

Abstract
Cyclosporine A (CsA) is widely used as an immunosuppressor in transplantation. Previous studies reported that CsA induces autophagy and that chronic treatment with CsA results in accumulation of autophagosomes and reduced autophagic clearance. Autophagy is a prosurvival process that promotes recovery from acute kidney injury by degrading misfolded proteins produced in the kidney. In the present study, we used TMBIM6-expressing HK-2, human kidney tubular cells (TMBIM6 cells) and Tmbim6 knockout (tmbim6−/−) mice. When exposed to CsA, the TMBIM6 cells maintained autophagy activity by preventing autophagosome accumulation. With regard to signaling, PRKKA/AMPK phosphorylation and mechanistic target of rapamycin (serine/threonine kinase) complex 1 (MTORC1) expression and its downstream target TFEB (transcription factor EB), a lysosome biogenesis factor, were regulated in the TMBIM6 cells. Lysosomal activity was highly increased or stably maintained in the presence of TMBIM6. In addition, treatment of tmbim6−/− mice with CsA resulted in increased autophagosome formation and decreased lysosome formation and activity. We also found that tmbim6−/− mice were susceptible to CsA-induced kidney injury. Taken together, these results indicate that TMBIM6 protects against CsA-induced nephrotoxicity both in vitro and in vivo by inducing autophagy and activating lysosomes.
Keywords: autophagy, Bax inhibitor-1, cyclosporine A, ER stress, nephrotoxicity
Abbreviations
- ACTB/β-actin
actin, β
- PRKKA/AMPK
protein kinase, AMP-activated, α 1 catalytic subunit/AMP-activated protein kinase
- Baf
bafilomycin A1
- BAX
BCL2-associated X protein
- Ca2+
calcium
- CsA
cyclosporine
- CTSB
cathepsin B
- EIF4EBP1
eukaryotic translation initiation factor 4E binding protein 1
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- GLB1/β-galactosidase
galactosidase, β 1; GUSB/β-glucuronidase, glucuronidase, β
- H&E
hematoxylin and eosin
- HK-2
human kidney 2
- LAMP2
lysosomal-associated membrane protein 2
- MAN/α-mannosidase
mannosidase, α
- MAP1LC3B/LC3B
microtubule-associated protein 1 light chain 3 β
- MTORC1
mechanistic target of rapamycin (serine/threonine kinase) complex 1
- NC
negative control
- PAS
periodic acid-schiff
- PBS
phosphate-buffered saline
- RPS6KB1
ribosomal protein S6 kinase, 70kDa, polypeptide 1
- SQSTM1/p62
sequestosome 1
- TFEB
transcription factor EB
- TMBIM6/BI-1
transmembrane BAX inhibitor motif containing 6
- V-ATPase
vacuolar-type H+-ATPase
- WT
wild type.
Introduction
Autophagy is a normal cellular process that may have a protective function during exposure to physiological or chemical stresses. Autophagy is activated during starvation to produce energy through lysosomal-mediated degradation of autophagosomes. Autophagosomes fuse with lysosomes and lysosomal hydrolytic enzymes break down the autophagosomes to produce amino acids, which are used for the synthesis of macromolecules and energy.1,2 MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 β) is ubiquitous in cells and tissues. LC3-II a cleaved and phosphatidylethanolamine–conjugated form of LC3 and SQSTM1/p62 (sequestosome 1) are markers of autophagy.3,4 Lysosomes are the main organelles involved in autophagosome processing. LC3-II, which is present in the lysosomal membrane and SQSTM1 are degraded by lysosomal acidic enzymes after fusion of the lysosome with the autophagosome. Incomplete maturation and decrease in the activity of lysosomes lead to decreased formation of autolysosomes and increased accumulation of SQSTM1 and LC3-II in autophagosomal membranes. In most cases, autophagy has protective effects,5 and impaired induction of autophagy may induce cell death.6,7
Cyclosporine A (CsA) is widely used as a potent immunosuppressive drug to improve organ transplantation success rates.8,9 However, nephrotoxicity is associated with this drug.10,11 Chronic transplant dysfunction, a transplantation-related condition, is a prevalent cause of late grafts attenuation that is mainly triggered by chronic CsA nephrotoxicity. Chronic transplant dysfunction is characterized by decreased glomerular filtration rate and increased proteinuria, and accounts for 30% to 40% of late graft loss.12 The question regarding CsA-induced chronic nephrotoxicity has not been satisfactorily answered.13,14 However, CsA-induced autophagy is reported to protect against immunosuppressant-induced nephrotoxicity.15,16 Lysosomal modulation has been emerged as a regulating approach in autophagy-associated pathological conditions, including nephrotoxicity.17,18 Lysosome activity and the fusion process contribute to the maintenance and enhancement of autophagy. Chronic CsA nephropathy also causes excessive autophagosome formation and decreases autophagic clearance by disturbing the autophagy process,19 suggesting that modulation of the autophagy process may counter CsA-induced nephrotoxicity.
On the considerations of the above findings, we have hypothesized that the TMBIM6 (transmembrane BAX inhibitor motif containing 6)-associated lysosomal activity is a protective mechanism against the altered autophagy process model, that is, CsA-induced nephrotoxicity. The hypothesis is covering the potentiality; TMBIM6 may become an important target protein in the renal transplantation field to develop therapeutic agents against the nephrotoxicity.
TMBIM620 inhibits BAX protein activation, and thereby regulates apoptosis. TMBIM6 may activate autophagy by regulating Ca2+ transfer from the endoplasmic reticulum (ER) to mitochondria in a PRKAA/AMPK-dependent manner.21 We have previously have reported that TMBIM6 overexpression enhances lysosome activity, including lowering the lysosome pH and activating lysosome V-ATPase.22,23 Indeed, TMBIM6 may exert regulatory activity that protects against ER stress and cell death by maintaining lysosomal activity. Another report suggests that decreased tubular lysosomal proteinase activity might play a role in CsA-induced nephrotoxicity.24 TMBIM6-associated lysosomal activity might be a protective mechanism against the altered autophagy process model, CsA-induced nephrotoxicity. This hypothesis implies potentiality: TMBIM6 may be an important target protein in the renal transplantation field for the development of therapeutic agents against nephrotoxicity.
In the present study, we observed that TMBIM6 induces autophagy by enhancing lysosome biogenesis and activity. Using CsA, an immune-suppressor, the link between TMBIM6 and autophagy was revised to explain TMBIM6 function. Following recent reports on TMBIM6 and lysosomal activity,22,23 we examined the effects of CsA treatment on TMBIM6-associated lysosome and autophagy regulation using both in vitro and in vivo models.
Results
TMBIM6 increases autophagy, cell viability, and suppresses ER stress in CsA-treated human kidney cells
CsA-induced renal injury has been linked to alterations of autophagic clearance.19 In this study, we examined the effect of TMBIM6, a recently established regulator of ER stress and autophagy, on CsA-treated renal proximal tubule cells (HK-2 cells). First, we confirmed TMBIM6 mRNA expression with or without CsA-treated proximal tubule cells. TMBIM6 mRNA expression was significantly decreased in the presence of CsA (Fig. S1A). Treatment with CsA induced LC3 cleavage and SQSTM1 accumulation, indicating an alteration of autophagic clearance (Fig. S1B). Next, to examine whether TMBIM6 regulates autophagy in a CsA-treated human kidney cell line, we determined the expressions of LC3-II and SQSTM1 in HK-2 cells. The immunoblotting results for the negative control (NC cells) and stable TMBIM6-overexpressing cells (TMBIM6 cells) treated with 20 μM CsA at various time points. A time-dependent increase in LC3-II expression was observed in TMBIM6 cells compared to NC cells. NC cells showed SQSTM1 accumulation after CsA treatment in a time-dependent manner, whereas TMBIM6 cells showed less SQSTM1 accumulation after CsA treatment (Fig. 1A). TMBIM6 mRNA expression was confirmed in the cells (Fig. 2S). Furthermore, cotreatment with bafilomycin A1 (Baf), a lysosome V-ATPase inhibitor, blocked LC3-II degradation by the lysosomes and enhanced CsA-induced LC3-II accumulation, especially in TMBIM6 cells, compared with NC cells (Fig. 1B). Although SQSTM1 accumulation in NC cells was not greatly affected by Baf, the relatively low expression level of SQSTM1 in TMBIM6 cells was reversed by Baf treatment, indicating that CsA-induced alteration of autophagic clearance occurs less in TMBIM6 cells. To confirm autophagosome and autolysosome formation, we employed tandem mCherry-GFP-LC3 fluorescence analysis: autophagosomes appear as yellow (mCherry and GFP) puncta, while autolysosomes appear as red (mCherry) puncta. The number of puncta based upon the color fluorescence was quantified in each cell. The autophagosomes were accumulated in the CsA-treated NC cells and was reduced in the CsA-treated TMBIM6 cells (Fig. 1C, quantified in the right panel). The number of autolysosomes was highly increased in the presence of TMBIM6. The ratio of red to yellow puncta after CsA treatment was also higher in TMBIM6 cells than in NC cells (data not shown). This suggests that autolysosome formation was greater in TMBIM6-overexpressing cells after CsA treatment than in control cells. Transmission electron microscopy was utilized to visualize the ultrastructures of autophagy organelles in HK-2 cells (Fig. 1D). TMBIM6 cells had greater numbers of lysosomes than NC cells. After treatment with CsA, the number of autophagosomes (double-membrane-bound vacuoles) increased and the number of autolysosomes (single-membrane-bound vacuoles) decreased in NC cells, while in TMBIM6 cells, the number of autophagosomes slightly decreased and autolysosomes increased. TMBIM6 cells also showed time-dependent resistance to CsA-induced cell death and apoptosis (Figs. S3A and SB). Consistently, ER stress was reduced in the CsA-treated TMBIM6 cells, compared with NC cells (Fig. S3C). Taken together, these results demonstrate that LC3-II conversion and SQSTM1 degradation occurred at higher levels in TMBIM6 cells than in NC cells, indicating increased formation of autolysosomes in TMBIM6 cells. Furthermore, these data indicate that TMBIM6 overexpression promotes autophagy, which might be involved in the regulatory effect on ER stress and cell death.
Figure 1.

TMBIM6 increases autophagy in HK-2 cells. (A) NC and TMBIM6 cells were treated with 20 μM CsA for 0, 6, 12, 24, or 48 h. Immunoblotting was performed with anti-LC3-II, SQSTM1, and ACTB antibodies. The lower panel shows the results of densitometric analysis. #P < 0.05 vs. NC cells for each period. Images shown are representative of 3 independent experiments. (B) NC and TMBIM6 cells were treated with 20 μM CsA in the presence or absence of 80 nM bafilomycin for 24 h or 48 h. Immunoblotting was performed with anti-LC3-II, SQSTM1, and ACTB antibodies. The lower panel shows the results of densitometric analysis. *, P < 0.05 vs. NC cells during each period. #, P < 0.05 CsA+Baf vs. CsA during each period. Images shown are representative of 3 independent experiments (C) NC and TMBIM6 cells were transiently transfected with mRFP-GFP-LC3 for 24 h, after which the cells were treated with 20 μM CsA for 24 h. Red and green puncta were visualized by confocal microscopy. The right panel shows quantification of the ratio of mRFP-GFP-LC3 red to yellow puncta. #P < 0.05 vs. NC cells after treatment with CsA. (D) Lysosome and autophagic vacuoles (AVs) were analyzed by electron microscopy in NC and TMBIM6 cells with or without CsA. Results were quantified by counting the number of autophagic vacuoles per slice of view. Representative transmission electron micrographs of cell-in-cell structure showing autophagosomes (black arrow), lysosomes (white arrow), and autolysosomes (white asterisk).#, P < 0.05 vs. TMBIM6 cells after treatment with CsA.
TMBIM6-induced autophagy is related to activation of PRKAA and suppression of MTORC1 in CsA-treated human kidney cells
In this study, CsA was shown to activate PRKAA in NC cells in a time-dependent manner. Likewise, CsA increased phospho-PRKAA levels in TMBIM6 cells more significantly than in the NC cells (Figs. 2A and B; quantified). A PRKAA-linked signaling protein, MTORC1, was also examined. CsA treatment resulted in a more significant decrease in the levels of phosphorylated (p-) RPS6KB1 (ribosomal protein S6 kinase, 70kDa, polypeptide 1) and EIF4EBP1 (eukaryotic translation initiation factor 4E binding protein 1), the downstream targets of MTORC1 in TMBIM6 cells than in NC cells. These results suggest that TMBIM6-induced autophagy is related to activation of PRKAA and inhibition of the MTORC1 pathway in CsA-treated kidney cells.
Figure 2.
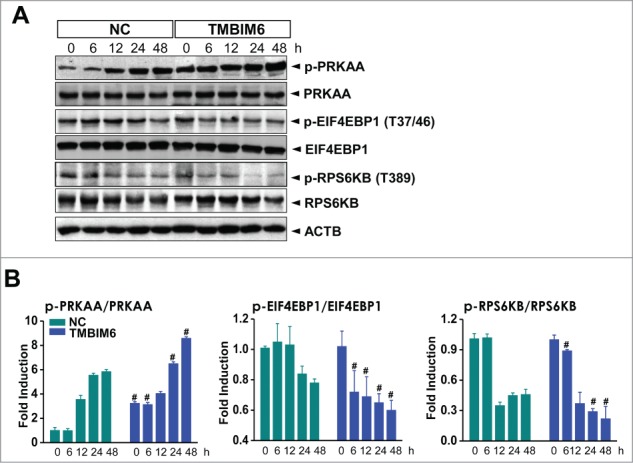
TMBIM6-induced autophagy is associated with inhibition of the MTORC1 pathway and activation of PRKAA in CsA-treated HK-2 cells. (A) Both NC and TMBIM6 cells were treated with 20 μM CsA for 0, 6, 12, 24, or 48 h. Western blotting was performed with p-PRKAA, PRKAA, p- EIF4EBP1, EIF4EBP1, p-RPS6KB, RPS6KB, and ACTB antibodies. (B) Densitometric quantification of selected western blot bands. #, P < 0.05 vs. NC cells for each period. Images shown are representative of 3 independent experiments.
TMBIM6 cells exhibit high lysosomal activity and biogenesis
Lysosomal activity is important for the autophagy degradation process,25,26 and we recently showed that TMBIM6 enhances lysosomal activity in other cell models.22,23 Based on these findings, we next tested TMBIM6-related lysosomal activity in CsA-treated cells. First, we measured the activities of the lysosomal enzymes, MAN/α-mannosidase, GLB1/β-galactosidase, and GUSB/β-glucuronidase in NC and TMBIM6 cells. Basal levels of these enzymes were higher in TMBIM6 cells than in NC cells. In the presence of CsA, these lysosomal enzymes were significantly decreased in the NC cells, while they were stably maintained in TMBIM6 cells (Fig. 3A). Using a specific lysosome dye, LysoTracker Red, we found that lysosomal fluorescence intensity was higher in CsA-treated TMBIM6 cells than in the treated NC cells (Fig. 3B). Staining with acridine orange also indicated that TMBIM6 cells have higher lysosomal V-ATPase activity after treatment with CsA than NC cells (Fig. 3C). We also analyzed protein expression and the activity of CTSB/cathepsin B, a lysosomal hydrolase. In NC cells treated with CsA, there was a transient increase in CTSB protein expression from 6 to 24 h, but levels of this protein decreased at 48 h (Fig. 3D). In contrast, protein expression levels of CTSB in TMBIM6 cells remained high and stable after treatment with CsA for 48 h. Similarly, the activity of CTSB was also higher in TMBIM6 cells than in NC cells, and was significantly higher than basal levels at 24 and 48 h of CsA treatment (Fig. 3E). We also measured the expression of the lysosomal-associated membrane protein 2 (LAMP2). In NC cells, LAMP2 expression was slightly increased by treatment with CsA (Fig. 3D). In TMBIM6 cells, LAMP2 levels were more significantly increased after CsA treatment in a time-dependent manner. Likewise, the fluorescence intensity of LAMP2 was greater in TMBIM6 cells than in NC cells after treatment with CsA (Fig. 3F). These findings suggest that TMBIM6 cells exhibit high lysosomal biogenesis and activity, probably linked to the enhanced autophagy clearance observed following CsA treatment.
Figure 3.

TMBIM6 cells exhibit high lysosomal activity following CsA treatment. (A) NC and TMBIM6 cells were treated with 20 μM CsA for 0, 6, 12, 24, or 48 h. Activities of MAN/α-mannosidase, GLB1/β-galactosidase, and GUSB/β-glucuronidase from lysosomal extracts were measured as described in Materials and Methods. #, P < 0.05 vs. NC cells for each period. Images shown are representative of 3 independent experiments. NC and TMBIM6 cells were treated with 20 μM CsA for 6 h and then exposed to 100 nM LysoTracker (B) or 1 µg/ml acridine orange (C) and photographed. Representative images are shown at × 600 magnification. Fluorescence intensities were quantified (right panel). *, P < 0.05 vs. NC cells without CsA; #, P < 0.05 vs. NC cells treated with CsA. (D) Western blotting was performed with CTSB, LAMP2, and LAMP1 antibodies. Images shown are representative of 3 independent experiments. (E) CTSB activity from lysosomal extracts was measured using a fluorospectrometer. #P < 0.05 vs. NC cells for each period. (F) LAMP2 immunocytochemical staining in NC and TMBIM6 cells treated with or without CsA for 6 h. DAPI was used to counterstain nuclei. Representative images are shown at ×600 magnifications. Fluorescence intensities were quantified (right panel). *, P < 0.05 vs. NC cells without CsA; #, P < 0.05 vs. NC cells treated with CsA.
TMBIM6 stimulates lyso-some biogenesis through TFEB activation
We examined TFEB, a master regulator of lysosomal biogenesis and autophagy, to determine the mechanisms involved in TMBIM6-associated lysosomal biogenesis and activation.27,28 We observed increased levels of TFEB mRNA after CsA treatment, especially in TMBIM6 cells (Fig. 4A). Treatment of cells with CsA for 6 h resulted in consistently stronger nuclear translocation of TFEB in TMBIM6 cells than in NC cells (Fig. 4B and C), suggesting that TMBIM6 stimulates lysosome biogenesis through TFEB activation to enhance autophagic clearance.
Figure 4.
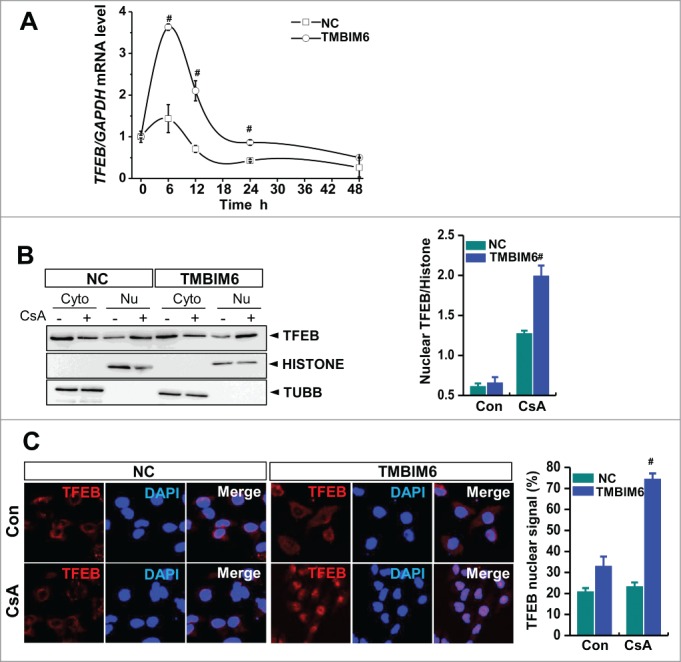
TMBIM6 induces TFEB activation. (A) TFEB mRNA levels were measured by real-time PCR, #, P < 0.05 vs. NC cells for each period. (B) Western blotting was performed with TFEB, histone (nuclear marker), and TUBB/tubulin (cytosol marker) antibodies. Densitometric quantification of western blot bands is shown in the right panel. #, P < 0.05 vs. NC cells treated with CsA. Cyto, cytosolic fraction; Nu, nuclear fraction. Images shown are representative of 3 independent experiments. (C) TFEB immunocytochemical staining of NC and TMBIM6 cells treated with or without CsA for 6 h. DAPI was used to counterstain nuclei. Representative images are shown at ×600 magnifications. Fluorescence intensities were quantified (right panel). #, P < 0.05 vs. NC cells treated with CsA.
Tmbim6-knockout increases CsA-induced kidney injury and ER stress
Before starting in vivo experiments, we evaluated Tmbim6 mRNA expression levels in the whole kidney, cortex, outer medulla, and inner medulla, and found that the expression of endogenous Tmbim6 was higher in the cortex than in the medulla (Fig. 5A). We also performed in situ hybridization analysis to confirm the localization of Tmbim6 in the kidneys of wild-type mice (WT) and Tmbim6-knockout mice (tmbim6−/−), and found that Tmbim6 was highly expressed as clusters of brown punctate signals in most cells in the cortex and medulla, with the signal being the strongest in the distal and proximal convoluted tubules in WT mice (Fig. 5B). The Tmbim6 signal was weaker in the collecting ducts, and very faint signals were found in mesangial cells and capillaries in the glomeruli. In the kidneys of tmbim6−/− mice, Tmbim6-positive signal was almost completely absent in the tubules and glomeruli. The signal from the housekeeping gene Ubc (ubiquitin C), was strong in all types of cells examined, including glomeruli, tubules, and collecting ducts, as well as endothelial cells. No signal was found in the negative control using the bacterial DapB gene. No differences in positive- or negative-control staining were identified when comparing kidney samples of WT and tmbim6−/− mice. Tmbim6 mRNA was also analyzed in the kidney tissues of CsA-treated mice. The expression of Tmbim6 was significantly decreased in WT mice (Fig. S4). To investigate the role of TMBIM6 in vivo, we examined CsA-induced nephrotoxicity in a tmbim6−/− mice model. WT and tmbim6−/− mice were treated with 15 mg/kg CsA by intraperitoneal injection, daily, for 30 d, after which kidneys and blood samples were collected. The kidney function of CsA-treated tmbim6−/− mice was significantly deteriorated as assessed by serum urea and creatinine concentrations (Fig. 5C and D). Upon histological examination with periodic acid-Schiff (PAS) staining, tmbim6−/− mice showed significant histological changes of the kidney, such as the loss of brush border cells and tubular dilation (Fig. 5E). The tmbim6−/− mice also exhibited higher ER stress response to CsA treatment than WT mice (Fig. 5F). After CsA treatment, tmbim6−/− mice showed greater levels of fibrosis than WT mice (Fig. 5G), suggesting that tmbim6−/− mice sustained more significant kidney injury and ER stress than did WT mice in response to CsA treatment.
Figure 5.

Tmbim6-knockout increases CsA-induced kidney injury and ER stress. (A) The mRNA expression of Tmbim6 was examined in whole kidney, renal cortex, and outer and inner medulla. Real-time PCR results from different sections of the kidney indicated that endogenous Tmbim6 mRNA levels were higher in the cortex region than in the outer and inner medullar regions. (B) In situ hybridization (ISH) analysis of Tmbim6, WT mice revealed clusters of brown punctate signals in most of the cells in the cortex and medulla, and signals were strongest in distal and proximal convoluted tubules. ((C)and D) WT mice and tmbim6−/− mice were treated with 15 mg/kg CsA by intraperitoneal injection daily for 30 d, and blood samples were collected for measurements of serum urea and serum creatinine. Each value represents the mean ± SEM of 3 independent experiments. #, P < 0.05 CsA tmbim6−/− mice vs. CsA WT mice. (E) Kidney tissue sections were used for PAS staining. Representative images are shown at × 600 magnification. (F) Immunoblotting of kidney lysates from WT mice and tmbim6−/− mice treated with or without CsA was performed using anti-HSPA5, -DDIT3, and -ACTB antibodies. Densitometric analysis results are shown in the right panel. #, P < 0.05 CsA tmbim6−/− mice vs. CsA WT mice. (G) Sirius Red staining was performed in the kidney tissue section to see the fibrosis.
TMBIM6 regulates CsA-induced autophagy in mice
To assess the biological function of autophagy in nephrotoxicity in vivo, we examined the expressions of several autophagy-related proteins in kidney tissue samples from CsA-treated mice, in particular LC3-II and SQSTM1. LC3-II protein expression was significantly higher in WT mice than in tmbim6−/− mice (Fig 6A). Similarly, WT mice had a greater number of GFP-LC3 puncta than did tmbim6−/− mice (Fig. 6B). To show more specifically show proximal tubule autophagy, LRP2 staining was performed in GFP-LC3-expressing WT and tmbim6−/− mice. Puncta formation was easier to be observed in proximal tubule brush border in the CsA-treated WT mice than the tmbim6−/− counter mice (Fig. 5S). Proximal tubule was positively stained with LRP2, a marker for proximal tubule brush border.2 SQSTM1 protein accumulation was also greater in tmbim6−/− mice after CsA treatment (Fig. 6A–C), indicating that stress may induce SQSTM1 accumulation in mouse tissues.19,29 To visualize the ultrastructure of autophagy organelles in CsA-treated mice, we examined tissue specimens by transmission electron microscopy (Fig. 6D). Control WT mice had greater numbers of lysosomes than did tmbim6−/− mice. Likewise, in CsA-treated tmbim6−/− mice, the abundance of autophagosomes (double-membrane-bound vacuoles) and autolysosomes (single-membrane-bound vacuoles) was decreased to a greater extent than in WT mice. Taken together, these results indicate that knockouts of Tmbim6 reduced the capacity for autophagic clearance in CsA-treated mice.
Figure 6.

TMBIM6 regulates CsA-induced autophagy in mice. (A) Immunoblotting of kidney lysates from WT mice and tmbim6−/− mice treated with or without CsA was performed using anti-LC3-II, -SQSTM1, and -ACTB antibodies. The right panel shows densitometric analysis results. #, P < 0.05 CsA tmbim6−/− mice vs. CsA WT mice. (B) Representative images of GFP-LC3 puncta in kidney tissues of GFP-LC3 WT and GFP-LC3 tmbim6−/− mice treated with or without CsA. GFP puncta were visualized by confocal microscopy. Representative images are shown at × 600 magnifications. (C) Kidney tissue sections were immunostained with an anti-SQSTM1 antibody. Representative images are shown at × 600 magnifications. (D) Lysosomes and autophagic vacuoles (AVs) were analyzed by electron microscopy in WT mice and tmbim6−/− mice treated with or without CsA. Results were quantified by counting the number of autophagic vacuoles per field of view (mean ± SEM of 3 mice per experimental group). Representative transmission electron micrographs of cell-in-cell structure showing autophagosomes (black arrow), lysosomes (white arrow) and autolysosomes (white asterisk). #, P < 0.05 CsA tmbim6−/− mice vs. CsA WT mice.
TMBIM6 regulates auto-phagy through the PRKAA-MTORC1 pathway in CsA-treated mice, enhancing lysosomal activity
We examined the effects of CsA on PRKAA and MTORC1 in WT and tmbim6−/− mice. PRKAA activation was higher in WT mice than in tmbim6−/− mice. The levels of p-RPS6KB1and p-EIF4EBP1 were also more significantly decreased in CsA-treated WT mice than in the tmbim6−/− mice (Fig. 7A), suggesting that TMBIM6 regulates CsA-induced activation of PRKAA and inhibition of MTORC1 in the nephrotoxicity model. Next, lysosomal enzymes, MAN/α-mannosidase, GLB1/β-galactosidase, and GUSB/β-glucuronidase were measured in this system. Similar to in vitro data, these enzymes were significantly reduced in the CsA-treated conditions (Fig. 7B). Compared to the knockout mice, these enzyme activities were decr-eased to a lesser extent in the WT mice, suggesting that TMBIM6 plays a regulatory role against CsA-induced alteration of lysosomal activity. In the WT mice, CTSB activity was less decreased than the tmbim6−/− mice, showing resistance against CsA (Fig. 7C). We next measured the expression levels of LAMP2 protein in kidney tissue. In CsA-treated WT mice, the level of LAMP2 protein expression were increased compared to CsA treated tmbim6−/− mice (Fig. 7D). To investigate a candidate mechanism to explain enhanced lysosomal biogenesis, we examined Tfeb transcription level and nuclear translocation. WT mice exhibited more elevated levels of Tfeb mRNA after CsA treatment compared with tmbim6−/− mice (Fig. 7E). In addition, nuclear translocation of TFEB was highly detected in the CsA-treated WT mice, compared with the treated tmbim6−/− mice (Fig. 7F). These observations suggest that loss of TMBIM6 inhibited lysosome biogenesis, which may be related to the decreased autophagic clearance ability of CsA-treated tmbim6−/− mice. We also confirmed the role of TMBIM6 in CsA-treated mice that were fed a low-salt diet. The result were similar to mouse fed with normal salt diet (Fig. 7). The kidneys of CsA-treated tmbim6−/− mice showed more significant destruction than those of WT mice, based on serum creatinine analysis and hematoxylin and eosin (H&E) staining (Figs. S6, S7C). Sirius red staining also demonstrated greater fibrosis in CsA-treated tmbim6−/− mice than in WT mice (Fig. S7G). In CsA-treated mice fed a low-salt diet, fibrosis patterns appeared more severe than those of CsA-treated mice fed a normal diet (Fig. 5G). However, all the low and normal salt diet-fed mice showed high levels of sensitivity against CsA, especially the tmbim6−/− mice. Other suggested mechanisms, including those involving PRKAA, MTORC1, or ER stress regulation, were also consistent with the finding with low salt diet (Fig. S7A to S7F).
Figure 7.

TMBIM6 regulates autophagy through the PRKAA-MTORC1 pathway in CsA-treated mice and regulates lysosomal activity. (A) Immunoblotting of kidney lysates from WT mice and tmbim6−/− mice treated with or without CsA was performed using p-PRKAA, PRKAA, p-EIF4EBP1, EIF4EBP1, p-RPS6KB, RPS6K, and ACTB antibodies. The right panel shows the densitometric analysis results. *, P < 0.05 tmbim6−/− mice vs. WT mice; #, P < 0.05 CsA tmbim6−/− mice vs. CsA WT mice. The activities of MAN/α-mannosidase, GLB1/β-galactosidase, and GUSB/β-glucuronidase (B) and CTSB activity (C) from lysosomal extracts were measured. *, P < 0.05 tmbim6−/− mice vs. WT mice; #, P < 0.05 CsA tmbim6−/− mice vs. CsA WT mice. (D) Kidney tissue sections were immunostained with an anti-LAMP2 antibody. Representative images are shown at × 600 magnification. (E) Tfeb mRNA levels were measured by real-time PCR; #, P < 0.05 CsA tmbim6−/− mice vs. CsA WT mice. (F) Western blotting was performed with antibodies to TFEB, histone (nuclear marker), and TUBB/tubulin (cytosol marker). Densitometric quantification of western blot bands is shown (lower). #, P < 0.05 CsA tmbim6−/− mice vs. CsA WT mice. Cyto, cytosolic fraction; Nu, nuclear fraction.
A general scheme describing TMBIM6-enhanced autophagy in response to CsA treatment is shown in Figure 8. CsA inhibits fusion between autophagosomes and lysosomes, leading to the accumulation of SQSTM1 and induction of ER stress, whereas TMBIM6 enhances PRKAA phosphorylation, thereby affecting MTORC1 status. Regulation of MTORC1 leads to TFEB nuclear translocation and lysosomal biogenesis, which is linked to autophagy. This mechanism is suggested to explain how TMBIM6 protects against CsA-induced nephrotoxicity.
Figure 8.

Proposed mechanism for TMBIM6-induced regulation against CsA-associated autophagy process alteration and renal injury. TMBIM6 protects against CsA toxicity by activating autophagy and enhancing autophagic degradation. TMBIM6 enhances PRKAA phosphorylation, regulates MTORC1 activity and increases TFEB activity. Resultantly, lysosome biogenesis, autolysosome formation, and autophagic flux are stimulated in the presence of TMBIM6, inhibiting CsA-induced autophagosome accumulation and its linked renal injury.
Discussion
In this study, we have showed that TMBIM6 enhances autophagy, which contributes to protection against CsA-induced renal toxicity. This effect appears to be mediated through p-PRKAA and MTORC1 signal transduction, in which enhanced autophagosome formation and lysosome activity are involved.
We observed that TMBIM6 overexpression led to increased expression of LC3-II, decreased expression of SQSTM1, and increased cell viability, suppressing the ER stress response in CsA-treated HK-2 cells (Fig. 1A, S3A, S3C). In TMBIM6 cells, LC3-II expression was significantly increased under cotreatment with CsA and Baf, compared with CsA alone, suggesting that TMBIM6 regulates the autophagy flux (Fig. 1B). However, LC3-II levels were not significantly affected in control cells. Considering that Baf, causes autophagosomes to accumulate by inhibiting lysosomal V-ATPase activity,30 the pattern of increased LC3-II in TMBIM6-expressing cells may represent both autophagosome formation and autophagy flux.31 We also showed that in TMBIM6 cells, the numbers of GFP-LC3-labeled autophagosomes and mCherry-LC3-labeled autolysosomes were more significantly increased after CsA stimulation compared with control cells, indicating enhanced autophagy in TMBIM6 cells (Fig. 1C). LC3-II is a marker of autophagosome formation;4 specifically, LC3-II binds SQSTM1, a receptor protein that targets ubiquitinated protein aggregates for lysosomal degradation, which are then selectively degraded via autophagy.32 Our results, therefore, strongly suggest that TMBIM6 enhances autophagy in the CsA-treated cells.
Several studies have reported that autophagy induced by CsA treatment protects renal tubular cells and kidneys.2,16 Considering that TMBIM6, suspected to act as an autophagy enhancer in this study, functionally inhibits BAX,20,33 the role of BAX in autophagy must be considered. Although neither overexpression of BAX or BAK1 nor ER-targeted BAK1 has been reported to affect autophagy,34 BAX has been shown to induce autophagy and autophagic cell death.35,36 If TMBIM6 affects autophagy through the inhibition of BAX, TMBIM6 might lead to the inhibition of autophagy. Recently, TMBIM6-induced inhibition of autophagy has been reported,37 but our data indicate that TMBIM6 enhances autophagy, suggesting that enhancement of autophagy is not tightly matched to BAX inhibition. Several mechanisms have been suggested regarding the role of TMBIM6 in the regulation of ER stress, including the interaction between TMBIM6 and ERN1/IRE-1α, the involvement of BCL2, intra-ER Ca2+ homeostasis, and highly enhanced lysosome activity. Our results suggest that lysosome-associated enhanced autophagy contributes to the regulation of ER stress and the resulting protective effect in the CsA nephropathy model.
TMBIM6 has been reported to have an antiapoptotic effect.20,33,38,39 There have been suggestive mechanisms about the protective properties against death stimuli.33,40 Among these suggested mechanisms, inhibition of ERN1/IRE-1α through interaction with TMBIM6, has been debated in studies of autophagy.21,37 The known characteristics of TMBIM6, such as a Ca2+ channel-like effect controlling intra-ER Ca2+ homeostasis, an indirect effect on mitochondrial Ca2+, and subsequent alterations in mitochondrial metabolism, seem to be more closely related to enhanced autophagy.21 The mitochondrial metabolism signaling may include PRKAA phosphorylation in TMBIM6-expressing cells. TMBIM6-associated enhanced autophagy appears to include the PRKAA-MTORC1 pathway (Fig. 2). Under stress, TMBIM6 maintains PRKAA phosphorylated and significantly inhibits the MTORC1 signaling pathway, which in turn, leads to the activation of autophagosome formation and subsequent enhanced lysosome degradation activity. We observed that TMBIM6 cells stably maintained lysosomal activity at high levels and also showed greatly enhanced lysosomal biogenesis following CsA treatment compared with control cells (Fig. 3A to F). We also found that TMBIM6 stimulated the activation of TFEB (Fig. 4B and C), a master regulator of lysosome biogenesis.27 TFEB activation is dependent on reduced MTORC1 activity.41 TMBIM6 cells also exhibited highly increased LAMP2 protein expression, suggesting that TMBIM6 promotes the fusion of autophagosomes with lysosomes, in addition to enhancing lysosome biogenesis. In this study, TMBIM6-mediated enhancement of lysosomal activity is suggested to protect cells against CsA-induced nephropathy. Other pathophysiological phenomena including aging, neurodegenerative disease, injuries, and infection have been suggested to require adaptive responses via lysosome activity.42-44 Abnormalities in lysosome action or biogenesis can lead to increased apoptosis.45,46 Lysosome impairment has also been suggested as a key mechanism in aminoglycoside antibiotic-induced nephrotoxicity.47-51 Renal cell-accumulated aminoglycosides are localized primarily in endosomal and lysosomal vacuoles. They typically induce clear shifts in the lysosomes of proximal tubular epithelial cells, followed by symptoms of tubular deterioration or malfunction such as increased excretion of filtered proteins, glucose, and discharge of the brush-border along with lysosomal enzymes. In this study, we found that CsA-induced nephropathy includes structural alterations and interstitial fibrosis (Figs. 5E, G, and S7G). The presence of TMBIM6 positively regulates autophagy process through the enhancement of lysosome biogenesis and functional maintenance, a suggestive protective mechanism against CsA-induced toxicity. The presence of TMBIM6 explains the biological role of lysosomes and of lysosome-related autophagy in nephrotoxic phenomena.
Autophagy signaling pathways under conditions of enhanced lysosome biogenesis are summarized (Fig. 8). We have reported that TMBIM6 regulated lysosome activity22 and this study also suggests that CsA increases autophagosome accumulation by inhibiting lysosomal fusion. The immunosuppressant, CsA decreases lysosome biogenesis thereby inducing autophagosome accumulation and alterations of autolysosome formation, which lead to renal injury (left panel in Fig 8). TMBIM6 overexpression increases phosphorylation of PRKAA that inhibits MTORC1 therewith TFEB activity is upregulated (right panel in Fig 8). The upregulation of TFEB activity increases lysosome biogenesis and lysosome activity, enhancing autophagosome fusion with lysosomes, which may be a protective mechanism against CsA cytotoxicity.
CsA-induced nephrotoxicity has been studied in the context of renal transplantation.52-55 Significant alterations of autophagy19,56 have been reported in CsA-associated nephrotoxicity. CsA-induced renal injury is frequently observed in renal tubular structure including proximal tubules.57-59 In addition, CsA downregulates endogenous TMBIM6 mRNA expression in renal proximal tubules cells (Fig. S1A). The endogenous expression of Tmbim6 mRNA is observed mainly in renal proximal and distal tubules (Fig. 5A and B). Other nephrotoxic agent, cisplatin was applied to the TMBIM6-knockout mice system. Cisplatin has been also known about inducing the impairment of renal proximal and distal tubular function.60 TMBIM6 showed protection effect against cisplatin-associated renal damage (data not shown). Moreover, accumulating evidence has indicated that TMBIM6 protects cells from death stimuli.20,33,38,39 Taken together, these observations suggest that the decrease of TMBIM6 expression is involved in decreased renal function, as well as the onset and exacerbation of complications of chronic renal failure through autophagy alteration. TMBIM6 enhancers or agonists may be useful as therapeutic agents in the complications of chronic renal failure. We screened natural products and chemical candidates through BAX inhibition screening assays, along with ER stress regulation and lysosome activity enhancement associated with TMBIM6. We observed that a few chemicals increased Tmbim6 gene expression and its related lysosome activity (data not shown). Considering that TMBIM6 may be useful as a new therapeutic mechanism against CsA-induced toxicity, extensive pharmaceutical studies of TMBIM6 should be performed in the near future.
Taken together, our results suggest that p-PRKAA-MTORC1-associated signaling explains TMBIM6-induced autophagy and the associated protective effects in CsA-induced nephrotoxic phenomena. More detailed studies are necessary to better explain the mechanism underlying the protective role of TMBIM6 against CsA-induced nephrotoxicity.
Materials and Methods
Reagents
Cyclosporine A (30024), bafilomycin A1 (B1793) and Hoechst stain (33342) were purchased from Sigma-Aldrich. Fluorescent probes, including LysoTracker Red DND-26 (L7528) and acridine orange (A3568), were obtained from Molecular Probes.
The antibody against LC3 (NB100-2220) was purchased from Novus Biological. The antibody against SQSTM1 (610832) was purchased from BD Biosciences and the antibody against LAMP2 (ab13524) was purchased from Abcam. The antibody against TFEB (MBS150693) was purchased from My Biosource. Antibodies against p-PRKAA (Thr172;2531), PRKAA (2532), pEIF4EBP1 (T37/46;2855), EIF4EBP1 (9644), p-RPS6KB/p70S6K (T389;9234), and RPS6KB/p70S6K (2708) were purchased from Cell Signaling Technology. Antibodies against ACTB/β-actin (sc-8432), CTSB/cathepsin B (sc-13985), LAMP1 (sc-18821), HSPA5/GRP78 (sc-13539), DDIT3/CHOP (sc-793), and GAPDH (sc-25778) were purchased from Santa Cruz Biotechnology. Anti-LRP2 antibody (ab184676) was purchased from Abcam.
Cell culture condition and stability
HK-2 cells from an immortalized human proximal tubular epithelial cell line were obtained from the Korean Cell Line Bank (Seoul National University, Seoul, Korea). Cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin, and incubated at 37°C in a humidified atmosphere containing 5% CO2. HK-2 cells were infected with lentivirus particles to introduce either a GFP expression vector (negative control, NC cells) or a TMBIM6-GFP expression vector (TMBIM6-overexpressing cells, TMBIM6 cells), both of which were obtained from Open Biosystems (GE Dharmacon, USA).
mCherry-GFP-LC3 plasmid transfection
Autophagy flux was monitored by fluorescence microscopy of the distribution and alteration of mCherry-GFP-LC3 fluorescent signals.61,62 The mCherry-GFP-LC3 plasmid encoding the tandem fluorescent reporter was generated in the laboratory of Dr. Terje Johansen63 and obtained from Add gene (plasmid 22418). Cultured HK-2 cells seeded in cover glass-bottom dishes were transfected with mCherry-GFP-LC3 plasmid using Lipofectamine 2000 (Invitrogen, 11668-019) for 24 h. After the designated treatments, live cell images were obtained using a DeltaVision microscope imaging system (Applied Precision, Issaquah, WA, USA). For the quantification of autophagic cells, GFP-LC3 and mCherry-LC3 punctate dots were determined from triplicate samples by counting a total of more than 30 cells.64
TFEB mRNA level detection by quantitative real-time PCR
Total RNA was extracted from HK-2 cells and mouse kidney tissue samples using TRIzol reagent (Invitrogen Life Technologies, 15596018). The ethanol-precipitated RNA fraction (500 ng) was reverse transcribed using a PrimeScriptTM RT reagent kit (Takara, RR037A) according to the manufacturer's protocol. Forward and reverse primer sequences for mouse Tfeb were 5′-AAC AAA GGC ACC ATC CTC AA-3′ and 5′-GGA GCC AGA GCT GCT TGT TA-3′, respectively. Forward and reverse primer sequences for human TFEB were 5′-TGT TGC TGC ATG CGC TC-3′ and 5′-CGG CAG TGC CTG GTA CAT-3′, respectively. Forward and reverse primer sequences for human TMBIM6 were 5′-AGC AGC ACC TGA AGG TC-3′ and 5′-TCA ATA TCA GGG AGC CCA AG-3′, respectively. Forward and reverse primer sequences for mouse Tmbim6 were 5′-TCC CTC GAC ACA GCA GCA CCT-3′ and 5′-CCC CAG AGA GGA CAG GAG CAT-3′, respectively. Forward and reverse primer sequences for mouse Gapdh were 5′-AAC TTT GGC ATT GTG GAA GG-3′ and 5′-GGA TGC AGG GAT GAT GTT CT-3′, respectively. Forward and reverse primer sequences for human GAPDH were 5′-AGG GCT GCT TTT AAC TCT GGT-3′ and 5′-CCC CAC TTG ATT TTG GAG GGA-3′, respectively. Relative gene expression was determined by real-time PCR using an Applied Biosystems 7900HT Fast Real-Time PCR System (Applied Biosystems, CA, USA) according to the manufacturer's recommended protocol. Each analysis was performed in triplicate.
Acridine orange staining
After the indicated treatments, HK-2 cells were washed with phosphate-buffered saline (PBS; Biosesang, P2007-1) and incubated with 1 µg/ml acridine orange (Molecular Probes, A1301) in normal medium for 30 min at 37°C. Subsequently, acridine orange was removed and cells were examined under an inverted fluorescence microscope. The cytoplasm and nucleus of stained cells fluoresced bright green, whereas the acidic autophagic vacuoles fluoresced bright red.65
LysoTracker Red staining
LysoTracker Red probes used for the assessment of lysosome activity have high selectivity for acidic organelles and are effective for labeling live cells.66 HK-2 cells were grown in culture dishes, rinsed with PBS, and stained with 100 nM LysoTracker Red DND-26 in serum-free medium for 30 min at room temperature. Cells were then washed with PBS. Lysosome size and staining intensity were analyzed by fluorescence microscopy at 590 nm. Cells were viewed and analyzed using SoftworxDeltaVision software (Applied Precision, Issaquah, WA, USA).
Isolation of lysosomes
Lysosomes were isolated from HK-2 cells to measure lysosomal enzyme activity and to perform western blotting analyses. Lysosomes were isolated by differential and density-gradient centrifugation techniques using an OptiPrep density gradient kit (Sigma-Aldrich, Lysosome Isolation Kit, LYSISO1) according to the manufacturer's instructions.
Lysosome enzyme activity assay
Activities of lysosomal enzymes were measured as described previously. Briefly, after isolation of lysosomes, assay mixtures were prepared in a final volume of 100 µL, as described below. The GLB1/β-galactosidase assay mixture consisted of 0.5 mM 4-methylumbelliferyl-β-galactoside (Matrix scientific, 098488) in 100 mM citrate-phosphate buffer (pH 4.35) with 0.4 M NaCl. The GUSB/β-glucuronidase assay mixture consisted of 1 mM 4-β-glucuronide in 100 mM acetate buffer (pH 4.0). Finally, the MAN/α-mannosidase assay mixture consisted of 4 mM α-mannopyranoside in 100 mM acetate-phosphate buffer (pH 4.0). Assay mixtures were incubated at 37°C for 30 min and measured at appropriate excitation wavelengths ranging from 360 nm to 410 nm. CTSB activity was measured in cell samples using a CTSB activity assay kit from Calbiochem (EMD Biosciences, CBA001) according to the manufacturer's instructions.
Western blot
Western blot analysis was performed as described previously with some modifications.33 Cells or tissue samples were lysed directly in culture dishes on ice with RIPA buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% sodium deoxycholate (ELPIS Biotech, EBA 1149), 1% Triton X-100 (Sigma,T8532) 5 mM EDTA, 0.1% SDS (Amresco, 0227-500G) containing a protease inhibitor cocktail and phosphatase inhibitor cocktail (Roche Diagnostics, 04906845001). Cell lysates were incubated for 30 min on ice, followed by centrifugation at 10,000 × g for 20 min at 4°C. The resulting supernatant fractions were collected and used as protein extracts. Protein extracts were added to sample buffer, boiled in a water bath for 5 min, and stored at −80°C until use. Protein extracts (30 µg) were separated on polyacrylamide gels and transferred to PVDF membranes for 40 min at 15 V using a semi-dry transfer system (Bio-Rad, Hercules, CA, USA). Membranes were blocked with 5% nonfat dry milk in 0.05% Tween-Tris-buffered saline (1x TBST; 20 mM Trizma base, pH 7.6, 136.9 mM NaCl, 500 µl Tween 20) for 60 min at room temperature. Blots were then probed overnight at 4°C with the relevant primary antibodies, washed, and probed again with species-specific secondary antibodies coupled to horseradish peroxidase (Santa Cruz Biotechnology, sc-2004, sc-2006, sc-2005 and sc-2768). Chemiluminescence reagents (GE Healthcare, RPN 2108) were added for detection, and immunoreactive bands were visualized using an LAS-3000 system (Fuji Film, Japan). Band intensities were measured and quantified using ImageJ software from the National Institutes for Health, as described by Miller (http://lukemiller.org/journal/2007/08/quantifying-western-blots-without.html).
LAMP2 and TFEB immunostaining
HK-2 cells were grown on cover glass-bottom dishes and treated with the indicated agents. Cells were then fixed with ice-cold methanol and permeabilized with PBST (PBS containing 0.25% Triton X-100). Following 30 min incubation in blocking buffer (1% BSA [Sigma Aldrich, SLBM27] in PBST), cells were incubated with rat anti-LAMP2 (1:100) or rabbit anti-TFEB (1:100) overnight at 4°C. Then, cells were washed twice and stained with either a FITC-conjugated anti-rat secondary antibody (1:100; Sigma, F6258) or a TRITC-conjugated anti-rabbit secondary antibody (1:100; Sigma, T5268) for 1 h. Finally, cells were mounted using VECTASHIELD® mounting medium (Vector Laboratories, H-1200) containing DAPI. Signals were examined by fluorescence microscopy using the FITC, TRITC, and DAPI channels.
Animal experiments
All animal experiments were approved by the Institutional Animal Care and Use Committee of Chonbuk National University, Jeon-ju, Korea. All procedures performed in this study followed ethical guidelines for animal studies. The WT and tmbim6−/− (knockout) mice used in this study were described previously.33 The GFP-LC3 transgenic mice used in this study were obtained from RIKEN BioResource Center, Japan.67 GFP-LC3 WT or GFP-LC3 tmbim6−/− mice were obtained by crossing tmbim6−/− mice and GFP-LC3 mice. All mice were housed in a temperature- and light-controlled environment.
In the first set of animal experiments to evaluate the effect of CsA on autophagy, mice were divided into 4 groups. (1) In the WT vehicle control group, WT (n = 6) and GFP-LC3 WT (n = 4) mice received a daily intraperitoneal injection of olive oil. (2) In the tmbim6−/− vehicle control group, tmbim6−/− (n = 6) and GFP-LC3 tmbim6−/− (n = 4) mice received a daily intraperitoneal injection of olive oil. (3) In the WT CsA group, WT (n = 6) and GFP-LC3 WT (n = 4) mice received a daily intraperitoneal injection of CsA (15 mg/kg/day in olive oil) for 30 d. Last, in the (4) tmbim6−/− CsA group, tmbim6−/− (n = 6) and GFP-LC3 tmbim6−/− (n = 4) mice received a daily intraperitoneal injection of CsA (15 mg/kg/d in olive oil for 30 d). The dosage and route of administration for CsA were selected based on previous studies in mice.19,68 Animals were anesthetized with ketamine one d after the treatment period was concluded, and blood and kidney tissue samples were collected.
GFP-LC3 WT mouse tissue samples for GFP observation were prepared as described previously.67,69 To prevent induction of autophagy during tissue preparation, mice were anesthetized with diethyl ether and immediately fixed by perfusion through the left ventricle with PBS. Tissues were harvested and further fixed with 4% paraformaldehyde for 24 h, followed by treatment with 15% sucrose in PBS for 4 h, which was increased to a 30% sucrose solution overnight. Tissue samples were embedded in Tissue-Tek OCT compound (Sakura Finetechnical, 14-373-65) and stored at −70°C. Samples were sectioned at a thickness of 5 μm with a cryostat (CM3050 S, Leica, and Deerfield, IL, USA), air-dried for 30 min, washed in PBS, and mounted on glass slides. The number of GFP-LC3 puncta per unit area of tissue was quantified by fluorescence microscopy. Autophagy in kidney tissues after CsA injection was also analyzed by western blot analysis of tissue extracts with antibodies against LC3 and SQSTM1.
Kidney function studies
Serum urea was measured using a Urea Colorimetric Assay Kit (BioVision, K375-100). Serum creatinine was measured using a Creatinine Colorimetric/Fluorometric Assay Kit (BioVision, K625-100).
Electron microscopy
Kidney tissues were fixed by a conventional method (2% glutaraldehyde and 2% formaldehyde in 0.05 M sodium cacodylate buffer, pH 7.2).70,71 Fixed samples were embedded in Spurr's resin (Electron Microscopy Sciences, 14300) and thin sections (80 nm) were then cut and stained with uranyl acetate and lead citrate for observation under a transmission electron microscope (H7650, accelerating voltage 100 kV, Hitachi, Tokyo, Japan) at the Center for University-wide Research Facilities (CBNU). The numbers of lysosomes and autophagic vacuoles were counted, and autophagy was quantified in each sample for at least 10 cells and confirmed by 2 additional independent experiments.
Histology and immunostaining
For H&E staining, kidney tissues were fixed in 4% paraformaldehyde for 24 h. Tissue was dehydrated in a graded ethanol series and left overnight in xylene. After embedding in paraffin, 4-μm-thick sections were cut on a rotary microtome (Leica Microsystems, Herlev, Denmark). H&E staining was performed to assess histological tissue injury. PAS staining of kidney sections was performed with a PAS kit (Sigma-Aldrich, 395B), followed by dehydration in an ascending ethanol series and then xylene for clearance and mounting. Sirius Red Stain kits (IHC WORLD, Nova Ultra special stain kit, IW-3012) were used to stain fibrosis in kidney sections.
In situ hybridization
In situ detection of Tmbim6 transcripts in WT and tmbim6−/− mouse kidney samples was performed using the RNAscope assay kit (Advanced Cell Diagnostics, 310035), as described previously.72 Briefly, 4-μm-thick paraffin-embedded tissue sections were deparaffinized and pretreated with heat and proteases, then hybridized with a target probe for the Tmbim6 gene. Next, an HRP-based signal amplification system was hybridized to the target probe before color development with 3, 3′-diaminobenzidine tetrahydrochloride (Advanced Cell Diagnostics, 310033). Positive staining was defined as the presence of brown punctate dots in the nucleus and/or cytoplasm. The housekeeping gene Ubc (ubiquitin C) served as a positive control. The DapB gene, which is derived from a bacterial gene sequence, was used as a negative control.
Statistical analysis
All data are presented as the mean ± SEM from several independent experiments, as specified in the figure legends. Differences between 2 groups were analyzed using 2-tailed Student t tests. Origin 8.0 software from OriginLab® Corp (Northampton, MA, USA) was used for statistical calculations. A value of P < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by funding from the Korean National Research Foundation (2015R1A2A1A13001849, 2012R1A2A2A01045214, 2008-0062279, NRF-2013R1A1A2006099). This work was partly supported by the Korea Healthcare Technology R&D Project (A121931), Ministry for Health and Welfare, Republic of Korea. This paper was partly supported by research funding of Chonbuk National University in 2014.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, et al.. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 2013; 32:2336-47; PMID:23921551; http://dx.doi.org/ 10.1038/emboj.2013.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, et al.. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 2011; 22:902-13; PMID:21493778; http://dx.doi.org/ 10.1681/ASN.2010070705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 2009; 452:181-97; PMID:19200883; http://dx.doi.org/ 10.1016/S0076-6879(08)03612-4 [DOI] [PubMed] [Google Scholar]
- 4.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720-8; PMID:11060023; http://dx.doi.org/ 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823-30; PMID:20811354; http://dx.doi.org/ 10.1038/ncb0910-823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kroemer G, Jaattela M. Lysosomes and autophagy in cell death control. Nat Rev Cancer 2005; 5:886-97; PMID:16239905; http://dx.doi.org/ 10.1038/nrc1738 [DOI] [PubMed] [Google Scholar]
- 7.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest 2005; 115:2679-88; PMID:16200202; http://dx.doi.org/ 10.1172/JCI26390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hariharan S, Johnson CP, Bresnahan BA, Taranto SE, McIntosh MJ, Stablein D. Improved graft survival after renal transplantation in the United States, 1988 to 1996. N Engl J Med 2000; 342:605-12. [DOI] [PubMed] [Google Scholar]
- 9.Sommerer C, Giese T, Meuer S, Zeier M. Pharmacodynamic monitoring of calcineurin inhibitor therapy: is there a clinical benefit? Nephrol Dial Transplant 2009; 24:21-7; PMID:18842676; http://dx.doi.org/ 10.1093/ndt/gfn556 [DOI] [PubMed] [Google Scholar]
- 10.Kopp JB, Klotman PE. Cellular and molecular mechanisms of cyclosporin nephrotoxicity. J Am Soc Nephrol 1990; 1:162-79; PMID:2104260 [DOI] [PubMed] [Google Scholar]
- 11.Busauschina A, Schnuelle P, van der Woude FJ. Cyclosporine nephrotoxicity. Transplant Proc 2004; 36:229S-33S; PMID:15041343; http://dx.doi.org/ 10.1016/j.transproceed.2004.01.021 [DOI] [PubMed] [Google Scholar]
- 12.Bakker RC, Scholten EM, de Fijter JW, Paul LC. Chronic cyclosporine nephrotoxicity in renal transplantation. Transplantation Reviews 2004; 18:54-64; http://dx.doi.org/ 10.1016/j.trre.2004.01.002 [DOI] [Google Scholar]
- 13.Nankivell BJ, Borrows RJ, Fung CL, O'Connell PJ, Allen RD, Chapman JR. The natural history of chronic allograft nephropathy. N Engl J Med 2003; 349:2326-33; PMID:14668458; http://dx.doi.org/ 10.1056/NEJMoa020009 [DOI] [PubMed] [Google Scholar]
- 14.Yoon HE, Yang CW. Established and newly proposed mechanisms of chronic cyclosporine nephropathy. Korean J Intern Med 2009; 24:81-92; PMID:19543484; http://dx.doi.org/ 10.3904/kjim.2009.24.2.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int 2012; 82:1271-83; PMID:22854643; http://dx.doi.org/ 10.1038/ki.2012.261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pallet N, Bouvier N, Legendre C, Gilleron J, Codogno P, Beaune P, Thervet E, Anglicheau D. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy 2008; 4:783-91; PMID:18628650; http://dx.doi.org/ 10.4161/auto.6477 [DOI] [PubMed] [Google Scholar]
- 17.Lee JK, Jin HK, Park MH, Kim BR, Lee PH, Nakauchi H, Carter JE, He X, Schuchman EH, Bae JS. Acid sphingomyelinase modulates the autophagic process by controlling lysosomal biogenesis in Alzheimer's disease. J Exp Med 2014; 211:1551-70; PMID:25049335; http://dx.doi.org/ 10.1084/jem.20132451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Appelqvist H, Waster P, Kagedal K, Ollinger K. The lysosome: from waste bag to potential therapeutic target. J Mol Cell Biol 2013; 5:214-26; PMID:23918283; http://dx.doi.org/ 10.1093/jmcb/mjt022 [DOI] [PubMed] [Google Scholar]
- 19.Lim SW, Hyoung BJ, Piao SG, Doh KC, Chung BH, Yang CW. Chronic cyclosporine nephropathy is characterized by excessive autophagosome formation and decreased autophagic clearance. Transplantation 2012; 94:218-25; PMID:22790389; http://dx.doi.org/ 10.1097/TP.0b013e31825ace5c [DOI] [PubMed] [Google Scholar]
- 20.Xu Q, Reed JC. Bax inhibitor-1, a mammalian apoptosis suppressor identified by functional screening in yeast. Mol Cell 1998; 1:337-46; PMID:9660918; http://dx.doi.org/ 10.1016/S1097-2765(00)80034-9 [DOI] [PubMed] [Google Scholar]
- 21.Sano R, Hou YC, Hedvat M, Correa RG, Shu CW, Krajewska M, Diaz PW, Tamble CM, Quarato G, Gottlieb RA, et al.. Endoplasmic reticulum protein BI-1 regulates Ca(2)(+)-mediated bioenergetics to promote autophagy. Genes Dev 2012; 26:1041-54; PMID:22588718; http://dx.doi.org/ 10.1101/gad.184325.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee GH, Kim DS, Kim HT, Lee JW, Chung CH, Ahn T, Lim JM, Kim IK, Chae HJ, Kim HR. Enhanced lysosomal activity is involved in Bax inhibitor-1-induced regulation of the endoplasmic reticulum (ER) stress response and cell death against ER stress: involvement of vacuolar H+-ATPase (V-ATPase). J Biol Chem 2011; 286:24743-53; PMID:21586565; http://dx.doi.org/ 10.1074/jbc.M110.167734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee MR, Lee GH, Lee HY, Kim DS, Chung MJ, Lee YC, Kim HR, Chae HJ. BAX inhibitor-1-associated V-ATPase glycosylation enhances collagen degradation in pulmonary fibrosis. Cell Death Dis 2014; 5:e1113; PMID:24625972; http://dx.doi.org/ 10.1038/cddis.2014.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ling H, Vamvakas S, Schaefer L, Schramm L, Paczek L, Daemmrich J, Schaefer RM, Heidland A. Dose-dependent stimulation/inhibition effects of cyclosporin A on lysosomal cathepsin activities in cultured proximal tubule cells. Res Exp Med (Berl) 1995; 195:355-64; PMID:8904025 [DOI] [PubMed] [Google Scholar]
- 25.Dehay B, Martinez-Vicente M, Caldwell GA, Caldwell KA, Yue Z, Cookson MR, Klein C, Vila M, Bezard E, et al.. Lysosomal impairment in Parkinson's disease. Mov Disord 2013; 28:725-32; PMID:23580333; http://dx.doi.org/ 10.1002/mds.25462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta 2009; 1793:664-73; PMID:18706940; http://dx.doi.org/ 10.1016/j.bbamcr.2008.07.014 [DOI] [PubMed] [Google Scholar]
- 27.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al.. A gene network regulating lysosomal biogenesis and function. Science 2009; 325:473-7; PMID:19556463 [DOI] [PubMed] [Google Scholar]
- 28.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al.. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429-33; PMID:21617040; http://dx.doi.org/ 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Riley BE, Kaiser SE, Shaler TA, Ng AC, Hara T, Hipp MS, Lage K, Xavier RJ, Ryu KY, Taguchi K, et al.. Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J Cell Biol 2010; 191:537-52; PMID:21041446; http://dx.doi.org/ 10.1083/jcb.201005012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 1998; 23:33-42; PMID:9639028; http://dx.doi.org/ 10.1247/csf.23.33 [DOI] [PubMed] [Google Scholar]
- 31.Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol 2004; 36:2491-502; PMID:15325587 [DOI] [PubMed] [Google Scholar]
- 32.Komatsu M, Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett 2010; 584:1374-8; PMID:20153326; http://dx.doi.org/ 10.1016/j.febslet.2010.02.017 [DOI] [PubMed] [Google Scholar]
- 33.Chae HJ, Kim HR, Xu C, Bailly-Maitre B, Krajewska M, Krajewski S, Banares S, Cui J, Digicaylioglu M, Ke N, et al.. BI-1 regulates an apoptosis pathway linked to endoplasmic reticulum stress. Mol Cell 2004; 15:355-66; PMID:15304216; http://dx.doi.org/ 10.1016/j.molcel.2004.06.038 [DOI] [PubMed] [Google Scholar]
- 34.Ullman E, Fan Y, Stawowczyk M, Chen HM, Yue Z, Zong WX. Autophagy promotes necrosis in apoptosis-deficient cells in response to ER stress. Cell Death Differ 2008; 15:422-5; PMID:17917679; http://dx.doi.org/ 10.1038/sj.cdd.4402234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang ZY, Lin JH, Muharram A, Liu WG. Beclin-1-mediated autophagy protects spinal cord neurons against mechanical injury-induced apoptosis. Apoptosis 2014; 19:933-45; PMID:24623173; http://dx.doi.org/ 10.1007/s10495-014-0976-1 [DOI] [PubMed] [Google Scholar]
- 36.Yee KS, Wilkinson S, James J, Ryan KM, Vousden KH. PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ 2009; 16:1135-45; PMID:19300452; http://dx.doi.org/ 10.1038/cdd.2009.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castillo K, Rojas-Rivera D, Lisbona F, Caballero B, Nassif M, Court FA, Schuck S, Ibar C, Walter P, Sierralta J, et al.. BAX inhibitor-1 regulates autophagy by controlling the IRE1alpha branch of the unfolded protein response. EMBO J 2011; 30:4465-78; PMID:21926971; http://dx.doi.org/ 10.1038/emboj.2011.318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim HR, Lee GH, Cho EY, Chae SW, Ahn T, Chae HJ. Bax inhibitor 1 regulates ER-stress-induced ROS accumulation through the regulation of cytochrome P450 2E1. J Cell Sci 2009; 122:1126-33; PMID:19339548; http://dx.doi.org/ 10.1242/jcs.038430 [DOI] [PubMed] [Google Scholar]
- 39.Bailly-Maitre B, Fondevila C, Kaldas F, Droin N, Luciano F, Ricci JE, Croxton R, Krajewska M, Zapata JM, Kupiec-Weglinski JW, et al.. Cytoprotective gene bi-1 is required for intrinsic protection from endoplasmic reticulum stress and ischemia-reperfusion injury. Proc Natl Acad Sci U S A 2006; 103:2809-14; PMID:16478805; http://dx.doi.org/ 10.1073/pnas.0506854103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lisbona F, Rojas-Rivera D, Thielen P, Zamorano S, Todd D, Martinon F, Glavic A, Kress C, Lin JH, Walter P, et al.. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha. Mol Cell 2009; 33:679-91; PMID:19328063; http://dx.doi.org/ 10.1016/j.molcel.2009.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM, et al.. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 2012; 5:ra42; PMID:22692423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol 2007; 8:622-32; PMID:17637737; http://dx.doi.org/ 10.1038/nrm2217 [DOI] [PubMed] [Google Scholar]
- 43.Ballabio A, Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta 2009; 1793:684-96; PMID:19111581; http://dx.doi.org/ 10.1016/j.bbamcr.2008.12.001 [DOI] [PubMed] [Google Scholar]
- 44.Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 2009; 10:623-35; PMID:19672277; http://dx.doi.org/ 10.1038/nrm2745 [DOI] [PubMed] [Google Scholar]
- 45.Parkinson-Lawrence EJ, Shandala T, Prodoehl M, Plew R, Borlace GN, Brooks DA. Lysosomal storage disease: revealing lysosomal function and physiology. Physiology (Bethesda) 2010; 25:102-15; PMID:20430954 [DOI] [PubMed] [Google Scholar]
- 46.Xiao H, Chen D, Fang Z, Xu J, Sun X, Song S, Liu J, Yang C. Lysosome biogenesis mediated by vps-18 affects apoptotic cell degradation in Caenorhabditis elegans. Mol Biol Cell 2009; 20:21-32; PMID:18923146; http://dx.doi.org/ 10.1091/mbc.E08-04-0441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watanabe M. Drug-induced lysosomal changes and nephrotoxicity in rats. Acta Pathol Jpn 1978; 28:867-89; PMID:735819 [DOI] [PubMed] [Google Scholar]
- 48.Quiros Y, Vicente-Vicente L, Morales AI, Lopez-Novoa JM, Lopez-Hernandez FJ. An integrative overview on the mechanisms underlying the renal tubular cytotoxicity of gentamicin. Toxicol Sci 2011; 119:245-56; PMID:20829429; http://dx.doi.org/ 10.1093/toxsci/kfq267 [DOI] [PubMed] [Google Scholar]
- 49.Mingeot-Leclercq MP, Brasseur R, Schanck A. Molecular parameters involved in aminoglycoside nephrotoxicity. J Toxicol Environ Health 1995; 44:263-300; PMID:7897692; http://dx.doi.org/ 10.1080/15287399509531960 [DOI] [PubMed] [Google Scholar]
- 50.Mingeot-Leclercq MP, Tulkens PM. Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother 1999; 43:1003-12; PMID:10223907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Olbricht CJ, Fink M, Gutjahr E. Alterations in lysosomal enzymes of the proximal tubule in gentamicin nephrotoxicity. Kidney Int 1991; 39:639-46; PMID:1711136; http://dx.doi.org/ 10.1038/ki.1991.76 [DOI] [PubMed] [Google Scholar]
- 52.Takeda A, Uchida K, Haba T, Tominaga Y, Katayama A, Yoshida A, Oikawa T, Morozumi K. Chronic cyclosporin nephropathy: long-term effects of cyclosporin on renal allografts. Clin Transplant 2001; 15 Suppl 5:22-9; PMID:11791791; http://dx.doi.org/ 10.1034/j.1399-0012.2001.0150s5022.x [DOI] [PubMed] [Google Scholar]
- 53.Hazzan M, Hertig A, Buob D, Copin MC, Noel C, Rondeau E, Dubois-Xu YC. Epithelial-to-mesenchymal transition predicts cyclosporine nephrotoxicity in renal transplant recipients. J Am Soc Nephrol 2011; 22:1375-81; PMID:21719789; http://dx.doi.org/ 10.1681/ASN.2010060673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tedesco D, Haragsim L. Cyclosporine: a review. J Transplant 2012; 2012:230386; PMID:22263104; http://dx.doi.org/ 10.1155/2012/230386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morozumi K, Takeda A, Uchida K, Mihatsch MJ. Cyclosporine nephrotoxicity: how does it affect renal allograft function and transplant morphology? Transplant Proc 2004; 36:251S-6S; PMID:15041348; http://dx.doi.org/ 10.1016/j.transproceed.2004.01.027 [DOI] [PubMed] [Google Scholar]
- 56.Lim SW, Doh KC, Jin L, Jin J, Piao SG, Heo SB, Chung BH, Yang CW. Ginseng treatment attenuates autophagic cell death in chronic cyclosporine nephropathy. Nephrology (Carlton) 2014; 19:490-9. [DOI] [PubMed] [Google Scholar]
- 57.Anderson R, Van Rensburg CE, Myer MS. Alpha-tocopherol prevents cyclosporin A-mediated activation of phospholipase A2 and inhibition of Na+, K(+)-adenosine triphosphatase activity in cultured hamster renal tubular cells. Toxicol Appl Pharmacol 1994; 125:176-83; PMID:8171426; http://dx.doi.org/ 10.1006/taap.1994.1062 [DOI] [PubMed] [Google Scholar]
- 58.Lally C, Healy E, Ryan MP. Cyclosporine A-induced cell cycle arrest and cell death in renal epithelial cells. Kidney Int 1999; 56:1254-7; PMID:10504468; http://dx.doi.org/ 10.1046/j.1523-1755.1999.00696.x [DOI] [PubMed] [Google Scholar]
- 59.Ortiz A, Lorz C, Catalan M, Ortiz A, Coca S, Egido J. Cyclosporine A induces apoptosis in murine tubular epithelial cells: role of caspases. Kidney Int Suppl 1998; 68:S25-9; PMID:9839279; http://dx.doi.org/ 10.1046/j.1523-1755.1998.06808.x [DOI] [PubMed] [Google Scholar]
- 60.Yao X, Panichpisal K, Kurtzman N, Nugent K. Cisplatin nephrotoxicity: a review. Am J Med Sci 2007; 334:115-24; PMID:17700201; http://dx.doi.org/ 10.1097/MAJ.0b013e31812dfe1e [DOI] [PubMed] [Google Scholar]
- 61.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID:17580304; http://dx.doi.org/ 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- 62.Gump JM, Staskiewicz L, Morgan MJ, Bamberg A, Riches DW, Thorburn A. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat Cell Biol 2014; 16:47-54; PMID:24316673; http://dx.doi.org/ 10.1038/ncb2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.N'Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, Brown EJ. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep 2009; 10:173-9; PMID:19148225; http://dx.doi.org/ 10.1038/embor.2008.238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM, Ding WX. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy 2011; 7:188-204; PMID:21107021; http://dx.doi.org/ 10.4161/auto.7.2.14181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene 2008; 27:6434-51; PMID:18955971; http://dx.doi.org/ 10.1038/onc.2008.310 [DOI] [PubMed] [Google Scholar]
- 66.Xu M, Liu K, Swaroop M, Sun W, Dehdashti SJ, McKew JC, Zheng W. A phenotypic compound screening assay for lysosomal storage diseases. J Biomol Screen 2014; 19:168-75; PMID:23983233; http://dx.doi.org/ 10.1177/1087057113501197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101-11; PMID:14699058; http://dx.doi.org/ 10.1091/mbc.E03-09-0704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang CW, Ahn HJ, Kim WY, Shin MJ, Kim SK, Park JH, Kim YO, Kim YS, Kim J, Bang BK. Influence of the renin-angiotensin system on epidermal growth factor expression in normal and cyclosporine-treated rat kidney. Kidney Int 2001; 60:847-57; PMID:11532080; http://dx.doi.org/ 10.1046/j.1523-1755.2001.060003847.x [DOI] [PubMed] [Google Scholar]
- 69.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, et al.. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012; 481:511-5; PMID:22258505; http://dx.doi.org/ 10.1038/nature10758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009; 461:654-8; PMID:19794493; http://dx.doi.org/ 10.1038/nature08455 [DOI] [PubMed] [Google Scholar]
- 71.Park KH, Kim BJ, Kang J, Nam TS, Lim JM, Kim HT, Park JK, Kim YG, Chae SW, Kim UH, et al.. Ca2+ signaling tools acquired from prostasomes are required for progesterone-induced sperm motility. Sci Signal 2011; 4:ra31; PMID:21586728 [DOI] [PubMed] [Google Scholar]
- 72.Jang BG, Lee BL, Kim WH. Distribution of LGR5+ cells and associated implications during the early stage of gastric tumorigenesis. PLoS One 2013; 8:e82390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.