

Abstract
Prostate cancer (PCa) is the second leading cause of cancer-related death in men; however, the molecular mechanisms leading to its development and progression are not yet fully elucidated. Of note, it has been recently shown that conditional stk11 knockout mice develop atypical hyperplasia and prostate intraepithelial neoplasia (PIN). We recently reported an inverse correlation between the activity of the STK11/AMPK pathway and the MAPK/p38 cascade in HIF1A-dependent malignancies. Furthermore, MAPK/p38 overactivation was detected in benign prostate hyperplasia, PIN and PCa in mice and humans. Here we report that STK11 expression is significantly decreased in PCa compared to normal tissues. Moreover, STK11 protein levels decreased throughout prostate carcinogenesis. To gain insight into the role of STK11-MAPK/p38 activity balance in PCa, we treated PCa cell lines and primary biopsies with a well-established MAPK14-MAPK11 inhibitor (SB202190), which has been extensively used in vitro and in vivo. Our results indicate that inhibition of MAPK/p38 significantly affects PCa cell survival in an STK11-dependent manner. Indeed, we found that pharmacologic inactivation of MAPK/p38 does not affect viability of STK11-proficient PCa cells due to the triggering of the AMPK-dependent autophagic pathway, while it induces apoptosis in STK11-deficient cells irrespective of androgen receptor (AR) status. Of note, AMPK inactivation or autophagy inhibition in STK11-proficient cells sensitize SB202190-treated PCa cells to apoptosis. On the other end, reconstitution of functional STK11 in STK11-deficient PCa cells abrogates apoptosis. Collectively, our data show that STK11 is a key factor involved in the early phases of prostate carcinogenesis, and suggest that it might be used as a predictive marker of therapeutic response to MAPK/p38 inhibitors in PCa patients.
Keywords: apoptosis, autophagy, AMPK, drug resistance, MAPK/p38, prostate cancer, STK11
Abbreviations
- ACACA/ACC
acetyl-CoA carboxylase alpha
- ADT
androgen deprivation therapy
- AICAR
5-aminoimidazole-4-carboxamide ribonucleotide
- AMPK
adenine monophosphate-activated protein kinase
- AR
androgen receptor
- BafA1
bafilomycin A1
- CASP3
caspase 3, apoptosis-related cysteine peptidase
- CS-FBS
charcoal stripped-fetal bovine serum
- DHT
dihydrotestosterone
- FOXO3/FOXO3A
forkhead box O3
- H&E
haematoxylin and eosin
- HIF1A/HIF1alpha
hypoxia inducible factor 1, alpha subunit (basic helix-loop-helix transcription factor)
- KLK3/PSA
kallikrein-related peptidase 3
- LDHA
lactate dehydrogenase A
- 3MA
3-methyladenine
- MAP1LC3A/LC3A
microtubule-associated protein 1 light chain 3 alpha
- MAPK
mitogen-activated protein kinase
- MAPK14/p38alpha
mitogen-activated protein kinase 14
- MAPK14/11i
MAPK14/p38alpha-MAPK11/p38beta inhibitor(s)
- MAPKAPK2/MK2
mitogen-activated protein kinase-activated protein kinase 2
- MTOR/mTOR
mechanistic target of rapamycin (serine/threonine kinase)
- PARP1/PARP
poly (ADP-ribose) polymerase 1
- PCa
prostate cancer
- PIN
prostate intraepithelial neoplasia
- PKM/PKM2
pyruvate kinase, muscle
- PRKAA1/2
protein kinase, AMP-activated, alpha 1/2 catalytic subunit
- RPS6KB1/p70S6K
ribosomal protein S6 kinase, 70kDa, polypeptide 1
- SB202190
4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)-1H-imidazole
- SLC2A1/GLUT1
solute carrier family 2 (facilitated glucose transporter), member 1
- STK11/LKB1
serine/threonine kinase 11
- ULK1
unc-51 like autophagy activating kinase 1.
Introduction
Prostate cancer (PCa) is the most frequently diagnosed cancer and the second leading cause of cancer death in males. In 2015 it is estimated that approximately 220,800 new cases will be diagnosed and approximately 27,540 men will die of PCa in the USA.1 Prostate-specific antigen (KLK3/PSA) is currently the only widely used serum biomarker for PCa. However, because of its low specificity, novel potential biomarkers have been studied.2-4 Despite the widespread use of KLK3 as a biomarker, testing has led to a significant migration in stage and grade of PCa; up to 30% of patients with clinically localized disease will undergo recurrence and metastatic disease after local therapy with curative intent.5 Androgen-deprivation therapy (ADT) has been the standard of care for this subset of patients with advanced PCa. The majority of patients will respond to this treatment, but most of them will become refractory to ADT within months. Therefore, there is a growing need for additional agents to be introduced in clinical practice for men with castration-resistant disease.
As a better understanding of PCa biology is achieved, providing further insight into the complex extracellular and intracellular signaling networks involved, drug development continues to improve, thanks to the identification of potential therapeutic targets.
Of note, it has been recently shown that conditional stk11/lkb1 knockout mice develop atypical hyperplasia and prostate intraepithelial neoplasia (PIN).6 In humans, STK11 germ line mutations cause Peutz-Jeghers syndrome, an inherited condition predisposing to hamartomas and cancer at different sites (breast, gastrointestinal and gynecological cancers).7,8 STK11 mutations have also been detected in sporadic cancers including non-small-cell lung cancer, cervical and pancreatic cancer, and endometrial carcinoma.9-14 Little is known about the possible involvement of the STK11 gene in human PCa: it is expressed in normal prostate secretory cells,15 while a homozygous deletion has been found in a PCa cell line (DU145).16 These findings suggest that STK11 may play an important role in human prostate carcinogenesis. STK11 encodes a tumor suppressor serine-threonine kinase which is involved in several cell functions, including proliferation, cell cycle arrest, differentiation, energy metabolism and cell polarity.17 The pivotal role of STK11 in controlling oncogenic pathways is mainly due to its downstream effectors, notably AMPK, which is a central metabolic mediator in normal and cancer cells owing to its crosstalk with the phosphoinositide 3-kinase, MTOR, and MAPK pathways.18
We recently reported an inverse correlation between the activity of the STK11-AMPK pathway and the MAPK/p38 signaling cascade in HIF1A/HIF1alpha-dependent malignancies such as colorectal and ovarian cancer.19-21 Indeed, inactivation of MAPK14/p38alpha causes HIF1A degradation and decreased expression of its target genes involved in glycolysis, thus reducing intracellular ATP levels. This acute energetic drop is sensed by AMPK, which promotes a FOXO3/FoxO3A-mediated autophagic response leading to cell survival. When inhibition of MAPK14 is protracted, autophagy is no longer able to sustain metabolism and cells undergo non-apoptotic cell death. Consistently, concomitant inhibition of MAPK14 and the autophagic machinery triggers apoptotic cell death.19,20,22,23 Of note, most prostate cancer deaths are due to the emergence of an androgen-resistant phenotype, which is dependent upon the activity of MAPKs, including MAPK/p38.24 In a study using transgenic adenocarcinoma of the mouse prostate (TRAMP) mice, strong epithelial MAPK/p38 activation was shown to be present in PIN and prostate tumors.25 In humans, overexpression of MAPK/p38 and overactivation of MAPK/p38 signaling occur in benign prostate hyperplasia and more markedly in prostate cancer patients, enhancing cell proliferation and cell survival.26 MAPK/p38 is able to sustain the expression of HIF1A also in prostate cancer cells, thus confirming our previous data obtained in colorectal and ovarian cancer.27 Importantly, a novel MAPK14/p38alpha-MAPK11/p38beta inhibitor (LY2228820 dimesylate) tested in phase I trials for advanced cancers showed early clinical activity in ovary, breast, and kidney cancer, and a phase II study of patients with ovarian cancer is underway.28
Here we show that STK11 is a key factor involved in the early phases of prostate carcinogenesis, and suggest that it might be used as a predictive marker of therapeutic response to MAPK/p38 inhibitors in PCa patients.
Results
STK11 expression is lost during PCa carcinogenesis
Evidence gathered from animal models and human subjects suggests that STK11 may be involved in PCa carcinogenesis. We therefore evaluated STK11 expression by immunoblot in 6 prostate specimens with no evidence of malignancy and in 22 prostate tumor samples. The results of this analysis are shown in Figure 1A. A full-length STK11 protein (52 kDa) was present in all benign samples examined. Densitometric analysis of immunoblotting data showed that STK11 expression in tumor samples was significantly reduced compared to normal tissues (Fig. 1B). Then, we evaluated the immunohistochemical pattern of STK11 expression in paraffin-embedded normal and cancer tissue samples. In non-neoplastic tissues, STK11 staining was limited to the cytoplasm of luminal cells lining the glandular acini (Fig. 1C, upper panel). Basal cells were invariably negative for STK11, as were stromal cells. In some cases, we were able to document the morphological transition of atrophic glands into high-grade PIN (Fig. 1C, middle panel). In these cases, atypical high-columnar cells of high-grade PIN lesions were uniformly negative for STK11, while atrophic luminal cells were positive. Twelve out of 22 tumor samples showed no staining at all for STK11 irrespective of grade, while sporadically positive cells (< 10%) were observed in the remaining 10 specimens (Fig. 1C, bottom panel).
Figure 1.
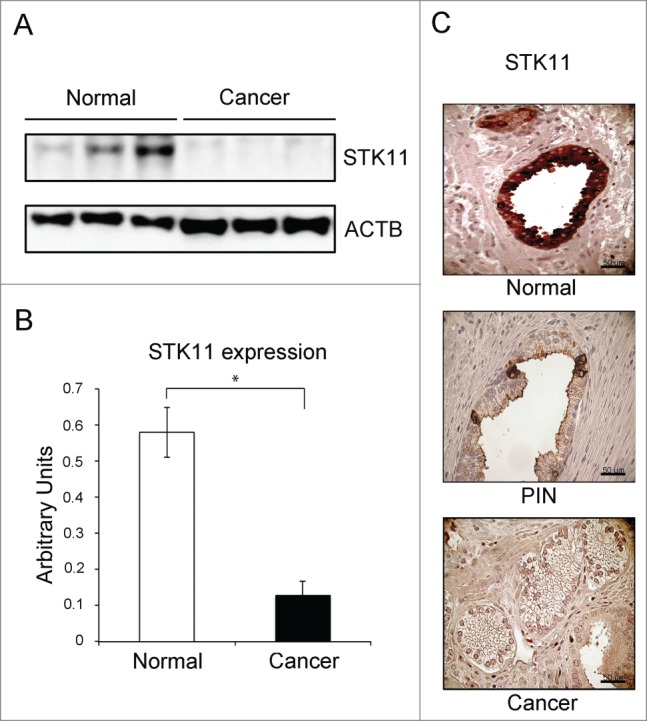
STK11 expression is lost during PCa carcinogenesis. (A) Immunoblot analysis of STK11 in normal and prostate cancer tissue samples. ACTB was used as a loading control. (B) Quantification of immunoblot band intensity by densitometry. (C) Immunohistochemical analysis of STK11 in normal and malignant prostate tissue: strong expression of STK11 in the luminal layer of atrophic glands (upper panel); moderate expression of STK11 in sporadic atypical cells of a high-grade PIN merging with an atrophic gland (middle panel); no expression of STK11 in prostate adenocarcinoma (lower panel). Original magnifications (40x), scale bar: 50 μm.
STK11-AMPK crosstalk with the MAPK/p38 pathway in PCa
In recent studies, we reported an inverse correlation between the activity of the STK11-AMPK and the MAPK/p38 pathways in HIF1A-dependent tumors.19-21 Moreover, MAPK/p38 is able to sustain the expression of HIF1A also in PCa cells.27 These observations prompted us to evaluate the relationship between STK11 expression and MAPK/p38 activation in a large number of PCa specimens by tissue microarray analysis. Our data did not reveal any staining for STK11 expression in any of the 130 PCa analyzed specimens, with the exception of rare intratumoral islets of normal glands, which displayed uniform positivity for this protein (Fig. 2A). Conversely, phospho-MAPK/p38 was globally expressed in 79 (60.7%) specimens, with a staining intensity ranging from weak (n = 39; 30%) to strong (n = 19; 14.6%) (Fig. 2B).
Figure 2.
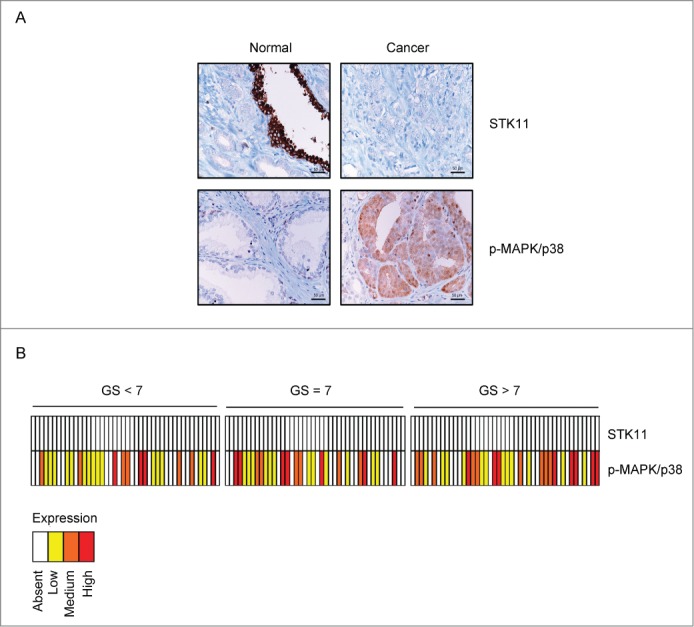
Immunohistochemical staining of STK11 and phospho-MAPK/p38 proteins in tissue microarrays of human prostate specimens. Tissue microarray sections were stained using monoclonal anti-STK11 and monoclonal anti phospho-MAPK/p38 antibodies in a dilution of 1:100 and 1:50, respectively. (A) STK11 staining was negative in prostate cancer and positive only in rare intratumoral islets of normal glands. Phospho-MAPK/p38 showed cytoplasmic and nuclear staining in 60.7% of tumor specimens, whereas it was weakly expressed in normal tissues, especially in basal cells. (B) Heat map summarizing STK11and phospho-MAPK/p38 staining in 130 prostate cancer patients.
To get insight into the role of the STK11-MAPK/p38 crosstalk in prostate cancer, we analyzed 2 cell lines, one harboring a homozygous STK11 deletion (DU145) and the other carrying STK11 wild-type (WT) alleles (PC3; Fig. 3A). These cell lines are both deficient for the expression of the androgen receptor (AR), thus assuring a more homogeneous genetic background, and express all MAPK/p38 isoforms with predominant expression of MAPK14 (Fig. 3B). During proliferation, DU145 cells showed high MAPK14 activity, as measured by the phosphorylation of its direct downstream target MAPKAPK2/MK2, and low PRKAA1/2 phospho-activation (Fig. 3C). Conversely, PC3 cells showed lower MAPK14 activity and higher PRKAA1/2 activation (Fig. 3C). Next, we treated both PCa cell lines with the MAPK14-MAPK11 inhibitor SB202190. Of note, MAPK14 blockade caused a time-dependent increase in phospho-PRKAA1/2 in PC3 cells, but not in DU145 cells (Fig. 3C), consistent with the role of STK11 as a main upstream activator of AMPK. Indeed, also the chemical activator AICAR failed to stimulate PRKAA1/2 in DU145 cells, but not in PC3 cells, as shown by immunoblot analysis of phospho-PRKAA1/2 and of the phospho-activated form of ACACA/ACC, a direct PRKAA1/2 downstream target (Fig. S1). These results suggest that STK11 status may be a key factor influencing PCa cellular response to the inhibition of MAPK14.
Figure 3.
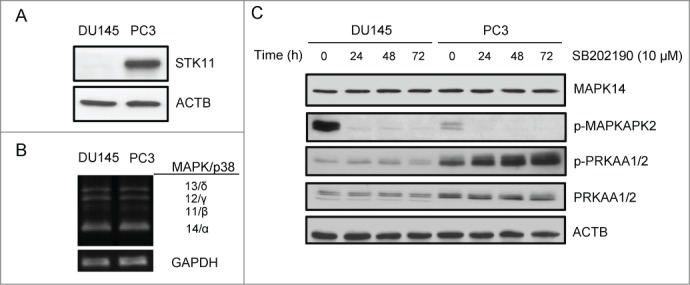
STK11-AMPK crosstalk with the MAPK/p38 pathway in prostate cancer. (A) Immunoblot analysis of STK11 in 2 cell lines, one carrying STK11 WT alleles (PC3) and the other harboring a homozygous deletion of STK11 (DU145). ACTB was used as a loading control. (B) Characterization of the expression of MAPK14, 11, 12, 13 (p38α, β, γ and δ) isoforms in DU145 and PC3 cell lines. Multiplex RT-PCR analysis was followed by separate PCR confirmation for each transcript and normalization to GAPDH signals. (C) DU145 and PC3 cells were treated with the MAPK14-MAPK11 inhibitor SB202190 for the indicated periods of time and total proteins were extracted for immunoblotting analysis with the indicated antibodies. ACTB was used as a loading control. The presented results are representative of at least 3 independent experiments.
STK11 status predicts cellular response to MAPK14 blockade, which is cytotoxic in DU145 and cytostatic in PC3 cells
Pharmacological manipulation of MAPK14 is emerging as a novel promising approach in cancer treatment and is currently in phase II clinical trial for advanced ovarian cancer.28,29 Thus, we treated both DU145 and PC3 cells with SB202190 for up to 96 h to measure cancer cell proliferation and death. Our data show that MAPK14 blockade differentially affected DU145 and PC3 cells. Indeed, after 48 h of treatment DU145 cells started to undergo cell death (Fig. 4A–C), while PC3 cells only reduced their proliferation rate (Fig. 4D–F).
Figure 4.

MAPK14 blockade differentially affects DU145 and PC3 cell growth and survival. DU145 and PC3 cells were treated with DMSO or SB202190 for the indicated periods of time. At the end of the treatment, relative cell viability (A, D), proliferative index (using the WST-1 assay) (B, E) and cell death (C, F) were determined at the indicated time points. Statistical analysis was performed using the Student t test; *, P<0.05, which was considered statistically significant. The presented results are representative of at least 3 independent experiments.
To gain insight into the molecular events leading to the differential response of PCa cells to MAPK14 blockade, we performed immunoblot analyses of apoptotic and autophagic markers. To this end, we evaluated the cleaved form of PARP1/PARP, one of the end products of the activated apoptotic effector CASP3/caspase 3, and the lipidated form of the autophagic protein MAP1LC3A/LC3A (LC3A-II), a key component in autophagosome biogenesis and function.30,31 STK11-deficient cells (DU145) clearly underwent apoptosis (Fig. 5A) when MAPK14 activity was blocked, while STK11-proficient cells (PC3) accumulated the autophagic marker LC3A-II in the same conditions (Fig. 5A). Importantly, pharmacological inhibition of MAPK14 triggered the phosphorylation of the AMPK-specific substrate ULK1 and decreased the phosphorylation of the MTOR substrate RPS6KB1/p70S6K in PC3 cells only (Fig. 5B). Indeed, AMPK regulates autophagy by promoting the initiation step through ULK1 activation and by repressing MTOR signaling.18,20,30,31 Evaluation of the autophagic flux by using specific inhibitors revealed that inhibition of MAPK14 promotes the activation of the autophagic machinery in PC3 cells. Indeed, 3-methyladenine (3MA), an inhibitor of the initial nucleation step of the autophagic process, or bafilomycin A1, an inhibitor of the maturation step that prevents formation of autolysosomes, significantly affected lipidation of LC3A or the subsequent degradation of LC3A-II, respectively, in SB202190-treated cells (Fig. 5C, upper panel). Since the autophagic pathway is involved in both cancer cell survival and death, we analyzed PARP1 cleavage in the same experiments finding that autophagy inhibition in SB202190-treated PC3 cells caused apoptosis (Fig. 5C, middle panel). Furthermore, inhibition of AMPK activity by compound C shifted the cellular response to MAPK14 blockade from autophagy to apoptosis induction in PC3 cells (Fig. 5D). These results indicate that MAPK14 inhibition triggers a stress response in PCa cells, which is buffered by AMPK-dependent autophagy in STK11-proficient cells (PC3), while resulting in apoptosis in STK11-deficient cells (DU145). Indeed, reconstitution of STK11 function in deficient DU145 cells restored the activation of phospho-PRKAA1/2 in response to MAPK14 blockade, while expression of a kinase-defective mutant failed to do so (Fig. 6A). PRKAA1/2 activation in SB202190-treated DU145 cells was sufficient to phosphorylate ULK1 with consequent reduction of PARP1 cleavage (Fig. 6A). Thus, reconstitution of WT STK11 function in DU145 cells promoted resistance to MAPK14 blockade by reducing apoptosis, as confirmed by transfection of a kinase-defective mutant (Fig. 6A–C). Conversely, genetic ablation of STK11 by specific siRNAs in PC3 cells treated with SB202190 prevented PRKAA1/2 activation, phosphorylation of ULK1 and lipidation of LC3A, thus blocking the autophagic response and leading to apoptosis (Fig. 6D).
Figure 5.
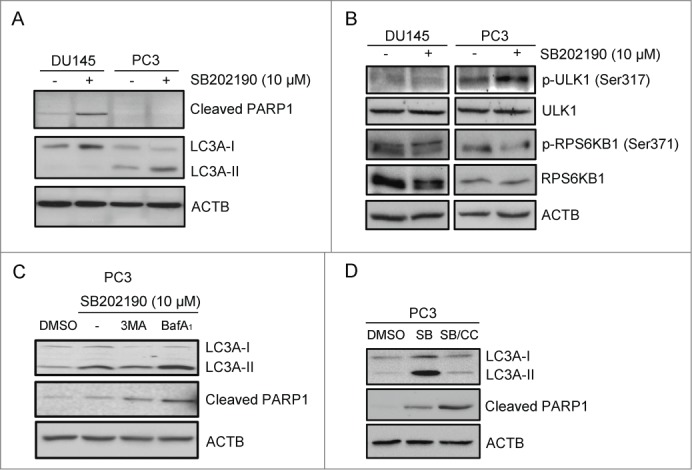
STK11 status predicts cellular response to MAPK14 blockade. (A) Immunoblot analysis of apoptotic and autophagic markers with the indicated antibodies in DU145 and PC3 cells treated with DMSO or SB202190 (10 μM) for 72 h. ACTB was used as a loading control. (B) Immunoblot analysis of DU145 and PC3 cells treated with DMSO or SB202190 (10 μM) for 72 h with the indicated antibodies. ACTB was used as a loading control. (C) STK11-proficient cells (PC3) were cultured in the absence or presence of SB202190 or a combination of SB202190 and 3-methyladenine (3MA; 10 mM) or bafilomycin A1 (BafA1; 0.1 nM) for 48 h. Immunoblot analysis was performed with the indicated antibodies. ACTB was used as a loading control. (D) PC3 cells were treated for 72 h with SB202190 in the presence or absence of the PRKAA1/2 inhibitor compound C (CC), and then autophagic and apoptotic markers were detected by immunoblotting. ACTB was used as a loading control. The presented results are representative of 3 or more independent experiments.
Figure 6.
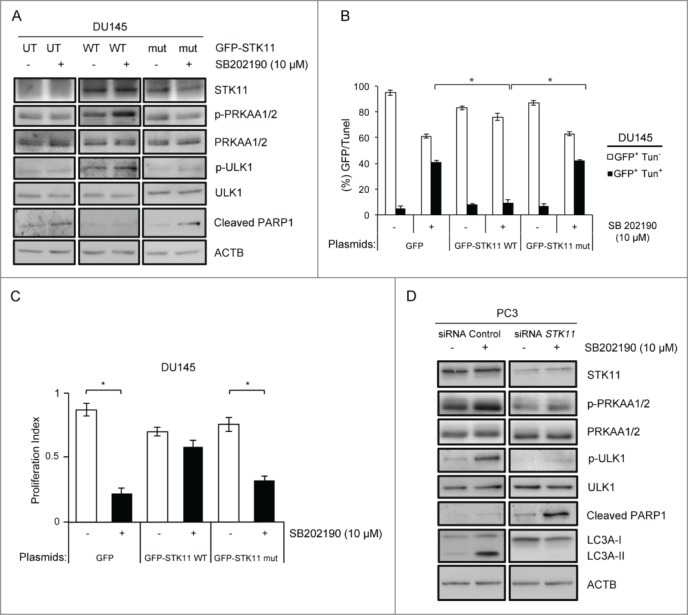
Cellular response upon MAPK14 blockade depends on STK11 status. (A) DU145 cells were transfected with the GFP-STK11 WT or the GFP-STK11 mut (K175_D176del) plasmid for 36 h and then treated with DMSO or SB202190 for an additional 48 h. At the end of the treatment, immunoblot analysis was performed with the indicated antibodies. ACTB was used as a loading control. (B) DU145 cells were transfected with the GFP, the GFP-STK11 WT or the GFP- STK11 mut (K175_D176del) plasmid and then stained with TUNEL assay. GFP+ Tun+ and GFP+ Tun− cells were detected by confocal immunofluorescence analysis and counted in each sample. Statistical analysis was performed using Student's t-tail test; *, P<0.05, which was considered statistically significant. (C) DU145 cells were transfected with the GFP, GFP- STK11 WT or GFP-STK11 mut (K175_D176del) plasmids and treated with DMSO or SB202190 (10 μM) for 48 h. Then, cells were analyzed by WST-1 assay to score the proliferation index. Statistical analysis was performed using the Student t test; *, P<0.05, which was considered statistically significant. (D) Cells transfected with STK11-specific and non-silencing siRNAs were treated with DMSO or SB202190 (10 μM) for 48 h. Then, cells were analyzed by immunoblotting with the indicated antibodies. ACTB was used as a loading control. The presented results are representative of 3 or more independent experiments.
MAPK14-MAPK11 inhibitor (MAPK14/11i) treatment of tumor biopsies showed an inverse correlation between STK11 levels and MAPK14/11i-dependent cell death irrespective of AR status
To get additional insight into the STK11-MAPK/p38 crosstalk in PCa, we cultured LNCaP cells (a hormone-sensitive cell line) in different conditions by increasing the androgen concentration of the culture medium (Fig. 7A). Of note, AR+ PCa cells also responded to MAPK/p38 inhibition in an STK11-dependent manner. In particular, MAPK14 blockade in LNCaP cells (AR+ and STK11-proficient) did not affect cell survival due to activation of PRKAA1/2 and induction of autophagy, irrespective of the presence and/or the concentration of androgens in the culture medium (Fig. 7B). Indeed, comparable results were obtained when LNCaP cells were cultured in the absence of androgens (charcoal stripped-fetal bovine serum [CS-FBS]), in the presence of basal levels of androgens (FBS) or by adding 2.9 mg/ml of dihydrotestosterone (DHT) to androgen-free medium (DHT final concentration 10 nM; Fig. 7A and B).
Figure 7.
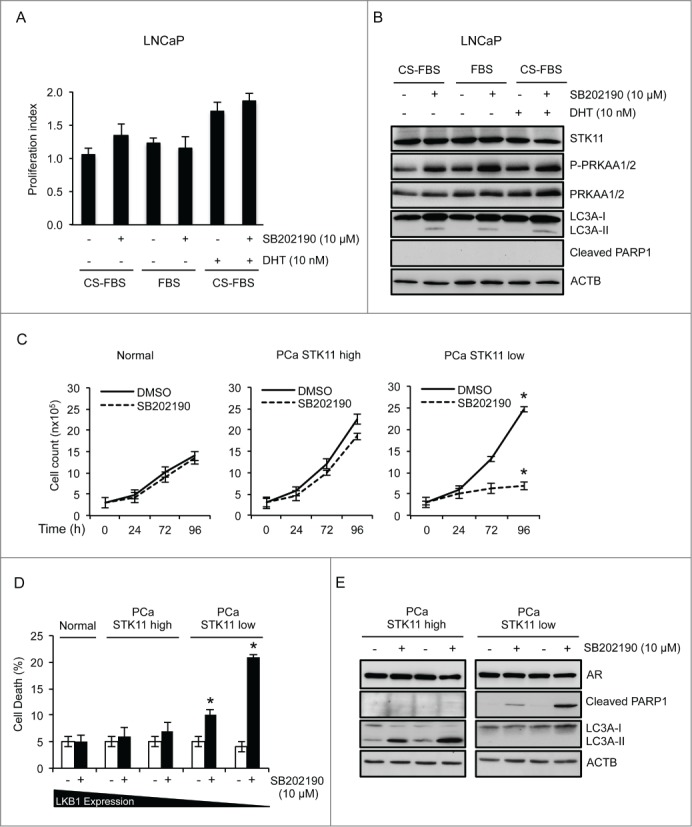
Role of STK11 as a predictive marker of MAPK14/11i-based therapy response in PCa irrespective of AR status. (A, B) LNCaP cells were cultured in phenol red–free RPMI containing 10% charcoal stripped-FBS (CS-FBS) or RPMI containing 10% FBS for 30 h before treatment with 10 nM DHT and/or 10 μM SB202190 (72 h). Cells were analyzed by WST-1 assay to score the proliferation index (A) and by immunoblot with the indicated antibodies for apoptotic and autophagic markers (B). (C, D) Primary cells from normal prostate and PCa were cultured in the presence or absence of SB202190 (10 µM) for up to 96 h. Cell number (C) and cell death (D) were determined at the indicated time points. (E) Immunoblot analysis of apoptotic and autophagic markers with the indicated antibodies in primary cells obtained from PCa biopsies and treated with DMSO or SB202190 (10 μM) for up to 96 h. ACTB was used as a loading control. The presented results are representative of 3 independent experiments. Statistical analysis was performed using the Student t test; *, P<0.05, which was considered statistically significant.
Recently, the importance of in vitro chemoresistance and chemosensitivity assays to select personalized chemotherapy drugs and kinase inhibitors has been appraised again.32 Thus, in order to get translational evidence supporting the role of STK11 as a predictive marker of MAPK14/11i-based therapy response in PCa, we treated 5 primary prostate biopsies obtained from hormone-sensitive PCa patients and controls with SB202190. To this end, we cultured in vitro primary cells from normal prostate and AR+ PCa specimens (2 expressing high/detectable levels of STK11 and 2 with low/absent STK11 expression) in the presence of SB202190 for up to 96 h. Our results show that normal prostate cells were mostly insensitive to MAPK14 inhibition, while AR+ PCa cells were sensitive to SB202190 in a STK11-dependent manner (Fig. 7C). Indeed, cells with low/absent STK11 expression significantly underwent cell death (Fig. 7D). Of note, molecular characterization of these cells revealed that AR+ PCa cells expressing high levels of STK11 reacted to SB202190 in the same way as PC3 cells, i.e., by activating autophagy, while AR+ PCa cells expressing low levels of STK11 triggered apoptosis, as shown by PARP1 cleavage (Fig. 7E), thus replicating the behavior of DU145 cells. These findings suggest that the balance between the apoptotic and autophagic machinery in PCa is regulated by the crosstalk between STK11 and MAPK14 independently from the hormonal status of the cells (Fig. 8).
Figure 8.
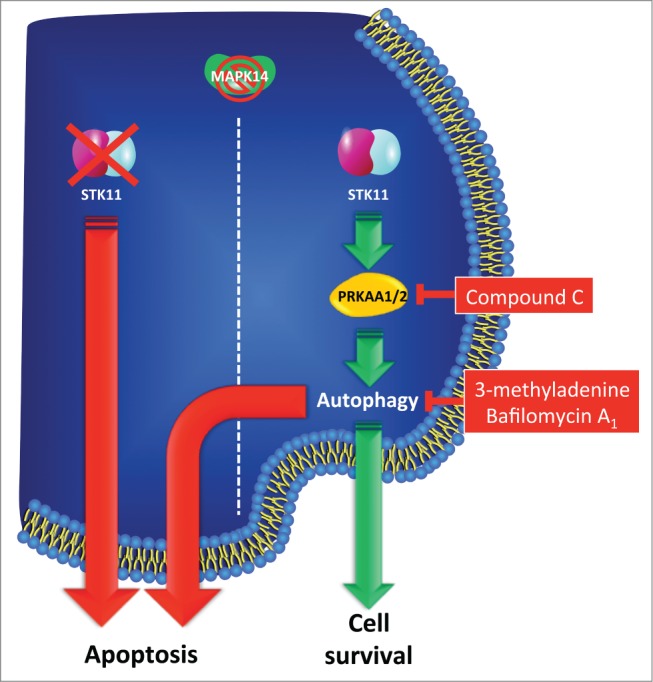
STK11 is a key factor involved in the early phases of prostate carcinogenesis, and it might be used as a predictive marker of therapeutic response to MAPK14-MAPK11 inhibitors in PCa patients. Representative scheme depicted to summarize the findings of this work. MAPK14-MAPK11 inhibitors differentially affected PCa cell growth, inducing apoptosis in STK11-deficient cells, while triggering autophagy in STK11-proficient cells. Reconstitution of WT STK11 function promoted resistance to MAPK14 blockade by reducing apoptosis in STK11 KO cells. Conversely, inhibition of autophagy or of PRKAA1/2 (AMPK) triggered apoptosis in STK11-proficient cells treated with MAPK14-MAPK11 inhibitors.
Collectively, these results confirm the possibility of using STK11 as a predictive marker for therapy response to MAPK14-MAPK11 inhibitors in PCa.
Discussion
Androgen deprivation therapy has been the standard of care for patients with advanced PCa for more than 50 y. Most patients will respond to this treatment by showing symptomatic improvement, metastasis regression, and a decline in KLK3 levels. However, the median duration of response to ADT is only 18-24 mo for patients with metastatic disease. About 30% to 40% of patients will respond to further hormonal manipulation, but the majority will become refractory to this treatment within months. Metastatic hormone-refractory prostate cancer is both morbid and rapidly progressive with a median survival of 18-20 mo.33 Until recently, no therapy had been shown to prolong life expectancy in this subset of patients, so there is a growing need for additional agents amenable to be used in prostate cancer. As we gain a better understanding of the various pathways involved in PCa and characterize their crosstalk, it becomes more challenging to rationally develop new specific agents. Targeting the androgen receptor pathway continues to have an important role in the treatment of PCa;34,35 however, mechanisms of drug resistance have been described also for novel agents recently introduced in the clinical practice (such as abiraterone and enzalutamide). Recent studies have shown that multiple mechanisms are involved in prostate cancer resistance to hormonal therapy. In particular, 3 general categories can be identified. The first includes genetic alterations in the AR gene, such as point mutations in the ligand-binding domain or AR amplification events. The second type of mechanism involves ligand-independent activation of AR by increased MAPK signaling or an altered balance between AR corepressors and coactivators. The third category encompasses mechanisms that promote prostate cancer cell survival through activation of alternative signaling pathways, thus bypassing AR functions.36
In this scenario, the focus should be moved toward the possibility to target alternative regulatory pathways involved in cancer-specific features such as glucose metabolism and autophagy. Of note, the MAPK/p38 pathway, which is involved in regulating cancer-specific glycolysis, autophagy, chemoresistance and molecularly targeted drug resistance in colorectal and ovarian cancer,29 has been shown to be one of the mediators of the androgen-resistant phenotype.24 Moreover, strong epithelial MAPK/p38 activation was shown in PIN and prostate tumors both in animal models and PCa patients,25,26 and its activity is required to sustain the expression of HIF1A also in PCa cells.27 These data suggest that MAPK/p38 may be the ideal candidate for targeted therapy in PCa patients. To verify this hypothesis, we treated PCa cell lines and primary biopsies with a well-established MAPK14-MAPK11 inhibitor (SB202190), that we and others have already extensively used in vitro and in in vivo preclinical mouse cancer models.29 Our results indicate that inhibition of MAPK14 significantly affects PCa growth depending on the expression or the mutational status of the STK11 gene (Fig. 6). Indeed, we found that pharmacological inactivation of MAPK14 does not affect survival of STK11-proficient AR− PCa cells (PC3) due to the triggering of the AMPK-dependent autophagic pathway, whereas it induces apoptosis in STK11-deficient AR− cells (DU145). Of note, PRKAA1/2 inactivation or autophagy inhibition in STK11-proficient cells sensitize SB202190-treated PCa cells to apoptosis. Moreover, reconstitution of wild-type STK11, but not expression of a kinase-defective mutant, significantly reduces the apoptotic response triggered by MAPK14/11i in STK11-deficient cells. These data support a model where an inverse correlation between activation of the STK11-AMPK and the MAPK/p38 pathways exists in HIF1A-dependent malignancies, as shown for colorectal and ovarian cancer, and now also in castration-resistant PCa (Fig. 8). To explore STK11-MAPK/p38 crosstalk in hormone-sensitive PCa, we cultured a PCa cell line expressing AR (LNCaP) in different conditions by increasing androgen concentration in the culture medium, and AR+ primary cells (expressing high/detectable or low/undetectable levels of STK11) obtained from patients with hormone-sensitive disease. Consistent with our data on AR− cells, our results indicated that hormone-sensitive PCa cells are also responsive to MAPK/p38 inhibition in an STK11-dependent manner. In particular, inactivation of MAPK14 in AR+ STK11-proficient PCa cells (LNCaP) does not affect cell survival irrespective of the presence and concentration of androgens in the culture medium. These findings confirm the existence of a general mechanism regulating the apoptotic and autophagic pathways in PCa, based on the crosstalk between STK11 and MAPK14 (Fig. 8). Collectively, our results suggest that this mechanism is independent from the hormonal status of the cells. However, we cannot rule out that androgens and/or AR can modulate the STK11-dependent therapeutic response to MAPK14/11i.
AMPK plays a critical role in regulating the cellular energetic state and, under conditions of energetic stress, one of its most important upstream activators is STK11, which is a tumor suppressor with a putative role also in the initial phases of prostate carcinogenesis in animal models.6 Here we show that STK11 expression is lost throughout PCa progression. Histologically, prostate carcinogenesis is characterized by a progressive transition from normal epithelium to high-grade PIN to invasive PCa. Moreover, recent findings suggest that certain focal atrophy lesions termed “proliferative inflammatory atrophy” could be pathogenetically associated with PCa, with frequent transitions from areas of proliferative inflammatory atrophy to high-grade PIN.37 In this setting, our results show a progressive loss of STK11 expression between normal and neoplastic tissue. Moreover, in some cases we identified areas of atrophic glands merged with high-grade PIN. In these lesions, STK11 expression was evident only in atrophic cells, while dysplastic cells showed no staining by immunohistochemistry.
Due to the inverse correlation between MAPK14 and AMPK activity, we suggest that STK11 might be used as a predictive marker of therapeutic response to MAPK14-MAPK11 inhibitors in PCa patients. In particular, subjects showing no STK11 expression could benefit from a therapeutic approach involving MAPK14-MAPK11 inhibitors alone or in combination with hormonal therapy, whereas subjects positive for STK11 expression would be suitable candidates for dual kinase inhibition targeting both MAPK14 and AMPK or for treatments based on MAPK14-MAPK11 inhibitors together with agents blocking the autophagic machinery (Fig. 8). The potential implications of our results are even more significant in light of the fact that a novel MAPK14-MAPK11 inhibitor (LY2228820 dimesylate) passed phase I trial for advanced cancers showing early clinical activity in ovary, breast, and kidney cancer and is currently being investigated in a phase II study of patients with ovarian cancer.
Materials and Methods
Tissue samples
This study was performed on tissue specimens derived from 22 radical retropubic prostatectomies from patients with prostate cancer. Written informed consent to take part was given by all participants. The median age at diagnosis was 68.5 y (range: 56–76), while the median preoperative KLK3 level was 8.65 ng/ml (95% confidence interval: 5.52–15.09). Patients were classified according to stage (TNM classification) and grade (Gleason score) based on prostatectomy specimens. All patients had hormone-sensitive disease and none was treated with androgen deprivation therapy before surgery. Presence of the androgen receptor was confirmed in all tissue specimens by immunohistochemistry. Prostate samples with no evidence of malignancy were obtained from 6 patients (median age: 67 y; range: 63-75) who underwent radical cystoprostatectomy for bladder carcinoma not involving the prostate and without foci of prostate adenocarcinoma.
Tumor samples were cut in 2 equal parts and one half was snap-frozen and cryopreserved in liquid nitrogen for western blot analysis. The remainder of tissue samples was fixed in 10% neutral-buffered formalin for 12 to 24 h, embedded in paraffin, and stained with haematoxylin and eosin (H&E; Sigma MHS16, 230251) for histological evaluation and immunohistochemistry. Tumor and normal tissue samples of prostate were collected and used for primary culture. Identification of tumor areas in the frozen tissue was achieved by matching this tissue with H&E-stained sections. Two different pathologists reviewed H&E-stained sections.
Immunohistochemistry and tissue microarray construction
Immunohistochemical evaluation of STK11 protein expression was carried out on paraffin-embedded tissue sections. Thin (3 µm) sections were deparaffinized and rehydrated through xylene and graded alcohol series. Slides were subjected to specific epitope unmasking by microwave treatment (700W) in citrate buffer (0.01 M, pH 6.0). After antigen retrieval, tissue samples were incubated for 10 min with 3% H2O2 to block endogenous peroxidase activity. Sections were treated with serum-free protein block (Dako, X0909) at room temperature (RT) for 10 min and then incubated for 1 h at RT, with a mouse anti-human STK11 monoclonal antibody (Santa Cruz Biotechnology, sc-32245). Binding of the secondary biotinylated antibody was detected using the Dako Real EnVision Detection System, Peroxidase/DAB kit (Dako, K5007), according to the manufacturer's instructions. Sections were counterstained with Mayer's haematoxylin (blue) and mounted with glycerol (Dako Cytomation, C0563). Negative controls were obtained incubating serial sections with the blocking solution and then omitting the primary antibody. Staining of histological sections was evaluated using an optical light microscope (a Leica microscope fitted with a Coolpix 990 digital camera [Nikon]). Positive staining corresponded to cells with dark-brown STK11 immunoreactivity.
Nine high-density tissue microarrays were used for STK11 and phospho-MAPK/p38 (Thr180/Tyr182; Cell Signaling Technology, 4631) immunostaining. Archived formalin-fixed paraffin-embedded prostatectomy tissue samples for 130 additional cases were obtained. All tumor cores were identified by a uro-pathologist (FS). These were selected by identifying representative tumor-containing slides and were used to assign the original tumor grade for each case. Three-mm cores were removed from the selected area (region of interest) by using a needle punch. Such 3-mm donor cores were subsequently embedded in previously arranged recipient paraffin blocks through a precisely spaced 15-hole array pattern. Core positions in the recipient paraffin block were noted on a tissue microarray map and a 2-mm pig kidney core was used as a marker for orientation. After cooling paraffin, recipient blocks were cut in the microtome and used for immunohistochemistry. Protein immunoreactivity was scored by the extent and intensity of staining, which was graded on an arbitrary scale ranging from 0 to 3, with 0 = negative, 1 = low, 2 = medium, and 3 = high expression.
Cell lines, primary culture of prostate biopsies and reagents
Prostate cancer cell lines, PC3, DU145 and LNCaP were grown in RPMI 1640 (Gibco, 11875–093) supplemented with 10% FBS (Gibco, 10270-106), 100 IU/ml penicillin and 100 μg/ml streptomycin in a humidified incubator at 37°C and 5% CO2 avoiding confluence at any time. For androgen-free conditions, LNCaP cells were cultured in RPMI 1640 containing 10% FBS for 24 h. Cells were switched into a phenol red-free RPMI 1640 medium (Gibco, 11835-030) containing either 10% FBS or 10% CS-FBS (Gibco, 12676-029) 30 h before treatments with 10 nM dihydrotestosterone (DHT; Sigma, T-1500), and/or 10 μM SB202190 (Sigma, S7067).
Human tumor and normal tissue samples of prostate were placed in a Petri dish with 1X phosphate-buffered saline (PBS; Sigma, D8537) and cut into small pieces of about 1 mm3. Each small piece of tissue was placed on the surface of a Petri dish which was previously wet with 1 ml of DMEM medium (Gibco, 11966-025) supplemented with 10% FBS, 100 IU/ml penicillin, 100 μg/ml streptomycin and 1% L-glutamine. Petri dishes were maintained for 2-3 h at 37°C with 5% CO2, after which the small pieces were covered with 4 ml of DMEM medium and placed back into the incubator. Cell proliferation was obtained around the prostate piece. After 7 d, cells were detached by trypsin/EDTA (Gibco, 15400-054) digestion and washed twice with ice-cold MACS buffer (PBS, pH 7.2, 0.5% BSA [Sigma, A7906], 2 mM EDTA). After being passed through a 30-µm filter to remove cell clumps, cells were counted and incubated for 30 min at 4°C in MACS buffer with the same volume (1 µl/105 cells) of FcR blocking reagent (Miltenyi Biotec, 130-059-901) and CD326 (EpCAM) Ab-conjugated magnetic microbeads (Miltenyi Biotec, 130-061-101). Cells were then washed, as per the manufacturer's instructions, and passed through an MS column (Miltenyi Biotec, 130-042-201) under the effect of a magnetic field generated by the Mini MACS Separation Unit (Miltenyi Biotec, 130-042-102; 130-042-303). EPCAM/CD326-positive cells retained inside the column were washed 3 times and then eluted once the magnetic field was removed. Eluted cells were resuspended in keratinocyte serum-free medium (Gibco, 10724-011) with 5 ng/ml recombinant EGF (Gibco, 10450-013), 50 µg/ml bovine pituitary extract (Gibco, 13028-014) and 30 ng/ml cholera toxin (Sigma, C8052) and incubated at 37°C with 5.0% CO2. These cells were then characterized for EPCAM and prostate-specific membrane antigen expression by immunocytochemistry to confirm their epithelial and prostatic lineage. Primary prostate cancer cells were used for proliferation studies at the second passage.
Bafilomycin A1 (B1793), compound C (P5499) and AICAR (A9978), were purchased from Sigma. 3-methyladenine (189490) was purchased from Calbiochem.
Immunoblotting
Immunoblotting analyses were performed according to Cell Signaling Technology instructions. Briefly, frozen prostate tissues and cell lines were homogenized in 1X lysis buffer (50 mM Tris-HCl, pH 7.4, 5 mM EDTA, 250 mM NaCl, 0.1% Triton X-100 [Sigma, T8787]) supplemented with protease and phosphatase inhibitors (1 mM PMSF [Sigma, P7626], 1.5 μM pepstatin A [Sigma, P5318], 2 μM leupeptin [Sigma, L2884], 10 μg/ml aprotinin [Sigma, A1153], 5 mM NaF [Sigma, S7920], 1 mM Na3VO4 [Sigma, S6508]). 20 μg of protein extracts from each sample were denatured in 5X Laemmli sample buffer and loaded into an SDS-polyacrylamide gel for western blot analysis. Western blots were performed using anti-ACTB/β-Actin (Sigma, A2066), anti-MAPK14 (Cell Signaling Technology, 9228), anti-phospho-MAPKAPK2/MK2 (Thr222; Cell Signaling Technology, 3316), anti-phospho-PRKAA1/2 (Thr172; Cell Signaling Technology, 2531), anti-PRKAA1/2 (Cell Signaling Technology, 2532), anti-phospho-ACACA/acetyl CoA carboxylase α (Ser79; Cell Signaling Technology, 3661), anti-PARP1 p85 (Promega, G7341), anti-MAP1LC3A (Novus Biologicals, NB100-2331), anti-STK11 (Abcam, ab15095), anti-phospho-ULK1 (Ser317; Cell Signaling Technology, 6887), anti-ULK1 (Cell Signaling Technology, 4773), anti-phospho-RPS6KB1 (Ser371; Cell Signaling Technology, 9208), anti-RPS6KB1 (Cell Signaling Technology, 9202), anti-AR (Dako, M3562). Western blots were developed with the ECL−plus chemiluminescence reagent (GE Healthcare, RPN2232) as per the manufacturer's instructions.
Semiquantitative multiplex PCR
Total RNAs were extracted using TRI Reagent (Sigma, T9424). Samples were treated with DNase I (Ambion, AM2224) and retro-transcribed using the High Capacity DNA Archive Kit (Applied Biosystems, 4368813). Semiquantitative multiplex PCRs were performed in triplicate using a C1000 Touch Thermal Cycler with 5 U/μl Taq DNA polymerase, 25 mmol/l MgCl2, 10X Taq buffer (5PRIME, 2200020), and 10 mmol/L deoxyribonucleotide triphosphates (Invitrogen, 10297-018). The following PCR conditions were used: 95°C for 15 min, followed by 28 cycles at 94°C for 30 sec, 59°C for 90 sec, 72°C for 60 sec, and in the final step, 72°C for 10 min. Primer sequences are available upon request.
Cell transfection and RNA interference
The transient transfection experiments were performed using TransIT-Prostate Transfection Kit (Mirus Bio LLC, 2130A) according to the manufacturer's instructions. Briefly, 10 μl of TransIT-Prostate Transfection Kit and 5 μl of Prostate Boost Reagent (Mirus Bio LLC, 2130B) were used to transfect 2.5 μg of the EGFP plasmid, the GFP-STK11 WT plasmid or the GFP-STK11 mut plasmid (STK11 K175_D176del). DU145 cells grown to 70% confluence were incubated for 24 h, and after addition of fresh medium, were cultured for an additional 36 h to express the plasmids before performing the experiments.
For RNA interference, cells were transfected with 5 nM siRNAs directed against STK11 (Ambion, validated oligos s13579) by using HiPerFect Transfection Reagent (Qiagen, 301707). On-TARGET-plus control 50 nM siRNAs (Thermo Scientific, D-001810-10-20) were used as control sequences. siRNA sequences are available upon request.
Apoptosis assay
For terminal uridine deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays (Roche, 11684795910) samples were fixed in 10% formalin for 12–24 h, dehydrated, and paraffin embedded. Detection of apoptosis at single cell level based on labeling of DNA strand breaks was performed using the In situ Cell Detection kit (Roche, 11684795910) following the manufacturer's instructions. Images were collected using a Zeiss LSM-5 Pascal confocal microscope and analyzed using the Zeiss LSM Image Browser software, version 4.2.0121. 100 cells were counted for each sample, and TUNEL-positive cells, GFP-positive cells and TUNEL/GFP double-positive cells were identified.
Microscopy quantification of viability and cell death
Cell viability and cell death of the reported cell lines were scored by counting. Supernatant fractions (containing dead/floating cells) were collected, and the remaining adherent cells were detached by trypsin/EDTA. Cell pellets were resuspended in 1X PBS and 10 μl were mixed with an equal volume of 0.01% trypan blue solution. Viable cells (unstained, trypan blue-negative cells) and dead cells (stained, trypan blue-positive cells) were counted with a phase contrast microscope. The percentages of viable and dead cells were calculated. The data shown in the Results section are representative of 3 or more independent sets of experiments.
Cell proliferation assay (WST-1)
Cell proliferation was determined using the Cell Proliferation Reagent WST-1 (Roche, 11644807001) as per the manufacturer's instructions. Briefly, cells were seeded into 96-well plates one day before treatment. After 24, 48, 72 or 96 h drug (or DMSO) exposure, or cell transfection with the GFP-STK11 WT or GFP-STK11 mut (K175_D176del) plasmids, 10 µl of the Cell Proliferation Reagent WST-1 were added to each well and incubated at 37°C in a humidified incubator for 1 h. Absorbance was measured on a microplate reader (MULTISCAN EX, Thermo Electron Corporation) at 450/655 nm. Each assay was performed in 6 replicates and the experiment was repeated twice. The proliferation index was calculated as the ratio of WST-1 absorbance of treated cells at the indicated time points to the WST-1 absorbance of the same experimental group at 0 h.
Statistical analysis
The statistical significance of the results was analyzed using the Student t test, and P < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank Dr Francesco Paolo Jori for his helpful discussion during the preparation of the manuscript and editorial assistance.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
A.P. is supported by an Italian Foundation for Cancer Research (FIRC) fellowship. This study was partially supported by an ‘Investigator Grant 2010’ from the Italian Association for Cancer Research (AIRC) (grant number: IG10177) (to C.S.) and FIRB – FUTURO IN RICERCA RBFR12VP3Q_003 from the Italian MIUR (to C.S.).
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin 2015; 65:5-29; PMID:25559415; http://dx.doi.org/ 10.3322/caac.21254 [DOI] [PubMed] [Google Scholar]
- 2.Lucarelli G, Fanelli M, Larocca AM, Germinario CA, Rutigliano M, Vavallo A, Selvaggi FP, Bettocchi C, Battaglia M, Ditonno P. Serum sarcosine increases the accuracy of prostate cancer detection in patients with total serum PSA less than 4.0 ng/ml. Prostate 2012; 72:1611-21; PMID:22430630; http://dx.doi.org/ 10.1002/pros.22514 [DOI] [PubMed] [Google Scholar]
- 3.Lucarelli G, Ditonno P, Bettocchi C, Spilotros M, Rutigliano M, Vavallo A, Galleggiante V, Fanelli M, Larocca AM, Germinario CA, Maiorano E, Selvaggi FP, Battaglia M. Serum sarcosine is a risk factor for progression and survival in patients with metastatic castration-resistant prostate cancer. Future Oncol 2013a; 9:899-907; PMID:23718310; http://dx.doi.org/ 10.2217/fon.13.50 [DOI] [PubMed] [Google Scholar]
- 4.Lucarelli G, Rutigliano M, Bettocchi C, Palazzo S, Vavallo A, Galleggiante V, Trabucco S, Di Clemente D, Selvaggi FP, Battaglia M, Ditonno P. Spondin-2, a secreted extracellular matrix protein, is a novel diagnostic biomarker for prostate cancer. J Urol 2013b; 190:2271-7; PMID:23665271; http://dx.doi.org/ 10.1016/j.juro.2013.05.004 [DOI] [PubMed] [Google Scholar]
- 5.Shao YH, Demissie K, Shih W, Mehta AR, Stein MN, Roberts CB, Dipaola RS, Lu-Yao GL. Contemporary risk profile of prostate cancer in the United States. J Natl Cancer Inst 2009; 101:1280-3; PMID:19713548; http://dx.doi.org/ 10.1093/jnci/djp262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pearson HB, McCarthy A, Collins CM, Ashworth A, Clarke AR. Lkb1 deficiency causesprostate neoplasia in the mouse.Cancer Res 2008; 68:2223-32; PMID:18381428; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-5169 [DOI] [PubMed] [Google Scholar]
- 7.Resta N, Pierannunzio D, Lenato GM, Stella A, Capocaccia R, Bagnulo R, Lastella P, Susca FC, Bozzao C, Loconte DC, et al.. Cancer risk associated with STK11/LKB1 germline mutations in Peutz-Jeghers syndrome patients: results of an Italian multicenter study. Dig Liver Dis 2013; 45:606-11; PMID:23415580; http://dx.doi.org/ 10.1016/j.dld.2012.12.018 [DOI] [PubMed] [Google Scholar]
- 8.Resta N, Simone C, Mareni C, Montera M, Gentile M, Susca F, Gristina R, Pozzi S, Bertario L, Bufo P, et al.. STK11 mutations in Peutz-Jeghers syndrome and sporadic colon cancer. Cancer Res 1998; 58:4799-801; PMID:9809980 [PubMed] [Google Scholar]
- 9.Launonen V. Mutations in the human LKB1/STK11 gene. Hum Mutat 2005; 26:291-7; PMID:16110486; http://dx.doi.org/ 10.1002/humu.20222 [DOI] [PubMed] [Google Scholar]
- 10.Matsumoto S, Iwakawa R, Takahashi K, Kohno T, Nakanishi Y, Matsuno Y, Suzuki K, Nakamoto M, Shimizu E, Minna JD, Yokota J. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene 2007; 26:5911-8; PMID:17384680; http://dx.doi.org/ 10.1038/sj.onc.1210418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hezel AF, Gurumurthy S, Granot Z, Swisa A, Chu GC, Bailey G, Dor Y, Bardeesy N, Depinho RA. Pancreatic LKB1 deletion leads to acinar polarity defects and cystic neoplasms. Mol Cell Biol 2008b; 28:2414-25; PMID:18227155; http://dx.doi.org/ 10.1128/MCB.01621-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wingo SN, Gallardo TD, Akbay EA, Liang MC, Contreras CM, Boren T, Shimamura T, Miller DS, Sharpless NE, Bardeesy N, et al.. Somatic LKB1 mutations promote cervical cancer progression. PLoS One 2009; 4:e 5137; PMID:19340305; http://dx.doi.org/ 10.1371/journal.pone.0005137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Contreras CM, Gurumurthy S, Haynie JM, Shirley LJ, Akbay EA, Wingo SN, Schorge JO, Broaddus RR, Wong KK, Bardeesy N, Castrillon DH. Loss of Lkb1 provokes highly invasive endometrial adenocarcinomas. Cancer Res 2008; 68:759-66; PMID:18245476; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-5014 [DOI] [PubMed] [Google Scholar]
- 14.Co NN, Iglesias D, Celestino J, Kwan SY, Mok SC, Schmandt R, Lu KH. Loss of LKB1 in high-grade endometrial carcinoma: LKB1 is a novel transcriptional target of p53. Cancer 2014; 120:3457-68; PMID:25042259; http://dx.doi.org/ 10.1002/cncr.28854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conde E, Suarez-Gauthier A, García-García E, Lopez-Rios F, Lopez-Encuentra A, García-Lujan R, Morente M, Sanchez-Verde L, Sanchez-Cespedes M. Specific pattern of LKB1 and phospho-acetyl-CoA carboxylase protein immunostaining in human normal tissues and lung carcinomas. Hum Pathol 2007; 38:1351-60; PMID:17521700; http://dx.doi.org/ 10.1016/j.humpath.2007.01.022 [DOI] [PubMed] [Google Scholar]
- 16.Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O'Meara S, Santarius T, Avis T, Barthorpe S, Brackenbury L, et al.. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther 2006; 5:2606-12; PMID:17088437; http://dx.doi.org/ 10.1158/1535-7163.MCT-06-0433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hezel AF, Bardeesy N. LKB1; linking cell structure and tumor suppression. Oncogene 2008a; 27:6908-19; PMID:19029933; http://dx.doi.org/ 10.1038/onc.2008.342 [DOI] [PubMed] [Google Scholar]
- 18.Chiacchiera F, Simone C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle 2010; 9:1091-6; PMID:20190568; http://dx.doi.org/ 10.4161/cc.9.6.11035 [DOI] [PubMed] [Google Scholar]
- 19.Chiacchiera F, Matrone A, Ferrari E, Ingravallo G, Lo Sasso G, Murzilli S, Petruzzelli M, Salvatore L, Moschetta A, Simone C. p38alpha blockade inhibits colorectal cancer growth in vivo by inducing a switch from HIF1alpha- to FoxO-dependent transcription. Cell Death and Diff 2009a; 16:1203-14; PMID:19343039; http://dx.doi.org/ 10.1038/cdd.2009.36 [DOI] [PubMed] [Google Scholar]
- 20.Chiacchiera F, Simone C. Inhibition of p38alpha unveils an AMPK-FoxO3A axis linking autophagy to cancer-specific metabolism. Autophagy 2009b; 5:1030-3; PMID:19587525; http://dx.doi.org/ 10.4161/auto.5.7.9252 [DOI] [PubMed] [Google Scholar]
- 21.Matrone A, Grossi V, Chiacchiera F, Fina E, Cappellari M, Caringella AM, Di Naro E, Loverro G, Simone C. p38alpha is required for ovarian cancer cell metabolism and survival. Int J Gynecol Cancer 2010; 20:203-11; PMID:20169663; http://dx.doi.org/ 10.1111/IGC.0b013e3181c8ca12 [DOI] [PubMed] [Google Scholar]
- 22.Comes F, Matrone A, Lastella P, Nico B, Susca FC, Bagnulo R, Ingravallo G, Modica S, Lo Sasso G, Moschetta A, Guanti G, Simone C. A novel cell type-specific role of p38alpha in the control of autophagy and cell death in colorectal cancer cells. Cell Death Differ 2007; 14:693-702; PMID:17159917; http://dx.doi.org/ 10.1038/sj.cdd.4402076 [DOI] [PubMed] [Google Scholar]
- 23.Simone C. Signal-dependent control of autophagy and cell death in colorectal cancer cell: the role of the p38 pathway. Autophagy 2007; 3:468-71; PMID:17495519; http://dx.doi.org/ 10.4161/auto.4319 [DOI] [PubMed] [Google Scholar]
- 24.Rodríguez-Berriguete G, Fraile B, Martínez-Onsurbe P, Olmedilla G, Paniagua R, Royuela M. MAP kinases and prostate cancer. J Signal Transduct 2012; 2012:169170; PMID:22046506; http://dx.doi.org/12661038 10.1155/2012/169170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uzgare AR, Kaplan PJ, Greenberg NM. Differential expression and/or activation of P38MAPK, erk1/2, and jnk during the initiation and progression of prostate cancer. Prostate 2003; 55:128-39; PMID:12661038; http://dx.doi.org/ 10.1002/pros.10212 [DOI] [PubMed] [Google Scholar]
- 26.Ricote M, García-Tuñón I, Bethencourt F, Fraile B, Onsurbe P, Paniagua R, Royuela M. The p38 transduction pathway in prostatic neoplasia. J Pathol 2006; 208:401-7; PMID:16369914; http://dx.doi.org/ 10.1002/path.1910 [DOI] [PubMed] [Google Scholar]
- 27.Khandrika L, Lieberman R, Koul S, Kumar B, Maroni P, Chandhoke R, Meacham RB, Koul HK. Hypoxia-associated p38 mitogen-activated protein kinase-mediated androgen receptor activation and increased HIF-1α levels contribute to emergence of an aggressive phenotype in prostate cancer. Oncogene 2009; 28:1248-1260; PMID:19151763; http://dx.doi.org/ 10.1038/onc.2008.476 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Eli Lilly and Company A phase 1 study of an oral p38 MAPK inhibitor in patients with advanced cancer In: ClinicalTrials.gov [Internet]. Bethesda (MD: ): National Library of Medicine (US) 2000- [cited 2014 Jan 21]. Available from: http://clinicaltrials.gov/archive/NCT01393990 NLM Identifier: NCT01393990. [Google Scholar]
- 29.Grossi V, Peserico A, Tezil T, Simone C. The p38α MAPK pathway: a key factor in colorectal cancer therapy and chemoresistance. World J Gastroenterol 2014; 20:9744-58; PMID:25110412; http://dx.doi.org/ 10.3748/wjg.v20.i29.9744; In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151-175; PMID:18188003; http://dx.doi.org/ 10.4161/auto.5338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Editorial: dishing out cancer treatment. Nat Biotechnol 2013; 31:85; PMID:23392482; http://dx.doi.org/ 10.1038/nbt.2516 [DOI] [PubMed] [Google Scholar]
- 33.Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013; 32:5501-11; PMID:23752182; http://dx.doi.org/ 10.1038/onc.2013.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med 2004; 10:33-39; PMID:14702632; http://dx.doi.org/ 10.1038/nm972 [DOI] [PubMed] [Google Scholar]
- 35.Fuzio P, Ditonno P, Lucarelli G, Battaglia M, Bettocchi C, Senia T, Perlino E. Androgen deprivation therapy affects BCL-2 expression in human prostate cancer. Int J Oncol 2011; 39:1233-42; PMID:21785821 [DOI] [PubMed] [Google Scholar]
- 36.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer 2001; 1:34-45; PMID:11900250; http://dx.doi.org/ 10.1038/35094009 [DOI] [PubMed] [Google Scholar]
- 37.De Marzo AM, Platz EA, Sutcliffe S, Xu J, Grönberg H, Drake CG, Nakai Y, Isaacs WB, Nelson WG. Inflammation in prostate carcinogenesis. Nat Rev Cancer 2007; 7:256-69; PMID:17384581; http://dx.doi.org/ 10.1038/nrc2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.