± 1.7 | 130.1 ± 1.5 | 113.0 ± 1.4 | <0.01 |
| Triglycerides (mg/dL) | 106.2 ± 8.8 | 150.4 ± 8.8 | 168.1 ± 8.8 | 150.4 ± 0.1 | <0.01 |
| HDL (mg/dL) | 57.9 ± 0.1 | 54.0 ± 0.1 | 50.2 ± 0.1 | 54.3 ± 0.1 | <0.01 |
| FPG (mg/dL) | 102.7 ± 1.8 | 104.5 ± 1.8 | 108.1 ± 1.2 | 106.3 ± 0.1 | NS |
| HbA1c (%) | 5.5 ± 0.1 | 5.7 ± 0.1 | 6.0 ± 0.1 | 5.8 ± 0.1 | <0.01 |
| Fasting insulin (pmol/L) | 5.9 ± 1.2 | 17.6 ± 2.0 | 21.1 ± 1.2 | 16.4 ± 0.9 | <0.01 |
| HOMA-IR | 13.2 ± 0.4 | 34.3 ± 0.6 | 42.0 ± 0.4 | 33.0 ± 0.3 | <0.01 |
| GIR | 89.0 ± 2.8 | 63.1 ± 3.5 | 49.2 ± 2.2 | 64.2 ± 1.8 | <0.01 |
| IL6 (pg/ml) | 1.6 ± 0.3 | 3.5 ± 0.4 | 6.3 ± 0.3 | 4.8 ± 0.2 | <0.01 |
| Visceral area (mm2) | 52.0 ± 2.9 | 165.7 ± 11.4 | 293.3 ± 10.7 | 222.6 ± 8.6 | <0.01 |
| Adipocyte diameter (μm) | 89.9 ± 1.4 | 99.9 ± 1.5 | 108.4 ± 1.0 | 102.4 ± 0.8 | <0.01 |
| Om-ATG5 | 2.2 ± 0.1 | 5.8 ± 0.5 | 7.2 ± 0.4 | 5.4 ± 0.3 | <0.01 |
| Om-MAP1LC3B | 0.0 ± 0.0 | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.1 ± 0.0 | <0.01 |
| Om-MKI67 | 4.4 ± 0.4 | 6.1 ± 1.1 | 4.1 ± 0.4 | 4.5 ± 0.3 | NS |
BMI, body mass index; FPG, fasting plasma glucose; GIR, glucose infusion rate during hyperinsulinemic euglycemic clamp; HDL, high density lipoprotein; HOMA-IR, homeostatic model assessment of insulin resistance; NS, nonsignificant (P>0.05).
Table 2.
Clinical and Biochemical Characteristics: Beer-Sheva Cohort (N=69)
| Lean | Overweight | Obese | |||
|---|---|---|---|---|---|
| BMI≤25 | 25<BMI<30 | BMI≥30 | Entire group | P ANOVA | |
| n | 14 | 11 | 44 | 69 | |
| Sex (male/female) | 4/10 | 6/5 | 6/38 | 16/53 | |
| Age (y) | 44.6 ± 5.3 | 47.5 ± 5.7 | 42.7 ± 2.3 | 43.9 ± 2.0 | NS |
| BMI (kg/m2) | 22.4 ± 0.6 | 27.7 ± 0.2 | 40.6 ± 0.8 | 34.8 ± 1.1 | <0.01 |
| Waist circumference (cm) | 68.9 ± 6.7 | 92.9 ± 2.9 | 114.1 ± 2.7 | 100.4 ± 3.3 | <0.01 |
| SBP (mm Hg) | 120.5 ± 3.4 | 121.8 ± 2.9 | 133.0 ± 2.6 | 128.3 ± 2.0 | <0.05 |
| DBP (mm Hg) | 73.1 ± 2.0 | 76.0 ± 2.0 | 82.3 ± 1.9 | 79.4 ± 1.4 | <0.05 |
| Triglycerides (mg/dL) | 105.3 ± 14.9 | 113.3 ± 17.6 | 158.0 ± 12.3 | 139.7 ± 9.1 | <0.05 |
| HDL (mg/dL) | 52.5 ± 3.6 | 41.6 ± 5.9 | 49.6 ± 2.2 | 48.7 ± 1.8 | <0.05 |
| FPG (mg/dL) | 95.2 ± 5.5 | 87.3 ± 3.5 | 104.1 ± 6.7 | 99.5 ± 4.4 | NS |
| HbA1c (%) | 5.6 ± 0.2 | 5.6 ± 0.2 | 5.9 ± 0.2 | 5.8 ± 0.1 | NS |
| hsCRP | 0.6 ± 0.4 | 0.9 ± 0.5 | 1.2 ± 0.3 | 1.1 ± 0.2 | <0.05 |
| Fasting insulin (pmol/L) | 3.2 ± 0.5 | 4.7 ± 0.9 | 12.7 ± 1.4 | 9.3 ± 1.1 | 0.001 |
| HOMA-IR | 0.8 ± 0.2 | 1.1 ± 0.2 | 3.3 ± 0.4 | 2.4 ± 0.3 | <0.001 |
BMI, body mass index; DBP, diastolic blood pressure; FPG, fasting plasma glucose; HbA1c, hemoglobin A1c ; HDL, high density lipoprotein; HOMA-IR, homeostatic model assessment of insulin resistance; hsCRP, high sensitivity c-reactive protein; SBP, systolic blood pressure; NS, nonsignificant (P>0.05).
Figure 1.
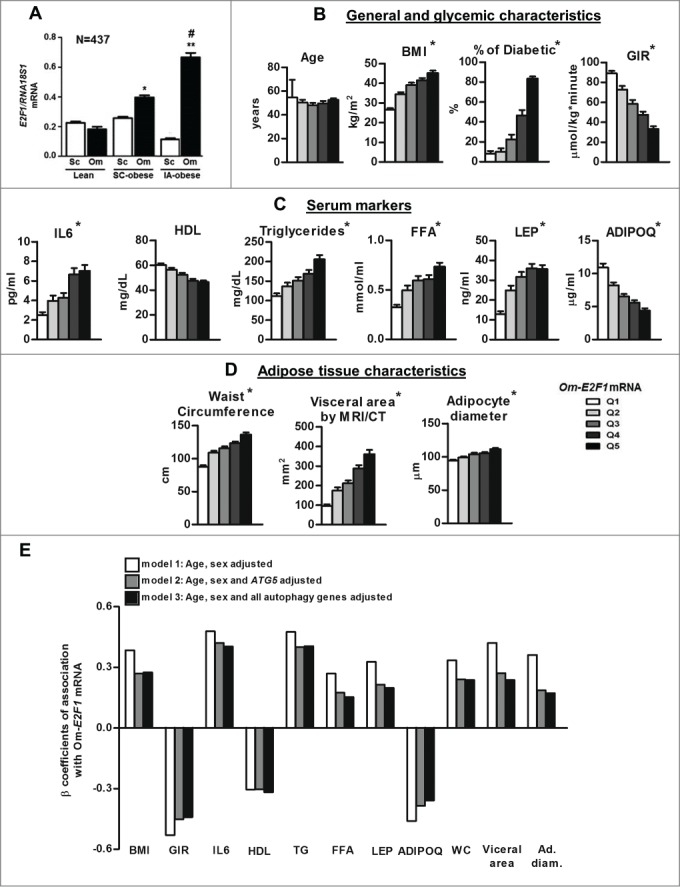
E2F1 mRNA expression in human adipose tissue and its correlation with clinical characteristics. (A) Paired (subcutaneous [Sc] and omental [Om]) fat tissue samples of 437 persons (Leipzig cohort, divided into 3 groups according to fat distribution pattern) were analyzed for the mRNA levels of E2F1 by quantitative real-time PCR, and presented as relative abundance to RNA18S1 (RNA, 18S ribosomal 1). *, P<0.05; ** P<0.01 versus Om of the lean. #, P<0.05 versus Om of Sc-obese. (B, C, D) Clinical parameters of 437 persons (Leipzig cohort) were cross-classified across quintiles of Om-E2F1 mRNA expression. White and black columns represent the lowest (Q1) and the highest (Q5) E2F1 mRNA levels, respectively, *, P of trend (by linear regression) <0.001. (E) Multivariate models to assess associations between Om-E2F1 mRNA levels and parameters shown in (B, C and D) as continuous variables. Values are the β coefficient of association, with model 1 adjusted for age and sex, model 2 for age, sex, and Om-ATG5 mRNA, and model 3 adjusted for age, sex, and omental mRNA expression of ATG5, MAP1LC3B and BECN1. All associations are with P values <0.05. BMI, body mass index; GIR, glucose infusion rate during hyperinsulinemic euglycemic clamp; HDL, high-density lipoprotein; TG, triglycerieds; WC, waist circumference.
Given that like E2F1, expression of autophagy genes in Om was similarly associated with obesity-related cardio-metabolic parameters,13 we assessed whether Om-E2F1 expression and autophagy could share a pathophysiological path. Indeed, in Om, E2F1 mRNA levels associated with the expression of ATG5 and MAP1LC3B mRNA levels (R = .387, P<0.001 and R = .161, P = 0.024, respectively, N=196). Moreover, we tested in multivariate models adjusted for age and sex, whether the association between Om-E2F1 mRNA levels and various clinical parameters was attenuated by adjusting also to the level of Om-ATG5 mRNA and autophagy genes (Fig. 1E). Notable attenuation was demonstrated in the association between Om-E2F1 mRNA, and BMI and waist circumference, visceral fat area, adipocyte diameter, and also with circulating parameters indicative of adipose tissue metabolic and endocrine function (FFA, LEP and ADIPOQ). Jointly, although this is a cross-sectional analysis where no causality or directionality could be inferred, these results provide clinical support for the notion that E2F1 and autophagy gene expression (at the mRNA level) share a common path connecting obesity and cardio-metabolic risk.
To substantiate these findings in a separate cohort, and to challenge the above notion with E2F1 expression assessed at the protein level, we utilized the Beer-Sheva bio-bank (N=69, Table 2). Paired Sc and Om adipose tissue samples from lean and obese persons revealed increased E2F1 protein content in obesity, particularly in the omental fat depot (Fig. 2A, B). Despite the lower sample size compared to the Leipzig cohort, in quintiles of Om-E2F1 protein content, significant P of trends was observed with several key parameters (given are mean ± SD of the low and top quintiles of E2F1 protein, respectively): Waist circumference: 87.1±3.4 and 115.2±5.7 (P =0.001); homeostatic model assessment-insulin resistance (HOMA-IR): 1.2±0.2 and 4.4±0.7 (P<0.001). These were also reflected in the mean number of positive criteria for the metabolic syndrome, being 1.5±0.3 in the lower Om-E2F1 protein quintile and 2.7±0.4 in the upper quintile (P of trend = 0.02). Furthermore, consistent with findings in the Leipzig cohort, statistically significant correlations between E2F1 protein expression in Om fat and the protein expression of autophagy genes (based on protein expression of LC3B-II and ATG5) were observed (Fig. 2C).
Figure 2.
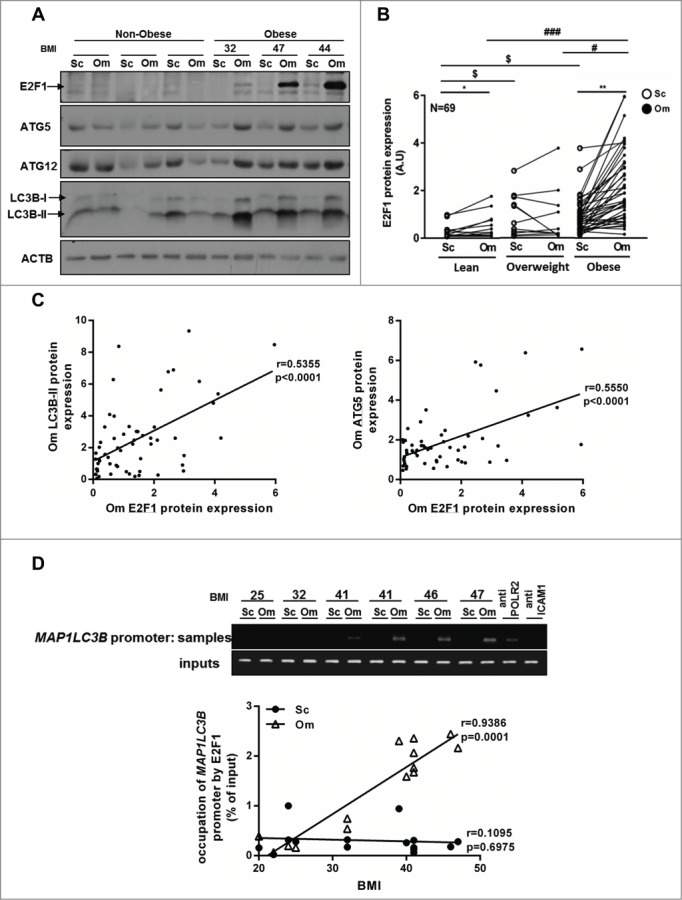
Protein expression of E2F1 and autophagy genes in human adipose tissue. (A) Representative blots of western blot analysis from paired (Sc and Om) fat tissue samples of 69 persons (Beer-Sheva cohort) of the protein levels of E2F1, autophagy genes and ACTB (actin, β) was used as a loading control. (B) E2F1 protein expression in paired fat tissues samples in lean, overweight and obese persons. A paired t test was performed to evaluate the differences between the expression of E2F1 in Sc and Om adipose tissues within each BMI group.*, P<0.05; **, P<0.01. One-way ANOVA was used to evaluate the differences in E2F1 expression between 3 groups (lean, overweight and obese) in Sc and Om depots. $, P<0.05 Sc of overweight or obese versus Sc of lean. #, P<0.05; ###, P<0.001 Om of overweight or lean versus Om of obese, respectively. (C) Linear regression analysis of E2F1 protein expression in omental fat and LC3B-II and ATG5 protein levels. (D) Formaldehyde cross-linked chromatin from 16 paired human (Om and Sc) adipose tissues was subjected to ChIP experiments. Immunoprecipitation of E2F1 containing complexes was performed using anti-E2F1 antibody. Anti-POLR2 (polymerase [RNA] II [DNA directed]), was used as positive control and anti-ICAM1 (intercellular adhesion molecule 1) was used as negative control. After isolation of bound DNA, end-point PCR and quantitative real time PCR were performed for a 750 bp region of the endogenous human MAP1LC3B promoter. Quantitative real-time PCR results were analyzed using linear regression. Inputs indicate PCR performed on DNA (diluted 1:300) without any immunoprecipitation.
To begin bridging the gap between mere clinical associations and more molecular support for a potential E2F1-mediated regulation of autophagy gene expression, we developed an E2F1 ChIP protocol for use in whole human adipose tissue.40 Fresh human adipose tissue samples were processed as described in Materials and Methods to obtain chromatin fragments cross-linked with chromatin-binding proteins, after which E2F1 ChIP was performed, and DNA sequences corresponding to the respective E2F1 binding sites in the MAP1LC3B promoter were amplified by PCR. Results demonstrate E2F1 binding, in vivo, to the MAP1LC3B promoter area in human adipose tissue, as shown in the end-point PCR results (Fig. 2D). For more quantitative analysis to complement the end-point PCR, quantitative real-time PCR was conducted, and association with BMI was assessed using linear regression (Fig. 2D). Positive correlations were observed between E2F1 binding to MAP1LC3B promoter in Om (but not SC) and BMI.
Collectively, results so far suggest that obesity, particularly visceral adiposity, associates with elevated E2F1 expression at the mRNA and protein levels, correlating with elevated obesity-associated cardio-metabolic risk signature and expression of autophagy genes. Furthermore, several E2F1-clinical associations were attenuated by adjusting for autophagy gene expression, and a novel ChIP assay in whole adipose tissue explants demonstrated BMI-associated increased occupancy of the MAP1LC3B promoter by E2F1.
Elevated adipose tissue E2F1 in obesity is likely unrelated to cell cycle regulation
Since E2F1 is best known for its cell-cycle regulation functions, and given that cell proliferation does occur in adipose tissue in obesity (adipocyte hyperplasia, macrophage proliferation),41 does E2F1 regulate adipose tissue cell cycle in obesity? And, which cellular component of adipose tissue contributes to increased E2F1 expression? We undertook several complementary approaches to address this question. Using whole adipose tissue ChIP assay, we determined whether similar to the MAP1LC3B promoter, increased E2F1 levels increase promoter occupancy of E2F1 classical target genes that mediate the cell-cycle regulatory function of E2F1. Intriguingly, E2F1 binding to the CCND1 promoter exhibited no BMI-dependent association (Fig. 3A). Next, the association between adipose tissue E2F1 and proliferation markers was studied. While increasing quintiles of Om-E2F1 mRNA levels readily associated with ATG5 mRNA, there was no similar association with mRNA of MKI67, a proliferating cell marker (Fig. 3B). At the protein level, PCNA (another commonly used marker of proliferating cells) tended to exhibit higher protein levels in Om than in Sc fat, but this was on average similarly observed in nonobese and obese (Fig. 3C). Similarly, MKI67 protein tended to be higher in obesity, consistent with cell proliferation as a contributor to adipose tissue expansion. Yet, there was no apparent association with E2F1 protein levels (Fig. 3C). Furthermore, a classical E2F1 target in cell cycle regulation, CDK4, was not found to exhibit similar differences in expression as E2F1, being higher in Sc than in Om fat (Fig. 3C). Finally, we assessed whether increased E2F1 expression could be contributed by the largely nonproliferating adipocyte cell fraction, or the stromal-vascular cells of adipose tissue, to which proliferating cells segregate. Strikingly, following collagenase digestion of the tissue, while E2F1 expression was mainly contributed by adipocytes, MKI67 was mainly increased in obesity in the stromal vascular fraction (SVF) (Fig. 3D). Jointly, E2F1 does not seem to correlate with proliferation markers' expression in human adipose tissue, or to be contributed by the proliferating cells fraction, suggesting that its elevated expression in obesity may indeed engage “nonclassical” E2F1 functions, such as the proposed regulation of autophagy gene expression.
Figure 3.
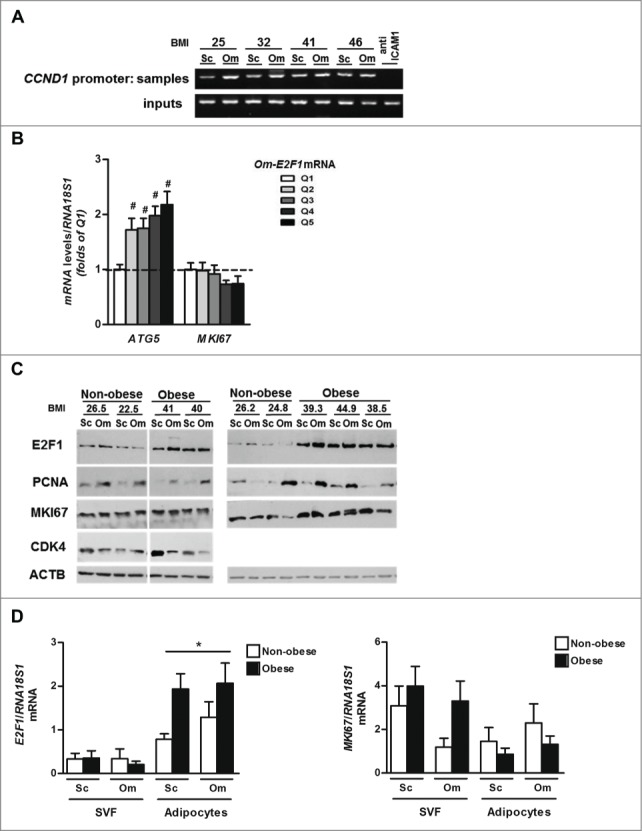
Expression of E2F1 and proliferation markers in whole human adipose tissue and in 2 fractions, adipocytes and SVF. (A) Formaldehyde cross-linked chromatin from 4 paired human (Sc and Om) adipose tissues was subjected to ChIP experiments. Immunoprecipitation of E2F1 containing complexes was performed using anti-E2F1 antibody. Anti- ICAM1 was used as negative control. After isolation of bound DNA, end-point PCR was performed for a 400-bp region of the endogenous human CCND1 promoter. Inputs indicate PCR performed on DNA (diluted 1:300) without any immunoprecipitation. (B) Om fat tissue samples of 437 persons (Leipzig cohort) were analyzed for the mRNA levels of ATG5 and MKI67 by quantitative real-time PCR and were cross-classified across quintiles of Om-E2F1 mRNA expression. White and black columns represent the lowest (Q1) and the highest (Q5) E2F1 mRNA levels, respectively, #, P<0.05. (C) Representative blots of western blot analysis from paired (Sc and Om) fat tissue samples of 9 persons; ACTB was used as a loading control. (D) Paired (Sc and Om) fat tissue samples from 15 persons were fractioned and mRNA levels of E2F1 and MKI67 were measured separately in adipocyte fraction and in SVF. *, P<0.05 versus SVF.
E2F1 overexpression activates autophagy gene promoter activity and sensitizes cells to stress-induced autophagy
E2F1 has been shown in cancer cells to activate the gene expression of several major autophagy genes.22,34 Yet, the interaction of E2F1 overexpression with a proinflammatory environment, as occurs in visceral adipose tissue in obesity, hasn't been previously explored. To functionally assess the effect of E2F1 overexpression on basal and cytokine-stimulated promoter activity of several autophagy genes, we utilized dual luciferase assay in HEK293 cells. These cells were utilized given the inherent difficulty in overexpression studies in adipocytes. Although limited, this approach as part of transcriptional regulation studies in adipocytes is common and has been used in multiple previous publications.42-46 Cells were transfected with plasmids encoding different human autophagy gene promoters (BECN1 [Beclin 1, autophagy related], ATG7 [autophagy-related 7], MAP1LC3B, ATG12 and DRAM1) governing the expression of luciferase, along with constitutive (RSV promoter) expression of Renilla luciferase (Fig. 4A). In the absence of E2F1 overexpression, TNF affected the promoter activity only of DRAM1 (Fig. 4A). E2F1 overexpression resulted in significant promoter activation of all autophagy genes excluding BECN1 (Fig. 4A). Yet, in addition, E2F1 overexpression significantly further activated the promoter activity of ATG12, MAP1LC3B and DRAM1 in response to TNF (Fig. 4A), suggesting a “TNF-sensitizing effect.” E2F1 overexpression sensitized HEK293 cells to TNF-induced autophagy as reflected by LC3-positive punctate assays, both by indirect immunofluorescence and by direct fluorescence microscopy in cells expressing EGFP-LC3 (Fig. S1A, S1B). In addition, since LC3-associated vesicles, which are not autophagosomes, have been described,47,48 we verified by electron microscopy that in E2F1-overexpressing cells TNF induced the generation of double-membrane vesicles that include diverse cytosolic constituents, consistent with autophagosomes (data not shown). Finally, we determined if, consistent with E2F1's sensitizing effect on TNF-induced autophagy, gene promoter activation translated into activated autophagic flux. Western blot analyses of LC3B-II and SQSTM1/p62 (sequestosome 1) accumulation with bafilomycin A1 (BafA1) revealed that E2F1 overexpression elevated autophagic flux in response to TNF. Similar sensitization was seen in response to oxidative stress induced by continuous H2O2 production in the medium (achieved by adding glucose oxidase). Jointly, overexpression of E2F1 not only activates autophagy gene promoter activity (as previously demonstrated), but may also sensitize cells to activation of autophagy in response to inflammatory or oxidative stress.
Figure 4.
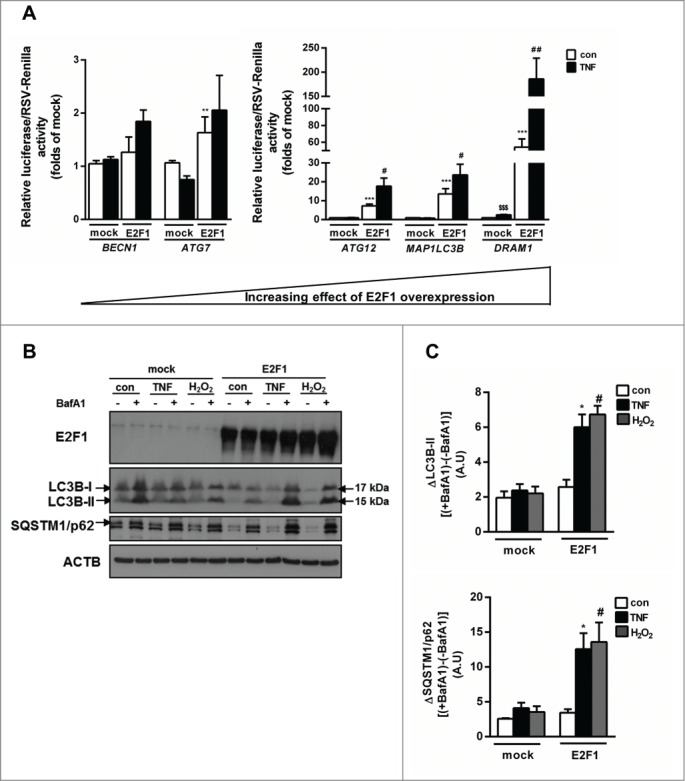
Overexpression of E2F1 increases autophagy gene expression and sensitizes cells to inflammation-induced autophagy. (A) HEK293 cells were transfected with pGL3-BECN1-luc, pGL3-ATG7-luc or similarly, MAP1LC3B, ATG12 or DRAM1-luc, and an empty pcDNA3 (mock), or a plasmid encoding wild-type E2F1. After normalization to endogenous control (RSV-Renilla), mock was determined as luciferase basal activity. Autophagy genes promoter activity was measured after treatment with TNF (10 ng/ml) for 24 h using Dual Luciferase Assay. (B) Autophagic flux measurements were performed in HEK293 cells as above, treated with or without BafA1 (0.1 µM), or H2O2 generated by adding 10 mU/ml glucose oxidase to the medium, and lysates were blotted for the autophagy client proteins LC3B-I/II and SQSTM1/p62. (C) Densitometry analysis of autophagic flux (delta in LC3B-II or SQSTM1/p62 in each condition between the BafA1 treated and untreated cells) in 3 independent experiments. *, P<0.05; **, P<0.005; ***, P<0.001 versus mock con. #, P<0.05; ##, P<0.01 versus mock TNF. $$$, P<0.001 versus mock con.
E2F1 and adipocyte autophagy—loss of function approach
To challenge a role for E2F1 overexpression in the regulation of autophagy gene expression and function in adipocytes, we utilized MEFs of wild-type (WT) and e2f1 knockout mice (e2f1−/−), which were differentiated into adipocyte-like cells. Previously, E2F1 was reported to be required for adipogenesis.28 Thus, we first assessed whether in vitro, using a stringent protocol to promote adipogenesis (see Materials and Methods for detail), we could achieve comparable adipogenesis of WT and e2f1−/− MEFs. Cellular morphology and lipid accumulation were determined using oil red O neutral lipid staining. MEFs from both WT and e2f1 knockout mice exhibited significant morphological changes typical of adipogenesis, including rounding up of the cells and massive lipid droplet accumulation (Fig. 5A). Although by phase contrast microscopy, differentiated e2f1−/− MEFs seemed to develop a smaller number of large lipid droplets, oil red O staining demonstrated similar degrees of lipid accumulation (Fig. 5A), which was verified in 3 independent experiments, in which MEFs were obtained from 2 different mice of each strain. Furthermore, e2f1−/− MEFs were indistinguishable from WT MEFs in the induction of a set of adipogenic and adipocyte-specific genes, including Pparg (peroxisome proliferator-activated receptor gamma), Cebpa (CCAAT/enhancer binding protein [C/EBP], α), Fabp4/aP2 (fatty acid binding protein 4, adipocyte) and Slc2a4 (solute carrier family 2 [facilitated glucose transporter], member 4) (Fig. 5B).
Figure 5.
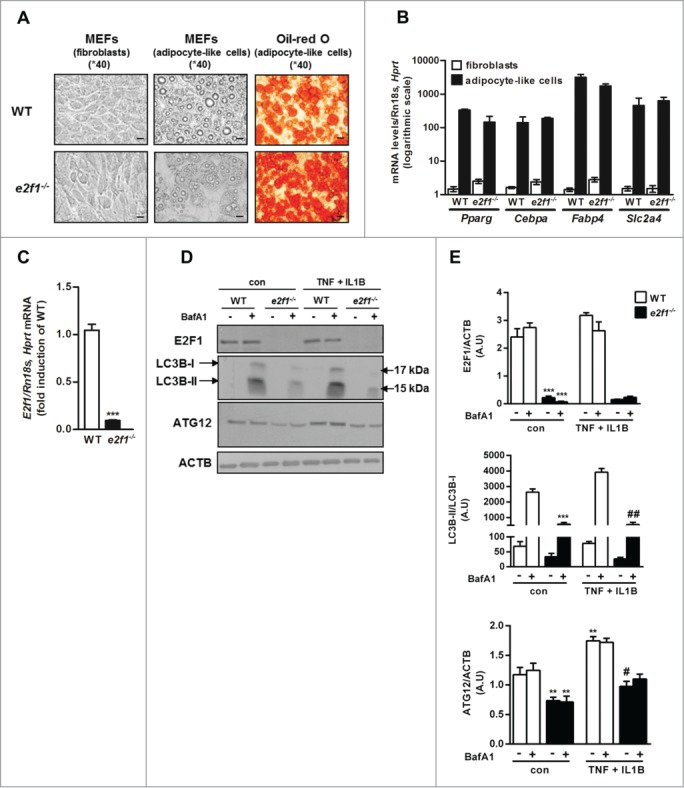
Attenuated basal and stimulated autophagy in e2f1−/− MEF-derived adipocytes. (A) Differentiation of primary mouse embryonic fibroblasts (MEFs) from WT or e2f1 knockout mice into adipocyte-like cells. Confluent MEFs were exposed to differentiation medium for 14 d. At the end of differentiation period adipocyte-like cells were stained with Oil red O, and representative images were obtained at 40X magnification by light microscopy. Scale bar: 20 µm. (B) Quantitative real-time PCR analysis of 5 adipogenic genes in fibroblasts or adipocyte-like cells from WT and e2f1−/−. Each transcript expression was normalized to Rn18s and Hprt/Hprt1 (hypoxanthine guanine phosphoribosyl transferase) mRNA levels. (C) Quantitative real-time PCR analysis of E2f1 mRNA levels in WT versus e2f1−/−. (D) e2f1−/− and WT adipocyte-like cells were treated, in full (serum-containing) medium, with TNF (10 ng/ml) + IL1B (10 ng/ml) for 24h and subjected to western blot analysis. Shown are representative blots of n=3 independent experiments, with their densitometry analysis (E). ACTB was used as a loading control. ***, P<0.001 (C) versus WT. **, P<0.05; ***, P<0.005 versus con WT. #, P<0.05; ## P<0.01 versus TNF + IL1B WT.
We next assessed how the absence of E2F1 affects autophagy gene expression and autophagic flux. e2f1−/− MEF-derived adipocytes were confirmed to express no E2f1 mRNA or protein levels, as expected (Fig. 5C, 5D to E, respectively). Serum and amino acid starvation (24 h), classical inducers of autophagy, increased Map1lc3b and Atg5 mRNA by 2.45±0.12 and 1.42±0.09 fold in WT MEF-adipocytes, and this response was significantly diminished in e2f1−/− MEF adipocytes (1.81±0.1 and 0.85±0.03 fold increase for Map1lc3b and Atg5 mRNA, respectively, both P<0.005 compared to their respective change in WT-MEF-adipocytes). At the protein level, in e2f1−/− MEF-derived adipocytes the autophagy-related proteins LC3B-I/-II and ATG12 were downregulated (Fig. 5D, 5E). Moreover, the lack of E2F1 markedly attenuated autophagic flux, as detected by the effect of BafA1 on LC3B-II. This was apparent both in the basal (unstimulated) state of the MEF-derived adipocytes, as well as in a “metainflammatory state” induced by treating the cells with TNF and IL1B, 2 prototypical proinflammatory cytokines implicated in adipose tissue inflammation in obesity (Fig 5D, 5E). Collectively, in adipocyte-like cells, the absence of E2F1 attenuated the expression of autophagy genes and markedly decreased autophagic flux in both basal state and in response to inflammatory stimulus.
To gain insight into the functional impact of absence of E2F1 in adipocytes, we further determined the metabolic and endocrine function of the cells, emphasizing functions most pertinent to changes known to occur in obesity. We again considered both basal state conditions, and the effect of “metainflammatory” environment mimicked by TNF and IL1B. To assess insulin response in the cells, MEF-derived adipocytes were acutely stimulated with submaximal (0.5 nM) and maximal (100 nM) insulin. e2f1−/− cells were more insulin-sensitive as determined by the effect of insulin on AKT phosphorylation (Fig. 6A, B). In WT MEF-derived adipocytes we could not detect an insulin-stimulated increase in GSK3B (glycogen synthase kinase 3 β) and RPS6KB1 (ribosomal protein S6 kinase, 70kDa, polypeptide 1) phosphorylation by 7 min of insulin stimulation, but this response was discernible in e2f1−/− cells (Fig. 6A, B).
Figure 6.
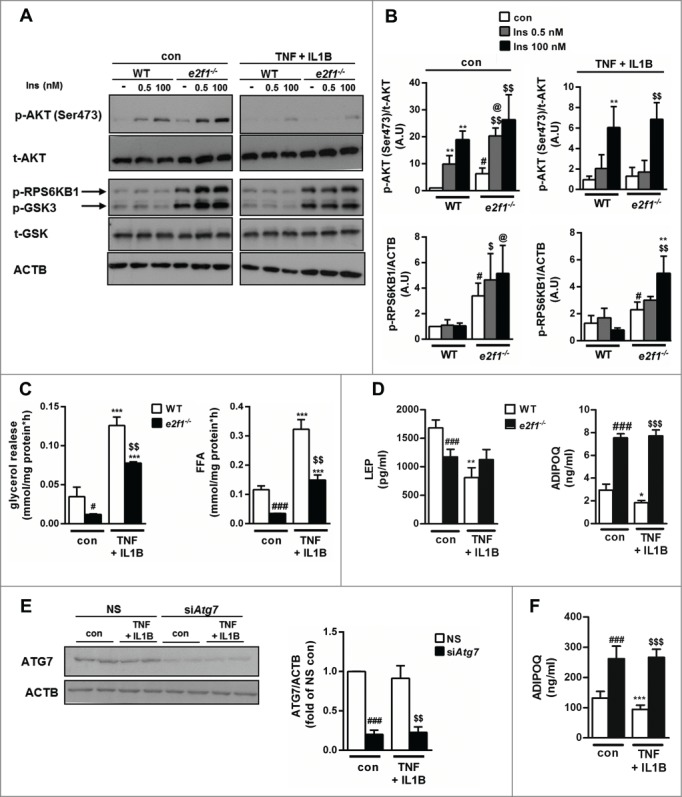
Metabolic and endocrine effects of E2F1 depletion in e2f1−/− adipocyte-like cells. Confluent MEFs were exposed to differentiation medium for 14 d. At the end of differentiation period e2f1−/− and WT adipocyte-like cells were treated with inflammatory stress inducers (TNF 10 ng/ml+IL1B 10 ng/ml) for 24 h. (A) To assess insulin signaling in e2f1−/− and WT adipocyte-like cells, they were treated with inflammatory stress inducers (TNF 10 ng/ml + IL1B 10 ng/ml) for 21 h, then serum starved (in the presence of the cytokines, as indicated) for 3 h, and then incubated with insulin (0.5 and 100 nM) for 7 min to assess insulin signaling. Shown are representative blots from western blot analysis of n=3 for the p-AKT, and for the p-RPS6KB1 and p-GSK3B n=2 independent experiments yielding identical results. Densitometry of p-AKT (Ser473) to total AKT and p-RPS6KB1 to ACTB ratios are shown in graphs next to the blots (B). (C) To evaluate lipolysis in e2f1−/− and WT adipocyte-like cells, they were treated as above, medium was changed for 1 h to KRBH buffer and lipolysis was measured by evaluating glycerol and FFA release to the medium. (D) To measure secreted levels of LEP and ADIPOQ, medium was analyzed by ELISA and corrected for cell protein concentrations. Protein concentrations of cell lysates were used to normalize the results. (E) Differentiated epididymal preadipocyte cell line was electroporated with either nonspecific sequence (NS) or with siRNA targeting Atg7. Cells were then treated, as above, with TNF (10 ng/ml) + IL1B (10 ng/ml), and lysates prepared to assess the efficiency of Atg7 knockdown. Densitometry is from 5 independent experiments. (F) ADIPOQ secretion to the medium of adipocytes without or with Atg7 siRNA-mediated knockdown was assessed as described above (in D). ADIPOQ secretion to the medium of adipocytes without or with Atg7 siRNA-mediated knockdown was assessed as described above (in D). *, P<0.05; **, P<0.005; ***, P<0.005 versus con WT/NS. #, P<0.05; ###, P<0.001 versus con WT/NS. $$, P<0.005; $$$, P<0.001 versus con e2f1−/−/siAtg7. @, P<0.05 versus Ins 0.5 WT.
In response to TNF and IL1B, e2f1−/− MEF-adipocytes exhibited a similar decline in insulin's capacity to stimulate signaling events, exhibiting a 4- and 2-fold decrease in insulin-stimulated AKT and GSK3B phosphorylation, respectively (Fig. 6A, B). We next assessed the effect of E2F1 absence on basal lipolysis. Adipocyte-like cells lacking E2F1 exhibited a markedly lower lipolytic flux, as indicated by both glycerol and FFA release into the medium. In response to TNF + IL1B, lipolysis rate significantly (>3-fold) increased, both in WT and e2f1−/− MEF-adipocytes (Fig. 6C). Yet, the level of FFA release by the e2f1−/− adipocytes stimulated by inflammatory cytokines was equivalent to the basal, unstimulated lipolysis in WT cells (Fig. 6C). Finally, adipocyte endocrine function was assessed by measuring LEP and total ADIPOQ secretion to the medium. e2f1−/− adipocyte-like cells secreted significantly less LEP (Fig. 6D). Importantly, in response to TNF and IL1B, WT MEF-adipocytes decreased the secretion of LEP, whereas e2f1−/− MEF-adipocytes were resistant to this effect of inflammatory cytokines. The lower LEP secretion by e2f1−/− MEF-adipocytes likely did not indicate a lower degree of adipocyte differentiation of these cells compared to the WT-MEF adipocytes, since the e2f1−/− MEFs secreted nearly 3-fold higher ADIPOQ levels (Fig. 6D). Interestingly, as was the case with LEP, in the absence of E2F1, cells were resistant to the ability of these cytokines to decrease ADIPOQ secretion (Fig. 6D). To determine if the metabolic and endocrine phenotype of e2f1−/− adipocytes could be mediated by lowered autophagy, we used siRNA-mediated knockdown of Atg7 in a differentiated epididymal preadipocyte cell-line.49 Atg7 knockdown efficiency was 80% as indicated by protein content (Fig. 6E), and resulted in a significant decrease in autophagic flux (52±11% decrease in delta SQSTM1/p62 with BafA1A, P<0.001, n=5 independent experiments). Remarkably, similarly to e2f1-KO MEF-derived-adipocytes, siRNA-mediated Atg7-knockdown cells exhibited a ˜2-fold increase in ADIPOQ secretion (Fig. 6F). Moreover, unlike cells treated with control siRNA (NS), cells were resistant to the ADIPOQ -lowering effect of the inflammatory cytokines (Fig. 6F). Collectively, these in vitro data on E2F1-depleted adipocyte-like cells are consistent with the clinical associations between E2F1 expression and circulating LEP, ADIPOQ, FFA levels, and insulin resistance (Fig. 1B, 1C and 1D). The similar effects observed in Atg7-knockdown adipocytes suggest a functional role for downregulated autophagy, which resembles the lack of E2F1. Moreover, resistance of e2f1−/− MEFs or Atg7-knockdown adipocytes to a cytokine-induced decrease in ADIPOQ secretion supports the “stress-sensitizing effect” of E2F1 overexpression (Fig. 4).
Discussion
Recent studies implicate proteins regulating the cell cycle in ‘non-cell-cycle-related functions', including in metabolism.33,50,51 In the present study we hypothesized that the transcription factor E2F1 may regulate autophagy gene expression in adipose tissue in obesity, and associate with cardio-metabolic risk. Our main findings are that E2F1 is upregulated in human adipose tissue in obesity, particularly in visceral fat of persons with the more dysmetabolic intraabdominal (visceral) adiposity. It correlates with indicators of obesity-associated dysmetabolism and inflammation, and with gene expression of several key autophagy genes (but not with cell proliferation markers). Statistical models could suggest E2F1 and autophagy genes on a joint path, and strikingly, ChIP studies could demonstrate in Om adipose tissue explants BMI-associated, elevated occupancy of the MAP1LC3B promoter by E2F1. In cells, as previously shown, overexpression of E2F1 activates promoter activity of several autophagy genes; yet, it also sensitizes cells to TNF-induced hyperactivation of ATG12, MAP1LC3B, and DRAM1 promoter activity. In adipocyte-like cells from e2f1 knockout mice, autophagy is downregulated in both basal and stimulated conditions, and their endocrine function is resistant to the disrupting effect of inflammatory cytokines.
Most studies on the regulation of autophagy frequently consider post-transcriptional modes of regulation.52 Here we propose a potential major contribution for regulation of autophagy at the gene expression level, particularly in genes involved in the later stages of autophagy (ATG12, MAP1LC3B and DRAM1), with lesser effect on genes that are involved in initiation of the process (BECN1 and ATG7). Interestingly, a similar mode of differential regulation of autophagy genes, favoring genes involved in later stages of the process, was reported for TP53 (tumor protein p53, note that the mouse nomenclature is TRP53, but we use TP53 hereafter to refer to both the human and mouse genes/proteins for simplicity) and HEY2 (hes-related family bHLH transcription factor with YRPW motif 2).53-55 Complementarily, while JUN and NR1H4/FXR (nuclear receptor subfamily 1, group H, member 4) may contribute preferentially to genes involved in autophagy initiation, TP63 (tumor protein p63), STAT3 (signal transducer and activator of transcription 3 [acute-phase response factor]) and FOXO3 (forkhead box O3) may regulate different autophagy genes without an obvious preference.56-66 Beyond the above, a novel aspect of our finding was that E2F1 sensitizes to the effects of proinflammatory cytokines: Functionally, in its absence, adipocyte-like cells do not diminish secretion of LEP and ADIPOQ in response to stimulation by TNF and IL1B. When overexpressed, E2F1 sensitizes to TNF-induced activation of promoter activity of ATG12, MAP1LC3B, and DRAM1, as well as to TNF-induced expression of the stress kinase MAP3K5 (mitogen-activated protein kinase kinase kinase 5, unpublished data), and autophagic flux. Further studies should unravel the molecular basis for this E2F1 inflammatory interaction in regulating autophagy genes. Inflammatory stress increases mRNA expression of ATG5 and ATG7 by the family of SMADs transcription factors in human hepatocellular-carcinoma cells.67 Similarly, mRNA levels of MAP1LC3B, ATG5, ATG12, ATG4B (autophagy-related 4B, cysteine peptidase) and BECN1 are elevated in response to oxidative stress by EGR1 (early growth response 1), FOXO1/2/3/4, and STAT3 in skeletal muscle cells, cardiomyocytes and hepatocytes.58,60,62,68-71 Thus, E2F1 may interact with other transcription factors to mediate transcriptional regulation of autophagy genes in response to the inflammatory environment of adipose tissue in obesity.
If extended to the clinical arena, these putative cellular and molecular interactions may be highly relevant: E2F1 is mainly increased in expression in omental adipose tissue of viscerally obese persons. This obesity phenotype is frequently associated with intraabdominal fat inflammation, suggesting that E2F1 overexpression frequently co-occurs in an inflammatory environmental milieu. Indeed, Om-E2F1 mRNA strongly associated with lower circulating ADIPOQ, higher LEP and FFA, and this association was markedly attenuated by further adjustment of the model to autophagy gene expression in the tissue. Moreover, E2F1 association with ATG5 was markedly attenuated by adjusting for circulating IL6, another indicator of obesity-related systemic inflammation (unpublished data). Jointly, these observations suggest a putative path leading from adipose E2F1, both directly and by sensitization to inflammatory pathways, via autophagy activation, to adipocyte dysfunction, which would occur in an obese proinflammatory environment.
Although mature adipocytes are largely considered ‘postmitotic’, there is increasing interest in cellular proliferation within adipose tissue in this tissue's adaptation to the obese state, involving diverse cell populations as preadipocytes, mesenchymal stem cells, immune cells and vascular cells.41,72 Our results suggest that E2F1, most studied as a cell-cycle and proliferation regulator, may not necessarily be primarily regulating these functions in human adipose tissue in obesity: First, while E2F1 mRNA or protein levels correlate with the expression of autophagy genes, this is not similarly observed with either proliferation markers like MKI67 or PCNA, nor with classical E2F1 target genes that are involved in cell cycle regulation, such as CDK4. Consistently, while BMI strongly correlates with E2F1 binding to the promoter of MAP1LC3B in omental human fat, such an association is not observed with CCND1. Additionally, while the increased expression of E2F1 in human adipose tissue in obesity could be attributed to the adipocyte cell fraction, MKI67 was mostly elevated in the stromal-vascular fraction. Thus, it is tempting to suggest that E2F1 overexpression may result in binding to low-affinity targets within the genome, such as autophagy genes, finally resulting in adipocyte dysfunction, including insulin resistance, elevated lipolysis, and altered adipokine release. All these are common features of dysfunctional adipose tissue in obesity.
Taken together, we propose that adipose tissue expression of E2F1 plays a key role in linking obesity with adipose tissue dysfunction, potentially by upregulating autophagy gene expression directly and by sensitizing cells to inflammation. Collectively, our results provide further support for the growing notion that classical cell-cycle regulation players may have “nonclassical” functions, contributing in this case to obesity-associated morbidity in humans.
Materials and Methods
Study population
Participants were from 2 cohorts, one in Beer-Sheva, Israel, and one in Leipzig, Germany, and adipose tissue biopsies were analyzed here exactly as described in previous publications.13,35-37,73 All procedures were approved, in advance, in both institutions separately, by the ethics committees of the 2 centers, and were conducted in accordance with Declaration of Helsinki guidelines. In brief, participants were recruited in both centers before undergoing abdominal surgery (primarily bariatric surgery and elective cholecystectomy) after providing written informed consent. Paired subcutaneous (Sc) and omental (Om) adipose tissue biopsies were obtained during surgery and immediately delivered to the laboratory where they were processed for mRNA or protein expression or chromatin immunoprecipitation (ChIP) studies, using methodology coordinated between the 2 centers.
Materials
Tissue culture medium (DMEM 01-055-1A, 01-170-1A), heat-inactivated fetal bovine serum (04-121-1A), antibiotic solutions (03-033-1B), L-glutamine solutions (03-020-1B), human recombinant insulin (01-818-1H), phosphate-buffered saline (PBS; 02-023-1A) and Earle's balanced salts solution (02-010-1A) were obtained from Biological Industries (Beit-Haemek, Israel). Indomethacin (I7378), dexamethasone (D4902), 3-isobutylmethylxanthine (IBMX; I7018), rosiglitazone (R2408) were obtained from Sigma-Aldrich. Recombinant murine/human TNF (410-MT and 210-TA, respectively) and murine IL1B (401-ML) were purchased from R&D Systems Inc.
Cell culture
Human embryonic kidney (HEK)293T cells (ATCC, ACS-4500) were grown in DMEM containing 4.5 mM glucose, 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml penicillin-streptomycin. Medium was changed every other day. For western blot analysis experiments, HEK293 were treated with TNF (10 ng/ml) or glucose oxidase (10 mU/ml; Sigma-Aldrich, G6125) without or with BafA1 (0.1 μM; LC Laboratories, BNB-1080) for 24 h. e2f1−/− and WT MEFs were kindly provided by Prof. Gustavo Leone (Department of Molecular Virology and Genetics, College of Medicine and Public Health, Ohio State University, Columbus, OH). MEFs were cultured in DMEM containing 4.5 mM glucose, 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin-streptomycin. Medium was changed every other day. Differentiation into adipocytes was performed as follows: MEFs were grown until 24 h postconfluence and differentiation was induced by adding 1 μM dexamethasone, 0.5 mM IBMX, 10 μg/ml of insulin and 10 μM of rosiglitazone to the medium for 3 d, after which cells were grown for an additional 10 d before being used. For quantitative real-time PCR experiments, adipocyte-like MEFs were serum and amino acid-starved 24 h, as indicated. For western blot analyses, adipocyte-like MEFs were treated with TNF (10 ng/ml) + IL1B (10 ng/ml) for 20 h and then BafA1 (0.1 μM) was added for additional incubation of 4 h. For lipolysis assessment, insulin signaling and LEP- ADIPOQ concentration measurements adipocyte-like MEFs were treated with TNF (10 ng/ml) + IL1B (10 ng/ml) for 20 h. Epididymal preadipocytes were grown and differentiated into mature adipocytes as previously described.49 Briefly, preadipocytes were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 4.5 g/L glucose, 20% FBS, 2 mM L-glutamine, 100 U/ml penicillin-streptomycin, 20 nM insulin, 1 nM triiodothyronine (Sigma-Aldrich, IRMM469). Medium was changed every other day. Differentiation into adipocytes was performed as follows: preadipocytes were grown until 24 h postconfluence and differentiation was induced by adding 0.125 mM indomethacin, 2 μg/ml dexamethasone, and 0.5 mM IBMX to the medium for 24 h, after which cells were grown for an additional 6 d before being used. For western blot analyses, differentiated epididymal preadipocytes were treated with TNF (10 ng/ml) + IL1B (10 ng/ml) for 24 h.
RNA extraction and quantitative real-time PCR
Total RNA from MEFs adipocyte-like cells was extracted using the RNeasy lipid tissue minikit (Qiagen, 74804) and quantified using nanodrop. Then 2 μg of RNA were reverse-transcribed with a high capacity cDNA reverse transcriptase kit (Life Technologies, 4374966). Taqman system (Life Technologies, 4369016) was used for real-time PCR amplification. Extraction of total RNA from paired human adipose tissue biopsies (Leipzig cohort) was done according to a previously described protocol.36 Relative gene expression was obtained after normalization to endogenous control genes, using the formula 2−ΔΔCt and specific primers (Table S1).
Cell lysates and western blot analysis
After treatments, mature adipocyte-like MEFs or HEK293 were rinsed 3 times with ice-cold PBS and scraped in an ice-cold lysis buffer containing 50 mM Tris-HCl, pH 7.5, 0.1% (w/v) Triton X-100 (Sigma-Aldrich, X100) 1 mM EDTA, 1 mM EGTA, 50 mM NaF, 10 mM sodium β-glycerophosphate, 5 mmol/liter sodium pyrophosphate, 1 mM sodium vanadate, 0.1% (v/v) 2-mercaptoethanol, and inhibitors (a 1:1,000 dilution of protease inhibitor cocktail; Sigma-Aldrich, P8340). The lysates were shaken for 20 min at 4°C, centrifuged (12,000 x g, 20 min at 4°C), and the supernatant fraction was collected. Preparation of total cell lysates from paired human adipose tissue biopsies was previously described.13,36 Protein concentration was determined using the BCA protein assay (Pierce, 23227). Protein samples were resolved on 10% SDS-polyacrylamide gel electrophoresis and subjected to western blot using specific antibodies (Table S2), followed by quantitation as described previously using ImageGauge 4.0 software (Ver.4.0, Fuji Photo Film).
Atg7 knockdown by small interfering RNA (siRNA) transfection
Atg7 and nonspecific sequence control (NS) siRNAs ON-TARGET plus smart pools were obtained from Dharmacon (Thermo Fisher Scientific Inc., Waltham, MA). Each siRNA was transfected into differentiated epididymal preadipocyte cell line at a concentration of 1 nmol/well by electroporation with GenePulser Xcell (Bio-Rad Laboratories Inc., Hercules, CA). Briefly, epididymal adipocytes were trypsinized on the third (of 6 d) d of differentiation process from 150-mm culture plate dish. Cells were rinsed 3 times with PBS, centrifuged (5 min, 890 x g, at room temperature) after last wash and supernatant was aspirated. Next, cells were suspended in 400 µl of Ingenio™ Electroporation Solution (Mirus Bio LLC, MC-MIR-50114) with 4 nmol (for 4 wells) of Atg7 or NS siRNA and electroporation was performed in accordance with the manufacturer's instructions using the optimal program (170 V, 10 ms, 2 pulses). After electroporation, cells were cultured in complete medium (DMEM, 20% fetal calf serum [FCS]). At 24 h post electroporation, cells were incubated with TNF (10 ng/ml) + IL1B (10 ng/ml) in DMEM 10% FBS for additional 24 h. Cell culture media was collected and ADIPOQ levels were evaluated using ELISA kit as described. Then cells were rinsed 3 times with ice cold PBS and subjected to western blot analysis.
Transient transfection and promoter activity assay
HEK293 cells were seeded in 24-well plates in 1 ml/well of DMEM 10% FCS. 24 h later, medium was changed to fresh 0.5 ml/well, and cells were transfected using jetPEI reagent (Poly-plus transfection, 101-01N), according to the manufacturer's instructions. Briefly, 200 ng/well of the expression plasmid (pCMV-empty/E2F1), 200 ng/well of the firefly luciferase reporter plasmids (Promega Corporation, E-1761) containing BECN1, ATG7, ATG12, DRAM1 or MAP1LC3B, and 2 ng/well of the Renilla luciferase reporter plasmid were used for each transfection. Four h after transfection, the medium was changed to fresh DMEM 10% with or without TNF (10 ng/ml). Cells were lysed 24 h after incubation with or without TNF 10 ng/ml treatment by applying 100 µl of Passive Lysis Buffer of the Dual Luciferase Reporter Assay Kit (Promega Corporation, E-1910) into each well of the 24-well plate. 20 µl of cell lysate were used for the luciferase reporter assay with the same kit, according to the manufacturer's protocol. Luminescence intensity was quantified in a GloMax 20/20n Luminometer (Promega Corporation, Madison, WI). The experiments were performed at least in triplicate. As a control for transfection efficiency, the firefly luciferase activity values were normalized to the Renilla luciferase activity values.
LEP and ADIPOQ concentrations
LEP and ADIPOQ were measured in cultured media with or without TNF (10 ng/ml) + IL1B (10 ng/ml) treatment for 20 h by ELISA (R&D Systems Inc, MOB00 and MRP300, respectively).
Lipolysis assay
e2f1−/− or WT MEF adipocyte-like cells were incubated without or with TNF (10 ng/ml) + IL1B (10 ng/ml) treatment for 20 h in DMEM 10% FCS. Next, cells were washed 3 times with PBS and incubated with Krebs-Ringer buffer (KRBH; 135 mM NaCl, 5 mM KCl, 1 mM MgSO4, 0.4 mM K2HPO4, 1 mM CaCl2, 20 mM HEPES, pH 7.4) for 1 h. Glycerol release was measured in KRBH buffer using Free Glycerol reagent (Sigma-Aldrich, F6428) and free fatty acids release was measured by NEFA-HR2 kit (Wako Chemicals, 999-34691). Results were normalized to mg protein cell lysate.
Oil red O staining for neutral lipids
In vitro differentiated e2f1−/− and WT MEFs into adipocyte-like cells were fixed with 4% formaldehyde for 30 min, then rinsed 3 times with 70% ethanol and stained with oil red O (Sigma-Aldrich, O0625) for 45 min. After staining, cells were washed 5 times with ddH2O, and lipid droplets were visualized under a light microscope (Nikon, ECLIPSE 80i, Melville, NY).
Chromatin immunoprecipitation
ChIP from fresh human adipose tissue explants was performed following a recently described protocol.40 Briefly, 1 gram of Sc or Om adipose tissue was minced into small pieces using 2 pairs of sterile scissors. Next, samples were cross-linked with 1% of formaldehyde at 37°C for 8 min. Cross-linking reaction was quenched using 0.125 M glycine. Samples were washed with ice-cold PBS 3 times, and following the last wash adipocyte lysis buffer was added. After 15 min of incubation on ice, samples were homogenized using a Dounce homogenizer (loose pestle, 20 strokes, Wheaton). Nuclei were released after 20 additional strokes using a tight pestle. After sonication, the protein-DNA complexes were immunoprecipitated using anti-ICAM1 (Santa Cruz Biotechnology, sc-7891) as a negative control, anti-POLR2/RNA polymerase 2 (Santa Cruz Biotechnology, sc-21750) as positive control) and anti-E2F1 antibodies (Santa Cruz Biotechnology, sc-193). After cross-linking reversal at 65°C for 4 h, DNA was purified using phenol-chloroform.74 Primers (5' to 3') for end-point PCR or quantitative real time PCR were as follows: MAP1LC3B- Fw - TGGAGGGGAAAGGATGGTCG, Rv – GGGGCGGAGCAGGTGTGTG. CCND1 – Fw – TAGAACAGGAAGATCGGAC, Rv – AGGGCCGCAAACCGCGGGC. Input (diluted 1/300) was used as a normalizing control.
Statistical analysis
Data are expressed as the mean ± SD (in Figs. 4–6) and SEM (in Fig. 1, Fig. 3, and Tables) and calculations were performed using GraphPad software. Statistically significant differences between 2 groups were evaluated using paired or unpaired Student t test, as indicated in the figure legends. Comparison of >2 groups was analyzed by ANOVA with Tuckey post-hoc analysis. Correlation between BMI and E2F1 binding to MAP1LC3B promoter was assessed by linear regression. Clinical and biochemical characteristics of the study populations were cross-classified across quintiles of visceral (Om) E2F1 mRNA expression in the Leipzig cohort and across quintiles of visceral (Om) E2F1 protein expression in Beer Sheva cohort and P of trend were calculated across quintiles, using SPSS statistical package.
All study protocols have been approved by the ethics committee of Soroka Academic Medical Center and Faculty of Health Sciences, Ben-Gurion University of the Negev (approval number 920061836) and the University of Leipzig (Registration number: 031-2006; 363-10-13122010; 017-12-23012012). All participants gave written informed consent before taking part in the 2 cohorts.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank the following colleagues for generously providing us with valuable tools to perform this study: Prof. Gustavo Leone, Department of Molecular virology and Genetics, College of Medicine and Public Health, Ohio State University, Columbus, OH, USA for the e2f1 knockout and WT- MEFs; Dr. Kenichi Yoshida, Department of Life Sciences, Meiji University, Kanagawa, Japan, and Prof. Eun-Kyeong Jo, Department of Microbiology, Chungnam National University School of Medicine, Daejeon, Korea for autophagy luciferase plasmids.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This study was supported in part by grants from the Deutsche Forschungsge-meinschaft (DFG): SFB 1052/1: “Obesity mechanisms” (project B2 to A.R., project B1 to M.B., and project B4 to N.K.), and the Israel Science Foundation (ISF 928/14 to A.R.).
References
- 1.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013; 368:651-62; PMID:23406030; http://dx.doi.org/ 10.1056/NEJMra1205406 [DOI] [PubMed] [Google Scholar]
- 2.Barnard RA, Wittenburg LA, Amaravadi RK, Gustafson DL, Thorburn A, Thamm DH. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy 2014; 10:1415-25; PMID:24991836; http://dx.doi.org/ 10.4161/auto.29165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, Mita AC, Curiel TJ, Espitia CM, Nawrocki ST, et al.. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014; 10:1403-14; PMID:24991835; http://dx.doi.org/ 10.4161/auto.29231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poklepovic A, Gewirtz DA. Outcome of early clinical trials of the combination of hydroxychloroquine with chemotherapy in cancer. Autophagy 2014; 10:1478-80; PMID:24991829; http://dx.doi.org/ 10.4161/auto.29428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, Schuchter LM, Torigian DA, Panosian JT, Troxel AB, et al.. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014; 10:1391-402; PMID:24991838; http://dx.doi.org/ 10.4161/auto.29119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, Kramer A, Tan KS, Heitjan DF, Rodgers G, Gallagher M, et al.. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014; 10:1369-79; PMID:24991839; http://dx.doi.org/ 10.4161/auto.29118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, et al.. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014; 10:1359-68; PMID:24991840; http://dx.doi.org/ 10.4161/auto.28984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, Rangwala R, Piao S, Chang YC, Scott EC, et al.. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014; 10:1380-90; PMID:24991834; http://dx.doi.org/ 10.4161/auto.29264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ, Yousem SA, et al.. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PloS one 2008; 3:e3316; PMID:18830406; http://dx.doi.org/ 10.1371/journal.pone.0003316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest 2009; 119:3329-39; PMID:19855132; http://dx.doi.org/ 10.1172/JCI35541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci U S A 2009; 106:19860-5; PMID:19910529; http://dx.doi.org/ 10.1073/pnas.0906048106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kovsan J, Bashan N, Greenberg AS, Rudich A. Potential role of autophagy in modulation of lipid metabolism. Am J Physiol Endocrinol Metab 2010; 298:E1-7; PMID:19887596; http://dx.doi.org/ 10.1152/ajpendo.00562.2009 [DOI] [PubMed] [Google Scholar]
- 13.Kovsan J, Bluher M, Tarnovscki T, Kloting N, Kirshtein B, Madar L, Shai I, Golan R, Harman-Boehm I, Schön MR, et al.. Altered autophagy in human adipose tissues in obesity. J Clin Endocrinol Metab 2011; 96:E268-77; PMID:21047928; http://dx.doi.org/ 10.1210/jc.2010-1681 [DOI] [PubMed] [Google Scholar]
- 14.Ost A, Svensson K, Ruishalme I, Brannmark C, Franck N, Krook H, Sandström P, Kjolhede P, Strålfors P. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol Med 2010; 16:235-46; PMID:20386866; http://dx.doi.org/ 10.2119/molmed.2010.00023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jansen HJ, van Essen P, Koenen T, Joosten LA, Netea MG, Tack CJ, Stienstra R. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology 2012; 153:5866-74; PMID:23117929; http://dx.doi.org/ 10.1210/en.2012-1625 [DOI] [PubMed] [Google Scholar]
- 16.Stienstra R, Haim Y, Riahi Y, Netea M, Rudich A, Leibowitz G. Autophagy in adipose tissue and the β cell: implications for obesity and diabetes. Diabetologia 2014; 57(8):1505-16; PMID:24795087 [DOI] [PubMed] [Google Scholar]
- 17.Maixner N, Kovsan J, Harman-Boehm I, Bluher M, Bashan N, Rudich A. Autophagy in adipose tissue. Obes Facts 2012; 5:710-21; PMID:23108431; http://dx.doi.org/ 10.1159/000343983 [DOI] [PubMed] [Google Scholar]
- 18.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 2010; 11:467-78; PMID:20519119; http://dx.doi.org/ 10.1016/j.cmet.2010.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res 2010; 20:748-62; PMID:20548331; http://dx.doi.org/ 10.1038/cr.2010.82 [DOI] [PubMed] [Google Scholar]
- 20.Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S, Sun X, Yoon G, Kang Y, Zhong W, et al.. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 2014; 516:108-11; PMID:25383523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, Moore DD. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014; 516:112-5; PMID:25383539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 2008; 27:4860-4; PMID:18408756; http://dx.doi.org/ 10.1038/onc.2008.117 [DOI] [PubMed] [Google Scholar]
- 23.Crosby ME, Almasan A. Opposing roles of E2Fs in cell proliferation and death. Cancer Biol Ther 2004; 3:1208-11; PMID:15662116; http://dx.doi.org/ 10.4161/cbt.3.12.1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berckmans B, De Veylder L. Transcriptional control of the cell cycle. Curr Opin Plant Biol 2009; 12:599-605; PMID:19700366; http://dx.doi.org/ 10.1016/j.pbi.2009.07.005 [DOI] [PubMed] [Google Scholar]
- 25.Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene 2005; 24:2810-26; PMID:15838517; http://dx.doi.org/ 10.1038/sj.onc.1208612 [DOI] [PubMed] [Google Scholar]
- 26.Fajas L. Re-thinking cell cycle regulators: the cross-talk with metabolism. Front Oncol 2013; 3:4; PMID:23355973; http://dx.doi.org/ 10.3389/fonc.2013.00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG Jr., Livingston DM, Orkin SH, Greenberg ME. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell 1996; 85:549-61; PMID:8653790; http://dx.doi.org/ 10.1016/S0092-8674(00)81255-6 [DOI] [PubMed] [Google Scholar]
- 28.Fajas L, Landsberg RL, Huss-Garcia Y, Sardet C, Lees JA, Auwerx J. E2Fs regulate adipocyte differentiation. Dev Cell 2002; 3:39-49; PMID:12110166; http://dx.doi.org/ 10.1016/S1534-5807(02)00190-9 [DOI] [PubMed] [Google Scholar]
- 29.Hausman DB, DiGirolamo M, Bartness TJ, Hausman GJ, Martin RJ. The biology of white adipocyte proliferation. Obes Rev 2001; 2:239-54; PMID:12119995; http://dx.doi.org/ 10.1046/j.1467-789X.2001.00042.x [DOI] [PubMed] [Google Scholar]
- 30.Rempel RE, Saenz-Robles MT, Storms R, Morham S, Ishida S, Engel A, Jakoi L, Melhem MF, Pipas JM, Smith C, et al.. Loss of E2F4 activity leads to abnormal development of multiple cellular lineages. Mol Cell 2000; 6:293-306; PMID:10983977; http://dx.doi.org/ 10.1016/S1097-2765(00)00030-7 [DOI] [PubMed] [Google Scholar]
- 31.Hsieh MC, Das D, Sambandam N, Zhang MQ, Nahle Z. Regulation of the PDK4 isozyme by the Rb-E2F1 complex. J Biol Chem 2008; 283:27410-7; PMID:18667418; http://dx.doi.org/ 10.1074/jbc.M802418200 [DOI] [PubMed] [Google Scholar]
- 32.Goto Y, Hayashi R, Kang D, Yoshida K. Acute loss of transcription factor E2F1 induces mitochondrial biogenesis in HeLa cells. J Cell Physiol 2006; 209:923-34; PMID:16972274; http://dx.doi.org/ 10.1002/jcp.20802 [DOI] [PubMed] [Google Scholar]
- 33.Blanchet E, Annicotte JS, Lagarrigue S, Aguilar V, Clape C, Chavey C, Fritz V, Casas F, Apparailly F, Auwerx J, et al.. E2F transcription factor-1 regulates oxidative metabolism. Nat Cell Biol 2011; 13:1146-52; PMID:21841792; http://dx.doi.org/ 10.1038/ncb2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kusama Y, Sato K, Kimura N, Mitamura J, Ohdaira H, Yoshida K. Comprehensive analysis of expression pattern and promoter regulation of human autophagy-related genes. Apoptosis 2009; 14:1165-75; PMID:19657746; http://dx.doi.org/ 10.1007/s10495-009-0390-2 [DOI] [PubMed] [Google Scholar]
- 35.Harman-Boehm I, Bluher M, Redel H, Sion-Vardy N, Ovadia S, Avinoach E, Shai I, Klöting N, Stumvoll M, Bashan N, et al.. Macrophage infiltration into omental versus subcutaneous fat across different populations: effect of regional adiposity and the comorbidities of obesity. J Clin Endocrinol Metab 2007; 92:2240-7; PMID:17374712; http://dx.doi.org/ 10.1210/jc.2006-1811 [DOI] [PubMed] [Google Scholar]
- 36.Bluher M, Bashan N, Shai I, Harman-Boehm I, Tarnovscki T, Avinaoch E, Stumvoll M, Dietrich A, Klöting N, Rudich A. Activated Ask1-MKK4-p38MAPK/JNK stress signaling pathway in human omental fat tissue may link macrophage infiltration to whole-body Insulin sensitivity. J Clin Endocrinol Metab 2009; 94:2507-15; PMID:19351724; http://dx.doi.org/ 10.1210/jc.2009-0002 [DOI] [PubMed] [Google Scholar]
- 37.Shapiro H, Pecht T, Shaco-Levy R, Harman-Boehm I, Kirshtein B, Kuperman Y, Chen A, Blüher M, Shai I, Rudich A. Adipose tissue foam cells are present in human obesity. J Clin Endocrinol Metab 2013; 98:1173-81; PMID:23372170; http://dx.doi.org/ 10.1210/jc.2012-2745 [DOI] [PubMed] [Google Scholar]
- 38.Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res 2005; 96:939-49; PMID:15890981; http://dx.doi.org/ 10.1161/01.RES.0000163635.62927.34 [DOI] [PubMed] [Google Scholar]
- 39.Manninen V, Tenkanen L, Koskinen P, Huttunen JK, Manttari M, Heinonen OP, Frick MH. Joint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki Heart Study. Implications for treatment. Circulation 1992; 85:37-45; PMID:1728471; http://dx.doi.org/ 10.1161/01.CIR.85.1.37 [DOI] [PubMed] [Google Scholar]
- 40.Haim Y, Tarnovscki T, Bashari D, Rudich A. A chromatin immunoprecipitation (ChIP) protocol for use in whole human adipose tissue. Am J Physiol Endocrinol Metab 2013; 305:E1172-7; PMID:24002573; http://dx.doi.org/ 10.1152/ajpendo.00598.2012 [DOI] [PubMed] [Google Scholar]
- 41.Haase J, Weyer U, Immig K, Kloting N, Bluher M, Eilers J, Bechmann I, Gericke M. Local proliferation of macrophages in adipose tissue during obesity-induced inflammation. Diabetologia 2014; 57:562-71; PMID:24343232; http://dx.doi.org/ 10.1007/s00125-013-3139-y [DOI] [PubMed] [Google Scholar]
- 42.Choi H, Kim SJ, Park SS, Chang C, Kim E. TR4 activates FATP1 gene expression to promote lipid accumulation in 3T3-L1 adipocytes. FEBS Lett 2011; 585:2763-7; PMID:21843524; http://dx.doi.org/ 10.1016/j.febslet.2011.08.002 [DOI] [PubMed] [Google Scholar]
- 43.Kim SJ, Nian C, McIntosh CH. GIP increases human adipocyte LPL expression through CREB and TORC2-mediated trans-activation of the LPL gene. J Lipid Res 2010; 51:3145-57; PMID:20693566; http://dx.doi.org/ 10.1194/jlr.M006841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hasan AU, Ohmori K, Konishi K, Igarashi J, Hashimoto T, Kamitori K, Yamaguchi F, Tsukamoto I, Uyama T, Ishihara Y, et al.. Eicosapentaenoic acid upregulates VEGF-A through both GPR120 and PPARgamma mediated pathways in 3T3-L1 adipocytes. Mol Cell Endocrinol 2015; 406:10-8; PMID:25697344; http://dx.doi.org/ 10.1016/j.mce.2015.02.012 [DOI] [PubMed] [Google Scholar]
- 45.Enomoto T, Ohashi K, Shibata R, Kambara T, Uemura Y, Yuasa D, Kataoka Y, Miyabe M, Matsuo K, Joki Y, et al.. Transcriptional regulation of an insulin-sensitizing adipokine adipolin/CTRP12 in adipocytes by Kruppel-like factor 15. PloS One 2013; 8:e83183; PMID:24358263; http://dx.doi.org/ 10.1371/journal.pone.0083183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laudes M, Bilkovski R, Oberhauser F, Droste A, Gomolka M, Leeser U, Udelhoven M, Krone W. Transcription factor FBI-1 acts as a dual regulator in adipogenesis by coordinated regulation of cyclin-A and E2F-4. J Mol Med 2008; 86:597-608; PMID:18368381; http://dx.doi.org/ 10.1007/s00109-008-0326-2 [DOI] [PubMed] [Google Scholar]
- 47.Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL Jr., Eissa NT. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med 2009; 15:267-76; PMID:19252503; http://dx.doi.org/ 10.1038/nm.1928 [DOI] [PubMed] [Google Scholar]
- 48.Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, Mizushima N, Grinstein S, Iwasaki A. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 2010; 32:227-39; PMID:20171125; http://dx.doi.org/ 10.1016/j.immuni.2009.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kovsan J, Osnis A, Maissel A, Mazor L, Tarnovscki T, Hollander L, Ovadia S, Meier B, Klein J, Bashan N, et al.. Depot-specific adipocyte cell lines reveal differential drug-induced responses of white adipocytes–relevance for partial lipodystrophy. Am J Physiol Endocrinol Metab 2009; 296:E315-22; PMID:19033543; http://dx.doi.org/ 10.1152/ajpendo.90486.2008 [DOI] [PubMed] [Google Scholar]
- 50.Lee Y, Dominy JE, Choi YJ, Jurczak M, Tolliday N, Camporez JP, Chim H, Lim JH, Ruan HB, Yang X, et al.. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature 2014; 510:547-51; PMID:24870244; http://dx.doi.org/ 10.1038/nature13267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dali-Youcef N, Mataki C, Coste A, Messaddeq N, Giroud S, Blanc S, Koehl C, Champy MF, Chambon P, Fajas L, et al.. Adipose tissue-specific inactivation of the retinoblastoma protein protects against diabesity because of increased energy expenditure. Proc Natl Acad Sci U S A 2007; 104:10703-8; PMID:17556545; http://dx.doi.org/ 10.1073/pnas.0611568104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takeda K, Noguchi T, Naguro I, Ichijo H. Apoptosis signal-regulating kinase 1 in stress and immune response. Annu Rev Pharmacol Toxicol 2008; 48:199-225; PMID:17883330; http://dx.doi.org/ 10.1146/annurev.pharmtox.48.113006.094606 [DOI] [PubMed] [Google Scholar]
- 53.Yu M, Xiang F, Beyer RP, Farin FM, Bammler TK, Chin MT. Transcription Factor CHF1/Hey2 Regulates Specific Pathways in Serum Stimulated Primary Cardiac Myocytes: Implications for Cardiac Hypertrophy. Curr Genomics 2010; 11:287-96; PMID:21119893; http://dx.doi.org/ 10.2174/138920210791233117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006; 126:121-34; PMID:16839881; http://dx.doi.org/ 10.1016/j.cell.2006.05.034 [DOI] [PubMed] [Google Scholar]
- 55.Eby KG, Rosenbluth JM, Mays DJ, Marshall CB, Barton CE, Sinha S, Johnson KN, Tang L, Pietenpol JA. ISG20L1 is a p53 family target gene that modulates genotoxic stress-induced autophagy. Mol Cancer 2010; 9:95; PMID:20429933; http://dx.doi.org/ 10.1186/1476-4598-9-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee J, Seok S, Yu P, Kim K, Smith Z, Rivas-Astroza M, Zhong S, Kemper JK. Genomic analysis of hepatic farnesoid X receptor binding sites reveals altered binding in obesity and direct gene repression by farnesoid X receptor in mice. Hepatology 2012; 56:108-17; PMID:22278336; http://dx.doi.org/ 10.1002/hep.25609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Williams JA, Thomas AM, Li G, Kong B, Zhan L, Inaba Y, Xie W, Ding WX, Guo GL. Tissue specific induction of p62/Sqstm1 by farnesoid X receptor. PloS one 2012; 7:e43961; PMID:22952826; http://dx.doi.org/ 10.1371/journal.pone.0043961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lipinski MM, Hoffman G, Ng A, Zhou W, Py BF, Hsu E, Liu X, Eisenberg J, Liu J, Blenis J, et al.. A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell 2010; 18:1041-52; PMID:20627085; http://dx.doi.org/ 10.1016/j.devcel.2010.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kreuzaler PA, Staniszewska AD, Li W, Omidvar N, Kedjouar B, Turkson J, Poli V, Flavell RA, Clarkson RW, Watson CJ. Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol 2011; 13:303-9; PMID:21336304; http://dx.doi.org/ 10.1038/ncb2171 [DOI] [PubMed] [Google Scholar]
- 60.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, et al.. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007; 6:458-71; PMID:18054315; http://dx.doi.org/ 10.1016/j.cmet.2007.11.001 [DOI] [PubMed] [Google Scholar]
- 61.Xiong X, Tao R, DePinho RA, Dong XC. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J Biol Chem 2012; 287:39107-14; PMID:22992773; http://dx.doi.org/ 10.1074/jbc.M112.412569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sengupta A, Molkentin JD, Yutzey KE. FoxO transcription factors promote autophagy in cardiomyocytes. J Biol Chem 2009; 284:28319-31; PMID:19696026; http://dx.doi.org/ 10.1074/jbc.M109.024406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang Y, Guerrero-Preston R, Ratovitski EA. Phospho-DeltaNp63alpha-dependent regulation of autophagic signaling through transcription and micro-RNA modulation. Cell Cycle 2012; 11:1247-59; PMID:22356768; http://dx.doi.org/ 10.4161/cc.11.6.19670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ming C, Song F, Li C, Yu Y, Zhang G, Yu H, Sun T, Tian J. Highly efficient downconversion white light in Tm(3)(+)/Tb(3)(+)/Mn(2)(+) tridoped P(2)O(5)-Li(2)O-Sb(2)O(3) glass. Opt Lett 2011; 36:2242-4; PMID:21685980; http://dx.doi.org/ 10.1364/OL.36.002242 [DOI] [PubMed] [Google Scholar]
- 65.Jia G, Cheng G, Gangahar DM, Agrawal DK. Insulin-like growth factor-1 and TNF-α regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol Cell Biol 2006; 84:448-54; PMID:16942488; http://dx.doi.org/ 10.1111/j.1440-1711.2006.01454.x [DOI] [PubMed] [Google Scholar]
- 66.Li DD, Wang LL, Deng R, Tang J, Shen Y, Guo JF, Wang Y, Xia LP, Feng GK, Liu QQ, et al.. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene 2009; 28:886-98; PMID:19060920; http://dx.doi.org/ 10.1038/onc.2008.441 [DOI] [PubMed] [Google Scholar]
- 67.Kiyono K, Suzuki HI, Matsuyama H, Morishita Y, Komuro A, Kano MR, Sugimoto K, Miyazono K. Autophagy is activated by TGF-β and potentiates TGF-β-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res 2009; 69:8844-52; PMID:19903843; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-4401 [DOI] [PubMed] [Google Scholar]
- 68.Sanchez AM, Csibi A, Raibon A, Cornille K, Gay S, Bernardi H, Candau R. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J Cell Biochem 2012; 113:695-710; PMID:22006269; http://dx.doi.org/ 10.1002/jcb.23399 [DOI] [PubMed] [Google Scholar]
- 69.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007; 6:472-83; PMID:18054316; http://dx.doi.org/ 10.1016/j.cmet.2007.11.004 [DOI] [PubMed] [Google Scholar]
- 70.Fiorentino L, Cavalera M, Menini S, Marchetti V, Mavilio M, Fabrizi M, Conserva F, Casagrande V, Menghini R, Pontrelli P, et al.. Loss of TIMP3 underlies diabetic nephropathy via FoxO1/STAT1 interplay. EMBO Mol Med 2013; 5:441-55; PMID:23401241; http://dx.doi.org/ 10.1002/emmm.201201475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, Liu Z, Cao W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem 2009; 284:31484-92; PMID:19758991; http://dx.doi.org/ 10.1074/jbc.M109.033936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sam S, Mazzone T. Adipose tissue changes in obesity and the impact on metabolic function. Transl Res 2014; 164(4):284-92; PMID:24929206 [DOI] [PubMed] [Google Scholar]
- 73.Bashan N, Dorfman K, Tarnovscki T, Harman-Boehm I, Liberty IF, Bluher M, Ovadia S, Maymon-Zilberstein T, Potashnik R, Stumvoll M, et al.. Mitogen-activated protein kinases, inhibitory-kappaB kinase, and insulin signaling in human omental versus subcutaneous adipose tissue in obesity. Endocrinology 2007; 148:2955-62; PMID:17317777; http://dx.doi.org/ 10.1210/en.2006-1369 [DOI] [PubMed] [Google Scholar]
- 74.Sambrook J, Russell DW. Purification of nucleic acids by extraction with phenol:chloroform. CSH protocols 2006; 472-2. [DOI] [PubMed] [Google Scholar]
- 75.Helin K, Harlow E, Fattaey A. Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol Cell Biol 1993; 13: 6501-08; PMID:8413249 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.