

Abstract
Autophagy is triggered in vascular smooth muscle cells (VSMCs) of diseased arterial vessels. However, the role of VSMC autophagy in cardiovascular disease is poorly understood. Therefore, we investigated the effect of defective autophagy on VSMC survival and phenotype and its significance in the development of postinjury neointima formation and atherosclerosis. Tissue-specific deletion of the essential autophagy gene Atg7 in murine VSMCs (atg7−/− VSMCs) caused accumulation of SQSTM1/p62 and accelerated the development of stress-induced premature senescence as shown by cellular and nuclear hypertrophy, CDKN2A-RB-mediated G1 proliferative arrest and senescence-associated GLB1 activity. Transfection of SQSTM1-encoding plasmid DNA in Atg7+/+ VSMCs induced similar features, suggesting that accumulation of SQSTM1 promotes VSMC senescence. Interestingly, atg7−/− VSMCs were resistant to oxidative stress-induced cell death as compared to controls. This effect was attributed to nuclear translocation of the transcription factor NFE2L2 resulting in upregulation of several antioxidative enzymes. In vivo, defective VSMC autophagy led to upregulation of MMP9, TGFB and CXCL12 and promoted postinjury neointima formation and diet-induced atherogenesis. Lesions of VSMC-specific atg7 knockout mice were characterized by increased total collagen deposition, nuclear hypertrophy, CDKN2A upregulation, RB hypophosphorylation, and GLB1 activity, all features typical of cellular senescence. To conclude, autophagy is crucial for VSMC function, phenotype, and survival. Defective autophagy in VSMCs accelerates senescence and promotes ligation-induced neointima formation and diet-induced atherogenesis, implying that autophagy inhibition as therapeutic strategy in the treatment of neointimal stenosis and atherosclerosis would be unfavorable. Conversely, stimulation of autophagy could be a valuable new strategy in the treatment of arterial disease.
Keywords: atherosclerosis, autophagy, neointima formation, senescence, sequestosome 1/p62, vascular smooth muscle cells
Abbreviations
- ACTA2
actin, α 2, smooth muscle, aorta
- ACTB
actin, β
- Apoe
apolipoprotein E
- ARE
antioxidant-response element
- ATG5
autophagy-related 5
- ATG7
autophagy-related 7
- ATG12
autophagy-related 12
- BMDM
bone marrow-derived macrophages
- BrdU
5-bromo-2′-deoxyuridine
- CASP3
caspase 3, apoptosis-related cysteine peptidase
- CDKN1A
cyclin-dependent kinase inhibitor 1A
- CDKN2A
cyclin-dependent kinase inhibitor 2A
- COL1A1/2
collagen, type I, α 1/2
- COL3A1
collagen, type III, α 1
- CXCL12
chemokine (C-X-C) motif ligand 12
- DCFDA
2′,7′-dichlorofluorescin diacetate
- EBSS
Earle's balanced salt solution
- GLB1
galactosidase, β 1
- GSTA
glutathione S-transferase, α
- KEAP1
kelch-like ECH-associated protein 1
- LAMP2
lysosomal-associated membrane protein 2
- LCCA
left common carotid artery
- MAP1LC3B
microtubule-associated protein 1 light chain 3, β
- MMP
matrix metalloproteinase
- NFE2L2
nuclear factor, erythroid 2-like 2
- NQO1
NAD[P]H dehydrogenase, quinone 1
- oxLDL
oxidized low-density lipoprotein
- PAS
periodic acid-Schiff
- RB
retinoblastoma
- ROS
reactive oxygen species
- SIPS
stress-induced premature senescence
- SQSTM1
sequestosome 1
- tBHP
tert-butyl hydroperoxide
- TEM
transmission electron microscopy
- TGFB
transforming growth factor, β
- TP53
tumor protein p53
- VSMC
vascular smooth muscle cell
- WD
western-type diet.
Introduction
Autophagy is a highly conserved and tightly regulated subcellular process involved in the degradation of long-lived proteins and damaged organelles. During this process, fragments of cytoplasmic material are sequestered in double-membrane vesicles, termed autophagosomes, and delivered to lysosomes for degradation.1 In this way, essential precursor molecules (e.g. amino acids) are recycled in order to replenish nutrient and energy supply.2 Basal autophagy is therefore an important housekeeping and life-sustaining process.
Vascular smooth muscle cells (VSMCs) are the major components of the blood vessel wall and play crucial roles in both physiological (e.g., regulation of blood pressure and vascular tone) and pathological processes (e.g. restenosis and atheroma formation). Accumulating evidence suggests that autophagy is activated in VSMCs in response to various stimuli including lipids, reactive oxygen species, cytokines, and growth factors and may act as an important mechanism for VSMC survival.3-9 Moreover, platelet-derived growth factor-induced autophagy promotes the development of a synthetic and proliferative VSMC phenotype,8 suggesting that autophagy may regulate VSMC phenotype and function.10 Because our knowledge of the regulation of auto-phagy in VSMCs is mainly based on cell culture experiments, we focused on the role of VSMC autophagy in arterial disease such as postinjury neointima formation and atherosclerosis. At this moment, the relation between autophagy and neointima formation is debatable because induction of autophagy by platelet-derived growth factor stimulates VSMC proliferation (vide supra) while the autophagy inducer rapamycin or derivatives thereof (e.g., everolimus) are essentially used to prevent restenosis.11,12 Even though autophagy occurs in VSMCs of atherosclerotic plaques,13,14 the role of VSMC autophagy in atherosclerosis has not been investigated. To study the role of autophagy in VSMCs in vitro and in vivo, a mouse model was constructed with defective autophagy in VSMCs only, by genetic deletion of the essential autophagy gene Atg7. Our data show that defective autophagy in VSMCs accelerates the development of stress-induced premature senescence (SIPS) and promotes both postinjury neointima formation and diet-induced atherogenesis.
Results
Autophagy is defective in Atg7F/FTagln-Cre+ (atg7−/−) VSMCs
The essential autophagy gene Atg7 was deleted in VSMCs by crossbreeding mice homozygous for the Atg7Flox allele (Atg7F/F) with a transgenic mouse strain that expresses Cre recombinase under control of the mouse Tagln/transgelin promoter. According to western blot analyses, the lack of ATG7 expression in isolated Atg7F/FTagln-Cre+ VSMCs (further referred to as atg7−/−) was associated with typical features of impaired autophagy such as severe SQSTM1 accumulation, decreased levels of the ATG12–ATG5 complex, an increased amount of unconjugated ATG5 and a marked decrease of MAP1LC3B-II levels with respect to Atg7+/+ VSMCs (Fig. 1A). Moreover, unlike Atg7+/+ VSMCs, atg7−/− VSMCs did not show enhanced processing of MAP1LC3B-I into MAP1LC3B-II and autophagosome formation upon starvation, a well-known stimulus of autophagy (Fig. 1B-C). These experiments confirm that atg7−/− VSMCs were unable to initiate autophagy.
Figure 1.

Autophagy is defective in Atg7F/FTagln-Cre+ VSMCs. (A) VSMCs were isolated from the aorta of Atg7+/+Tagln-Cre+ (+/+) and Atg7F/FTagln-Cre+ (−/−) mice. Western blot analysis of ATG7, SQSTM1, ATG12–ATG5 and MAP1LC3B in untreated Atg7+/+ and atg7−/− VSMCs. ACTB was used as loading control. Bands are shown in duplicate. (B,C) Atg7+/+ and atg7−/− VSMCs were treated with EBSS for 48 h, followed by western blot analysis of MAP1LC3B (B) or transmission electron microscopy (C). Autophagic vesicles, characterized by incorporated dense degraded material and indicated by arrows, were quantified per cell (***, P < 0.001 vs. atg7−/−; ###, P < 0.001 vs. control; two-way ANOVA with genotype and treatment as category factors). Scale bar: 1 µm. The right panel shows a high-power image of autophagosomes in EBSS-treated Atg7+/+ VSMCs. Scale bar: 500 nm.
Defective autophagy in VSMCs triggers an antioxidative backup mechanism
ROS production, oxidative damage and cell death are major events in cardiovascular disease15,16 and may be regulated by autophagy.17,18 To test the role of autophagy in VSMC survival against oxidative stress, Atg7+/+ and atg7−/− VSMCs were treated with 25 µmol/l tBHP or 50 µg/ml oxLDL for 24 h. atg7−/− VSMCs were much more resistant to oxidative stress-induced cell death than Atg7+/+ VSMCs (Fig. 2A). Along these lines, treatment with 100 µmol/l tBHP for 6 h stimulated ROS production in Atg7+/+ VSMCs but not in atg7−/− VSMCs (Fig. 2A). Interestingly, atg7−/− VSMCs treated with 10 µmol/l puromycin for 12 h or exposed to UV-irradiation for 10 min did not reveal improved protection against apoptosis (Fig. 2B).
Figure 2.

Defective autophagy in VSMCs results in increased protection against oxidative stress-induced cell death. (A) Atg7+/+Tagln-Cre+ (+/+) and Atg7F/FTagln-Cre+ (−/−) VSMCs were treated with 25 µmol/l tBHP or 50 µg/ml oxLDL for 24 h (***, P < 0.001 vs. Atg7+/+; ###, P < 0.001 vs. control; n = 3 experiments in triplicate; two-way ANOVA with genotype and treatment as category factors and Dunnett Post Hoc test). To measure ROS production, Atg7+/+ and atg7−/− VSMCs were left untreated or treated with 100 µmol/l tBHP for 6 h, followed by a DCFDA staining (***, P < 0.001 vs. Atg7+/+; n = 200 cells/condition in duplicate; two-way ANOVA with genotype and treatment as category factors). (B) Atg7+/+ and atg7−/− VSMCs were treated with 10 µmol/l puromycin (PM) for 12 h or exposed to UV-irradiation for 10 min (NS, not significant vs. Atg7+/+; ###, P < 0.001 vs. control; n = 2 experiments in triplicate; two-way ANOVA with genotype and treatment as category factors). (C) Analysis of Gsta and Nqo1 expression in Atg7+/+ and atg7−/− VSMCs by real time RT-PCR (***, P < 0.001; n = 2 experiments in duplicate; Student t test) and western blotting. (D) Western blot analysis of NFE2L2 in cytoplasmic and nuclear fractions of Atg7+/+ and atg7−/− VSMCs. (E) atg7−/− VSMCs were transfected with 100 nmol/l siRNA against Nfe2l2 (siNfe2l2) or nontargeting siRNA (siCtrl). After 72 h, silencing efficiency was confirmed by western blotting by assessment of NFE2L2, GSTA and NQO1 expression and VSMCs were treated with tBHP or oxLDL for 24 h (***, P < 0.001 vs. siCtrl; ###, P < 0.001 vs. control; n = 3 experiments in duplicate; two-way ANOVA with genotype and treatment as category factors and Dunnett Post Hoc test).
To explain the observed cytoprotection against oxidative stress, RNA of untreated Atg7+/+ and atg7−/− VSMCs was analyzed. Microarray analysis revealed an upregulation of the genes encoding several antioxidative enzymes such as Gsta1/3/4 (glutathione S-transferase, α 1/3/4) and Nqo1 (NAD[P]H dehydrogenase, quinone 1) in atg7−/− VSMCs. The microarray data are available via the National Center for Biotechnology Information Gene Expression Omnibus at: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ytuzumoktdqzpifandacc=GSE54019. Upregulation of GSTA and NQO1 was confirmed by real-time RT-PCR and western blotting (Fig. 2C). Because accumulation of SQSTM1 induces nuclear factor erythroid 2-like 2 (NFE2L2)-dependent transcription of detoxifying enzymes such as GSTA,19 this pathway was further studied by western blot analysis. Cytoplasmic and nuclear fractions of atg7−/− VSMCs showed enhanced translocation of NFE2L2 into the nucleus (Fig. 2D). Silencing of Nfe2l2 completely suppressed GSTA and NQO1 expression and abolished the protection of atg7−/− VSMCs against tBHP and oxLDL (Fig. 2E). These experiments indicate that the NFE2L2 signaling pathway is activated in atg7−/− VSMCs as a backup mechanism to protect against oxidative stress. Overall, Atg7 deletion in VSMCs did not result in a general prosurvival status but only protected VSMCs against oxidative stress-mediated cell death via upregulation of antioxidative enzymes.
Defective autophagy in VSMCs elicits cellular hypertrophy, and increases migration capacity and total collagen amount
Microscopic analysis of atg7−/− VSMCs did not reveal a typical spindle-shaped phenotype as compared to Atg7+/+ VSMCs. Instead, atg7−/− VSMCs were characterized by a more rhomboid shape and an increase in cellular size (Fig. 3A). Importantly, atg7−/− VSMCs showed a significant increase in protein content as compared to Atg7+/+ VSMCs, indicating that the hypertrophic phenotype of atg7−/− VSMCs is not just a result of cytosolic dilation but is associated with increased protein quantity (Fig. S1A). In vivo, the medial thickness of atg7−/− aorta was significantly increased as compared to Atg7+/+ aorta (Fig. 3B). In addition, TEM images of atg7−/− aorta showed an increase in size of the individual VSMCs (Fig. S1B). However, the number of VSMC layers of atg7−/− aorta was not altered. All together, these findings suggest that defective autophagy in VSMCs results in cellular hypertrophy in vitro and in vivo.
Figure 3.

Defective autophagy in VSMCs elicits cellular hypertrophy, and increases migration capacity and total collagen amount. (A) VSMCs isolated from Atg7+/+Tagln-Cre+ (+/+) and Atg7F/FTagln-Cre+ (−/−) aorta were labeled with calcein AM and visualized by confocal fluorescence microscopy. Scale bar: 10 µm. Cell size was measured using z-stack images (*, P < 0.05; n = 2 experiments; Student t test). (B) Thoracic aorta of Atg7+/+ and atg7−/− mice were stained with H&E to measure the width of the media (white arrows) (**, P < 0.01; n = 6 regions/aorta; Univariate). Scale bar: 25 µm. Note that the number of VSMC layers between Atg7+/+ and atg7−/− aorta was not different. (C) Migratory capacity of Atg7+/+ and atg7−/− VSMCs was analyzed using an Oris Migration Assay (***, P < 0.001; n = 2 experiments in triplicate; Student t test). Western blot analysis of TGFB and CXCL12 in Atg7+/+ and atg7−/− VSMCs. (D) Atg7+/+ and atg7−/− VSMCs were left untreated or treated with 10 ng/ml TGFB for 48 h and stained with Sirius red to examine total collagen amount (***, P < 0.001 vs. Atg7+/+; ###, P < 0.001 vs. control; n = 4 experiments in triplicate; two-way ANOVA with genotype and treatment as category factors).
Because VSMC migration and collagen deposition play an important role in the development of neointimal and atherosclerotic lesions, the capacity of atg7−/− VSMCs to migrate and to synthesize collagen was examined. atg7−/− VSMCs showed enhanced spontaneous migration as compared to Atg7+/+ VSMCs (Fig. 3C). Moreover, western blot analysis revealed a substantial upregulation of different promigratory growth factors such as TGFB (transforming growth factor β) and CXCL12 (chemokine [C-X-C] motif ligand 12) in atg7−/− VSMCs (Fig. 3C). To test the influence on collagen content, Atg7+/+ and atg7−/− VSMCs were treated with 10 ng/ml TGFB for 48 h. atg7−/− VSMCs showed a significant increase in collagen amount in both control and TGFB-treated conditions as compared to Atg7+/+ VSMCs (Fig. 3D). Further analysis of COL1A1/2 (collagen, type I, α 1/2) and COL3A1 (collagen, type III, α 1) expression by immunofluorescence showed that atg7−/− VSMCs express less COL1A1/2 but more COL3A1 as compared to Atg7+/+ VSMCs (Fig. S1C). TGFB treatment augmented COL1A1/2 and COL3A1 expression in both genotypes.
Defective autophagy in VSMCs accelerates senescence
A BrdU incorporation assay was performed to examine the proliferation capacity of Atg7+/+ and atg7−/− VSMCs. A nearly 2-fold reduction in proliferation of atg7−/− VSMCs was observed as compared to Atg7+/+ VSMCs (Fig. 4A). Moreover, atg7−/− VSMCs were characterized by a significant increase in nuclear size (Fig. 4A). According to a cell cycle analysis, the percentage of atg7−/− nuclei in G1-phase was increased (36±5% vs. 55±1%) while the percentage of atg7−/− nuclei in G2/M-phase was strongly decreased (38±2% vs. 15±1%), suggestive of a G1-cell cycle arrest and senescence (Fig. 4B). Importantly, cell cycle analysis did not show signs of VSMC polyploidy in both cell types. Staining for senescence-associated GLB1 activity confirmed the presence of senescent cells in atg7−/− VSMC cultures (Fig. 4C). Because cellular senescence can be established by the activation of different tumor suppressor pathways including CDKN2A-RB (stress response) and TP53-CDKN1A (DNA damage response) pathways,20,21 these proteins were examined via western blotting. CDKN2A was highly upregulated in atg7−/− VSMCs and accompanied by hypophosphorylation and activation of RB while acetylated TP53 and CDKN1A levels remained unaltered (Fig. 4D), indicating stress-induced premature senescence. Next, we performed a comet assay to determine possible DNA damage. Neither Atg7+/+ nor atg7−/− VSMCs showed comet tail formation. In contrast, Atg7+/+ VSMCs treated with 0.5 mmol/l H2O2 for 10 min showed severe comet tail formation representing a large number of DNA strand breaks (Fig. 4E). Given that the DNA damage response pathway was not activated and DNA strand breaks were absent, senescence in atg7−/− VSMCs was not characterized by DNA damage.
Figure 4.

Defective autophagy in VSMCs accelerates senescence. (A) Atg7+/+Tagln-Cre+ (+/+) and Atg7F/FTagln-Cre+ (−/−) VSMCs were treated with BrdU to examine proliferation (***P < 0.001; n = 9 counting regions of 300 cells/region/condition; Student t test). The size of BrdU-positive nuclei was measured to detect nuclear hypertrophy (**, P < 0.01; n = 50 nuclei; Student t test). (B) DNA cell cycle analysis of Atg7+/+ (blue) and atg7−/− (red) VSMCs. The bar graph shows the percentage of Atg7+/+ and atg7−/− nuclei in the G1 and G2/M phase of the cell cycle (**, P < 0.01; ***, P < 0.001; Student t test). (C) Atg7+/+ and atg7−/− VSMCs were stained with X-gal mixture for 24 h, followed by Nuclear Fast Red staining. Scale bar: 125 µm. The number of senescence-associated GLB1-positive VSMCs was quantified. (***, P < 0.001; n = 2 counting regions of 200 cells/region/condition; Student t test) (D) Western blot analysis of CDKN2A, phospho RB, total RB, acetylated TP53 and CDKN1A in Atg7+/+ and atg7−/− VSMCs. (E) Detection of DNA damage in Atg7+/+ and atg7−/− VSMCs by comet assay. Atg7+/+ VSMCs treated with .5 mmol/l H2O2 for 10 min were used as positive control. Scale bar: 25 µm.
Furthermore, we investigated a possible link between senescence and the NFE2L2 pathway. Nfe2l2 silencing in atg7−/− VSMCs did not affect CDKN2A protein expression (Fig. S2A). Overexpression of CDKN2A in Atg7+/+ VSMCs induced senescence as illustrated by increased GLB1 activity but altered neither NQO1 expression nor NFE2L2 activation (Fig. S2B). These experiments suggest that the senescence and NFE2L2 pathway act independently in autophagy deficient VSMCs.
To investigate the role of SQSTM1 accumulation (typical of defective autophagy, vide supra) in the induction of VSMC senescence, Atg7+/+ VSMCs were transiently transfected with SQSTM1-encoding plasmid DNA. Overexpression of SQSTM1 in Atg7+/+ VSMCs resulted in increased CDKN2A expression and a slight decrease in RB phosphorylation (Fig. 5A). Moreover, SQSTM1 overexpressing Atg7+/+ VSMCs showed reduced proliferation (Fig. 5B) and increased senescence-associated GLB1 activity (Fig. 5C). These experiments indicate that SQSTM1 mediates the induction of senescence in autophagy defective VSMCs.
Figure 5.

SQSTM1 accumulation links defective VSMC autophagy to senescence. (A) Atg7+/+ VSMCs were transfected with 5 µg plasmid DNA encoding SQSTM1 (SQSTM1+). Four d after transfection, VSMCs were analyzed for SQSTM1, CDKN2A, phospho RB and total RB expression by western blotting. (B,C) SQSTM1 overexpressing VSMCs were incubated with BrdU (B) to examine proliferation capacity (***, P < 0.001; n = 2 counting regions of 1000 cells/region/condition in duplicate; Student t test) or (C) stained with X-gal mixture for 24 h to quantify the number of senescence-associated GLB1-positive VSMCs (***, P < 0.001; n = 3 counting regions of 150 cells/region/condition in duplicate; Student t test). Scale bar: 50 µm.
Defective VSMC autophagy promotes neointima formation after ligation-induced injury
To evaluate the consequences of defective VSMC autophagy in vivo, neointimal lesion formation was induced by ligation of the left common carotid artery (LCCA) in Atg7+/+Tagln-Cre+ (Atg7+/+) and Atg7F/FTagln-Cre+ (atg7−/−) mice. Five d after ligation, the activity of the gelatinase MMP9 was already strongly elevated in atg7−/− LCCA as shown by zymographic analysis (Fig. 6A). Moreover, western blot analysis showed a significant increase in TGFB and CXCL12 expression in the LCCA of atg7−/− mice (Fig. 6B). To investigate the effect on neointimal thickening, LCCA of Atg7+/+ and atg7−/− mice were analyzed 5 wk after ligation. atg7−/− mice showed a dramatic increase in stenosis (90±3%) as compared to Atg7+/+ mice (54±6%) (Fig. 7A). In addition, the total collagen content was significantly elevated in the neointima of atg7−/− mice (Fig. 7B). The level of apoptosis was similar in lesions of atg7−/− mice versus Atg7+/+ mice (Fig. S3A). Also the percentage of macrophages within the neointima was not different between both groups (Fig. S3B). SQSTM1 staining confirmed the presence of autophagy deficient VSMCs in media and lesions of atg7−/− mice (Fig. S4A). Furthermore, neointimal atg7−/− VSMCs were characterized by CDKN2A upregulation (Fig. 7C), senescence-associated GLB1 activity (Fig. 7D) and nuclear hypertrophy (Fig. S3C), indicating that neointima formation in atg7−/− mice was associated with VSMC senescence.
Figure 6.
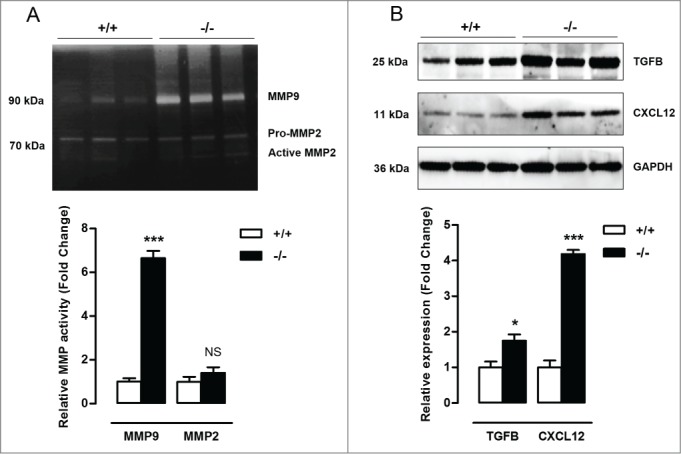
Defective VSMC autophagy promotes upregulation of MMP9, TGFB and CXCL12, 5 d after ligation-induced injury. (A) The left common carotid artery (LCCA) of Atg7+/+Tagln-Cre+ (+/+) and Atg7F/FTagln-Cre+ (−/−) mice (n = 3) was ligated for 5 d. Gelatin zymographic analysis of the LCCA to detect MMP9 and MMP2 activity followed by densitometric analysis (***, P < 0.001; NS, not significant; Student t test). (B) Western blot analysis of the LCCA for TGFB, CXCL12 and GAPDH. Relative expression of TGFB/GAPDH and CXCL12/GAPDH was determined by densitometric analysis (*, P < 0.05; ***, P < 0.001; Student t test).
Figure 7 (See previous page).

Defective VSMC autophagy promotes neointima formation 5 wk after ligation-induced injury. (A,B) The left common carotid artery (LCCA) of Atg7+/+Tagln-Cre+ (+/+) (n = 10) and Atg7F/FTagln-Cre+ (−/−) mice (n = 12) was ligated for 5 wk. Sections of the LCCA were stained with anti-ACTA2 antibody (A) or Sirius red (B) to quantify the degree of stenosis and total collagen deposition, respectively. Scale bar: 100 µm. (**P < 0.01; *P < 0.05; Student t test). (C) Sections of the LCCA were immunostained for CDKN2A to quantify CDKN2A-positive nuclei (black arrowheads). Scale bar: 25 µm. (**, P < 0.01; Mann Whitney test). (D) The LCCA was stained for senescence-associated GLB1 activity ex vivo (right panel). Scale bar: 250 µm. Sections of the LCCA were then counterstained with periodic acid-Schiff (PAS) to identify senescent neointimal VSMCs (left panel). Note that the neointimal VSMCs are surrounded by a cage of PAS-positive basal lamina. Scale bar: 10 µm.
Defective VSMC autophagy accelerates atherogenesis
To investigate the role of VSMC autophagy in atherosclerosis, Atg7+/+Tagln-Cre+ and Atg7F/FTagln-Cre+ mice were crossbred with apoe−/− mice and fed a western-type diet (WD) for 10 or 14 wk. Body weight, total cholesterol and lipoproteins were not different between both groups after 10 and 14 wk on WD (Table S1, Fig. S5), indicating that Atg7 deletion in VSMCs does not influence normal growth and lipid metabolism. Plaques of Atg7F/FTagln-Cre+,apoe−/− (atg7−/−,apoe−/−) mice were characterized by increased accumulation of SQSTM1 as compared to Atg7+/+Tagln-Cre+,apoe−/− (Atg7+/+,apoe−/−) mice, confirming autophagy deficiency (Fig. S4B). Plaques in the brachiocephalic artery of 10-wk WD-fed atg7−/−,apoe−/− mice showed a 3-fold increase in size as compared to Atg7+/+,apoe−/− mice (58 ± 9µm2 vs. 156 ± 19µm2; P < 0.001). Furthermore, plaques of atg7−/−,apoe−/− mice were characterized by elevated plaque necrosis, plaque apoptosis, macrophage content, total collagen content, and fibrous cap thickness (Fig. 8A-E). After 14 wk on WD, plaque size (171 ± 11 µm2 vs. 166 ± 9 µm2; P > 0.05), macrophage content as well as the degree of necrosis and apoptosis was similar in Atg7+/+,apoe−/− and atg7−/−,apoe−/− mice (Fig. 9A-C). However, total collagen content and fibrous cap thickness were still elevated in plaques of atg7−/−,apoe−/− mice (Fig. 9D-E). Interestingly, the amount of COL1A1/2 was significantly decreased in atg7−/−,apoe−/− plaques while the amount of COL3A1 was increased (Fig. S6A and S6B). Plaques located in the aortic root showed a similar plaque composition as compared to plaques in the brachiocephalic artery (data not shown). Next, we investigated whether VSMCs in atg7−/−,apoe−/− plaques revealed characteristics of senescence. VSMCs within the fibrous cap of atg7−/−,apoe−/− plaques were characterized by enhanced senescence-associated GLB1 activity (Fig. 10A), hypophosphorylation and activation of RB (Fig. 10B), and nuclear hypertrophy (Fig. S6C), indicative of senescence. Importantly, plaque formation in atg7−/−,apoe−/− mice was not attributed to increased proliferation of medial VSMCs as shown by severe hypophosphorylation of RB in the media (Fig. 10C).
Figure 8.
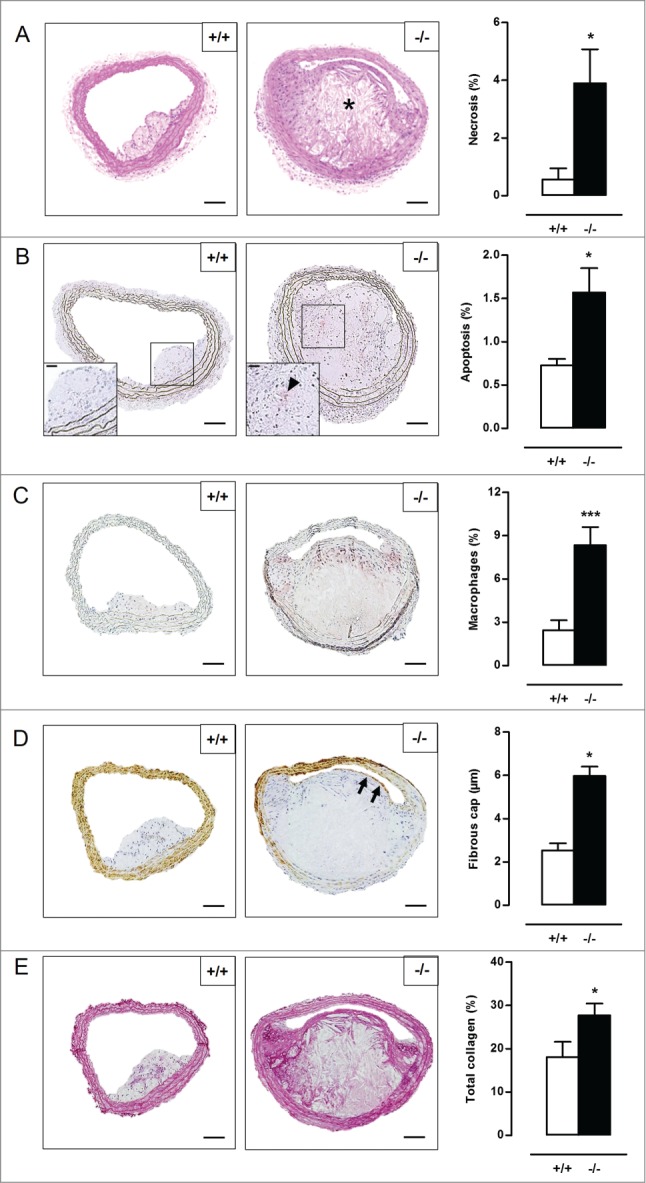
Defective VSMC autophagy accelerates atherogenesis after 10 wk of western-type diet. (A) Atg7+/+Tagln-Cre+ apoe−/− (+/+) and Atg7F/FTagln-Cre+,apoe−/− (−/−) mice (n = 16) were fed a western-type diet for 10 wk. Sections of the brachiocephalic artery were stained with H&E to quantify plaque size and percentage of necrosis. (*, P<0.05; Student t test). (B to D) Consecutive sections were immunostained for cleaved CASP3 (B), LAMP2 (C) and ACTA2 (D) to measure the percentage of apoptosis (indicated by a black arrowhead in the high-power photograph of the boxed area in the left corner of each image), percentage of macrophages and fibrous cap thickness (indicated by black arrows), respectively (*, P < 0.05; Student t test (B); ***, P < 0.001; Student t test (C); *P < 0.05; n = 10 measurements/mouse, Repeated Measure (D)). (E) Consecutive sections were stained with Sirius red to quantify total collagen. (*, P < 0.05; Student t test). *, necrotic core. Scale bar: 100 µm.
Figure 9.
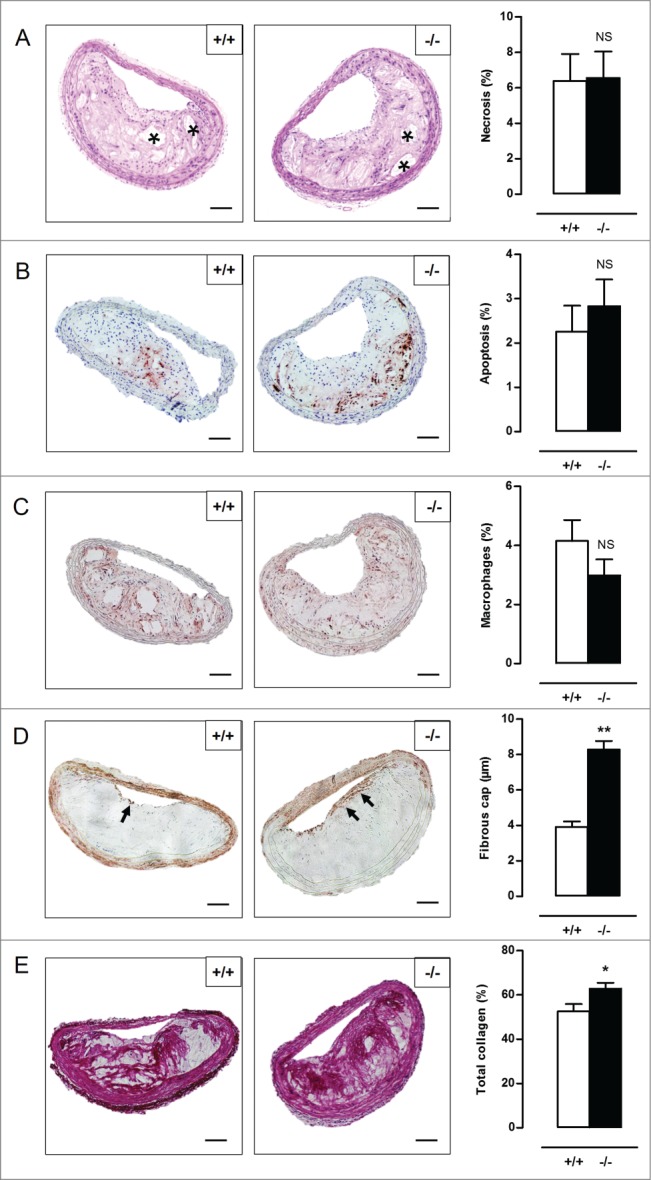
Defective VSMC autophagy promotes formation of a thick fibrous cap and enhances total collagen deposition after 14 wk of western-type diet. (A) Atg7+/+Tagln-Cre+,apoe−/− (+/+) and Atg7F/FTagln-Cre+,apoe−/− (−/−) mice (n = 16) were fed a western-type diet for 14 wk. Sections of the brachiocephalic artery were stained with H&E to quantify plaque size and percentage of necrosis. (NS, not significant; Student t test). (B to D) Consecutive sections were immunostained for cleaved CASP3 (B), LAMP2 (C) and ACTA2 (D) to measure the percentage of apoptosis, percentage of macrophages and fibrous cap thickness (indicated by black arrows), respectively. (NS, not significant; Student t test (B,C); **, P < 0.01; n = 10 measurements/mouse, Repeated Measure (D)). (E) Consecutive sections were stained with Sirius red to quantify total collagen. (*, P < 0.05; Student t test). Scale bar: 100 µm. *, necrotic core.
Figure 10.

Atherosclerotic plaques of Atg7F/FTagln-Cre+,apoe−/− mice show several features of VSMC senescence. (A) Sections of the aortic root were stained for senescence-associated (black arrowheads) and compared with serial TAGLN staining (not shown) to locate the fibrous caps. Scale bar: 10 µm. (B) Consecutive sections of the aortic root were double stained for phospho RB (red; white arrowheads) and ACTA2 (green). Scale bar: 25 µm (C) Sections of the brachiocephalic artery were double stained with phospho RB and periodic acid-Schiff (PAS) to quantify phospho RB-positive VSMC nuclei in the media (**, P < 0.01; Mann Whitney test). Scale bar: 50 µm.
Activation of the NFE2L2 and senescence pathway occurs in autophagy defective VSMCs but not in macrophages
Because it was previously demonstrated that defective autophagy in macrophages led to increased plaque destabilization after 12 and 16 wk of WD,22 antioxidative mechanisms and cellular senescence were further examined in autophagy deficient macrophages (Fig. 11A). Unlike atg7−/− VSMCs, atg7−/− macrophages did not overexpress GSTA and NQO1 and were much more sensitive to tBHP-induced cell death as compared to Atg7+/+ macrophages (Fig. 11B-C). This effect was attributed to the lack of NFE2L2 activation in atg7−/− macrophages (Fig. 11D). Furthermore, besides the unaltered proliferation (Fig. 11E), atg7−/− macrophages showed neither cell cycle arrest nor upregulation of CDKN2A (Fig. 11F-G), indicating that defective autophagy in macrophages does not result in cellular senescence.
Figure 11.

Defective autophagy in macrophages triggers neither an antioxidative backup mechanism nor senescence. Bone marrow-derived macrophages were isolated from Atg7+/+Lysm-Cre+ (+/+) and Atg7F/FLysm-Cre+ (−/−) mice. (A) Western blot analysis of ATG7 and SQSTM1 in untreated Atg7+/+ and atg7−/− macrophages. (B) Real-time RT-PCR and western blot analysis of GSTA and NQO1 in Atg7+/+ and atg7−/− macrophages (NS, not significant; n = 2 experiments in duplicate; Student t test). (C) Atg7+/+ and atg7−/− macrophages were treated with 50 µmol/l tBHP for 24 h (***, P < 0.001 vs. Atg7+/+; ###, P < 0.001 vs. control; n = 2 experiments in duplicate; Two-way ANOVA with genotype and treatment as category factors). (D) Western blot analysis of NFE2L2 in cytoplasmic and nuclear fractions of Atg7+/+ and atg7−/− macrophages. (E) Proliferation of Atg7+/+ and atg7−/− macrophages were examined by BrdU incorporation assay (NS, not significant; n = 6 counting regions of 500 cells/region/condition; Student t test) (F) DNA cell cycle analysis of Atg7+/+ (blue) and atg7−/− (red) macrophages. The bar graph shows the percentage of Atg7+/+ and atg7−/− nuclei in G1 and G2/M phase of the cell cycle (NS, not significant; Student t test). (G) Western blot analysis of CDKN2A in Atg7+/+ and atg7−/− macrophages.
Discussion
In this study, we provide novel evidence for a prominent role of autophagy in the regulation of VSMC survival and phenotype. Defective autophagy has major effects on the mechanistic, functional and morphological properties of VSMCs, with significant implications for the development of cardiovascular diseases such as postinjury neointima formation and diet-induced atherosclerosis (Fig. 12).
Figure 12.
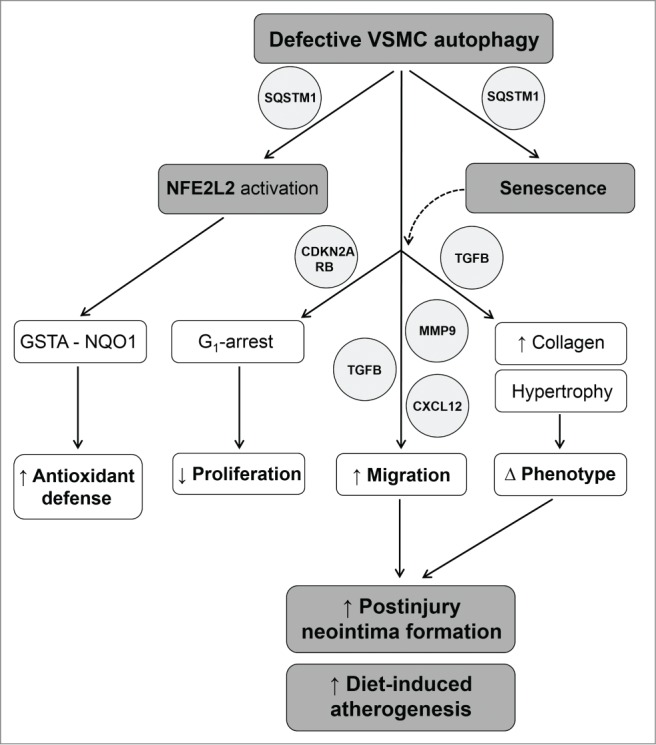
Overview of the mechanisms by which defective VSMC autophagy accelerates senescence and promotes postinjury neointima formation and diet-induced atherogenesis. SQSTM1 accumulation in autophagy defective VSMCs triggers NFE2L2 activation and transcription of multiple antioxidative enzymes including GSTA and NQO1. Upregulation of GSTA and NQO1 promotes VSMC survival against oxidative stress under defective autophagy conditions. SQSTM1 accumulation in autophagy defective VSMCs triggers the development of stress-induced premature senescence. Autophagy defective VSMCs are characterized by CDKN2A-RB-mediated G1 proliferation arrest, increased migration and changes in VSMC phenotype. Enhanced migration is associated with increased secretion of MMP9, TGFB and CXCL12. The phenotype of autophagy defective VSMCs is defined by nuclear and cellular hypertrophy, and by increased collagen content. Defective autophagy in VSMCs accelerates postinjury neointima formation and diet-induced atherogenesis.
The relationship between autophagy and cell survival has been thoroughly investigated by several research groups. Because autophagy is considered as an important mechanism for the survival of VSMCs,5,6,9 it is plausible to assume that defects in the autophagic machinery could aggravate VSMC death. Surprisingly, our data indicate that atg7−/− VSMCs were highly resistant to oxidative stress as compared to Atg7+/+ VSMCs. The protection against oxidative stress-mediated cell death was attributed to increased expression of different antioxidative enzymes such as GSTA and NQO1. These phase II enzymes serve as a detoxification mechanism to protect cells against electrophilic insults and oxidative stress. Accumulation of the protein SQSTM1, one of the hallmarks of defective autophagy,23 plays a central role in the upregulation of these antioxidative enzymes. SQSTM1 accumulation triggers nuclear translocation of the transcription factor NFE2L2,19,24 a well-known key regulator of the intracellular redox balance. Under basal conditions, NFE2L2 is immobilized in the cytoplasm by binding to its inhibitor KEAP1 (kelch-like ECH-associated protein 1). However, when SQSTM1 accumulates, it will interfere with the NFE2L2-KEAP1 binding complex by sequestrating KEAP1 into aggregates.19,24 As a result, NFE2L2 is released from its inhibitor and translocated to the nucleus where it promotes transcription of multiple antioxidative genes by binding to the antioxidant response element (ARE) in their promoter region. Therefore, SQSTM1 is considered to be a crucial activator of NFE2L2.19 In the present study, atg7−/− VSMCs showed increased nuclear translocation of NFE2L2. Silencing of Nfe2l2 suppressed the expression of GSTA and NQO1 and abolished the protection of atg7−/− VSMCs against oxidative stress. Importantly, atg7−/− VSMCs did not reveal improved protection against ROS-independent apoptosis. Hence, Atg7 deletion in VSMCs did not result in a general prosurvival status but only protected VSMCs against oxidative stress-mediated cell death. Interestingly, this antioxidative backup mechanism seems to be cell-type specific. atg7−/− macrophages revealed neither NFE2L2 activation nor elevated phase II enzyme expression, which is in line with previous reports showing that various cell types including hepatic cells, neurons, pancreatic β cells, and macrophages are more susceptible to (oxidative stress-mediated) cell death under defective autophagy conditions.22,25-27
Besides an improved antioxidant defense, atg7−/− VSMCs were characterized by cellular hypertrophy, nuclear hypertrophy, a decline in proliferative capacity due to a G1-mediated cell cycle arrest and senescence-associated GLB1 activity, indicative of cellular senescence.28 Moreover, atg7−/− VSMCs show an increase in TGFB and CXCL12 expression, which is most likely related to the increase in migration potential and the development of a senescence-associated secretory phenotype.29,30 Cellular senescence can be established by the activation of 2 different tumor suppressor pathways: TP53-CDKN1A and CDKN2A-RB.20,21 The TP53 pathway is triggered by DNA damage and generally associated with CDKN1A upregulation to initiate growth arrest. The CDKN2A-RB (retinoblastoma) pathway is engaged when cells are exposed to cellular stress, leading to the development of stress-induced premature senescence (SIPS).31 During SIPS, the increased expression of CDKN2A leads to hypophosphorylation and activation of RB, resulting in prevention of transcription of proliferation-promoting genes.20 In the present study, senescence in atg7−/− VSMCs was associated with the activation of the CDKN2A-RB pathway but not with DNA damage-mediated activation of the TP53-CDKN1A pathway. Importantly, the senescence pathway acts independently of the NFE2L2-ARE pathway as shown by CDKN2A overexpression and Nfe2l2 silencing experiments.
Several research groups have shown that senescence contributes to the aging process and to the development of age-related pathologies.29,32 Given that CDKN2A expression is elevated in aged VSMCs33 and CDKN2A is considered as a biomarker of aging,29,34 we define here a possible link between autophagy deficiency and VSMC aging. The relationship between autophagy and aging has been strongly investigated over the last 10 y. Induction of autophagy by caloric restriction, spermidine, resveratrol or rapamycin extends life span in different organisms.35 For example, the autophagy-inducers rapamycin and resveratrol suppress cellular senescence by partially preventing loss of proliferative potential,36,37 whereas inhibition of autophagy by knockdown of ATG7 or ATG12 induces premature senescence.38 In the present study, we dissected out the molecular link between defective autophagy and VSMC senescence. Overexpression of SQSTM1 induced senescence in Atg7+/+ VSMCs as shown by CDKN2A accumulation, RB hypophosphorylation, reduced proliferation capacity and increased senescence-associated GLB1 activity. This experiment indicates that severe accumulation of SQSTM1, which occurs during defective VSMC autophagy, mediates the induction of senescence in VSMCs. Although some researchers have suggested that SQSTM1 could play a role in the induction of senescence in other pathologies,39,40 this is the first study that addresses SQSTM1 accumulation as the direct link between defective autophagy and VSMC senescence. The inverse relationship between autophagy and senescence seems logical if they are both considered as 2 cytoprotective pathways.41 Indeed, autophagy is an important homeostatic regulator by eliminating damaged intracellular components and may play a critical role in the prevention of cellular senescence.42 When autophagy is impaired, however, senescence can be engaged as a backup mechanism to protect the cell.41
Because changes in VSMC survival, phenotype, proliferation and migration are critical factors in the development of arterial vascular disease, we investigated their functional significance in 2 mouse models for postinjury neointima formation and atherosclerosis. Postinjury neointimal lesions are formed by the accumulation of VSMCs and extracellular matrix.43 Five wk after carotid ligation, atg7−/− mice developed larger and more collagen-rich neointimal lesions as compared to Atg7+/+ mice. The mechanism behind this excessive neointimal formation includes high MMP9 activity in the ligated LCCA of atg7−/− mice already 5 d after ligation. Matrix metalloproteinases facilitate migration of VSMCs by remodeling the extracellular matrix.44 In particular MMP9 and MMP2 are upregulated in response to vascular injury and to trigger intimal thickening.45 Because MMP2 levels rise at a later time and remain lower than MMP9 levels,46 we failed to detect MMP2 activity 5 d after ligation. Moreover, the expression of TGFB and CXCL12 was significantly increased in the LCCA of atg7−/− mice. TGFB plays a crucial role in intimal thickening and may stimulate MMP9 and MMP2 expression after arterial injury which in turn may augment the bioavailability of TGFB, thereby creating a positive feedback loop promoting intimal thickening.47 Given that TGFB is also involved in collagen synthesis, RB-dependent G1-growth arrest and induction of several senescence markers (e.g. GLB1 activity),48-50 this growth factor most likely plays a central role in both the development of senescence and the formation of postinjury neointima in atg7−/− mice. CXCL12 is an important chemoattractant that is released by VSMCs after mechanical injury and triggers the recruitment of SMC progenitor cells to the lesion.51,52 All together, the excessive neointimal thickening in atg7−/− mice could be attributed to a combination of factors: cellular hypertrophy of neointimal VSMCs, increased deposition of collagen, improved VSMC migration, and recruitment of smooth muscle progenitor cells due to increased MMP9 activity and elevated levels of TGFB and CXCL12. These findings support our in vitro data that autophagy defective VSMCs exhibit improved migration potential and develop a senescence-associated secretory phenotype which is characterized by the increased secretion of growth factors (TGFB), cytokines (CXCL12) and proteases (MMPs).29,30 Furthermore, neointimal atg7−/− VSMCs were characterized by CDKN2A upregulation, senescence-associated GLB1 activity and nuclear hypertrophy, indicating that neointima formation in atg7−/− mice was associated with VSMC senescence.
Atherosclerotic lesions are characterized by the accumulation of VSMCs, immune cells, lipids and extracellular matrix within the arterial wall. Ten-wk-old WD-fed atg7−/−,apoe−/− mice developed larger and more advanced plaques as compared to Atg7+/+,apoe−/− mice, due to an increase in necrotic core size, fibrous cap thickness, macrophage and total collagen content. After 14 wk on WD, the differences in plaque size, macrophage content, necrosis and apoptosis between both groups were no longer significant. Still, the fibrous cap thickness and total collagen content were significantly elevated in plaques of 14-wk WD-fed atg7−/− ,apoe−/− mice. To assess the contribution of collagen to plaque stability, the amount of COL1A1/2 (more rigid) and COL3A1 (less rigid) was measured. In accordance with our in vitro findings, plaques of atg7−/−,apoe−/− mice showed a significant decrease in COL1A1/2 while COL3A1 was increased. Taken together, we cannot define that plaque stability was improved in atg7−/−, apoe−/− mice because several important criteria such as a sufficient decrease in plaque necrosis and plaque apoptosis and high amounts of COL1A1/2 were not evident. Moreover, VSMCs within the fibrous cap of atherosclerotic plaques of atg7−/−, apoe−/− mice were characterized by several senescence markers including RB hypophosphorylation, senescence-associated GLB1 activity and nuclear hypertrophy. Different research groups have shown that VSMC senescence is present in atherosclerotic plaques and contributes to the pathogenesis of atherosclerosis.21,32,53,54 Our observations confirm once more the dual role of VSMCs in atherosclerosis. Although VSMCs are considered beneficial in terms of plaque stability due to the formation of a thick fibrous cap that safeguards plaques from rupturing, VSMCs also play a fundamental role in promoting plaque development.55
Because defective autophagy in VSMCs accelerates atherogenesis, we do not recommend inhibition of VSMC autophagy but rather suggest controlled stimulation of autophagy as a therapeutic strategy to treat atherosclerosis as recently suggested.56-58 Caloric restriction for example, is a powerful tool to induce autophagy and has both life-span-prolonging and antiatherogenic properties in mice.59 Also SIRT1 (sirtuin 1) deacetylase, an inducer of autophagy and a negative regulator of cellular aging, plays a protective role in atherosclerosis. VSMC-specific deletion of SIRT1 deacetylase in apoe−/− mice promotes atherosclerosis associated with increased DNA damage and senescence.60 Finally, it is worthwhile to mention that MTOR (mechanistic target of rapamycin [serine/threonine kinase])-inhibitors such as rapamycin (or rapalogs) are able to stimulate autophagy and have significant promise to treat patients with unstable plaques, particularly in combination with statins or metformin.61
In conclusion, defective autophagy in VSMCs accelerates senescence and promotes ligation-induced neointima formation and diet-induced atherogenesis. Although neointimal and atherosclerotic lesions of VSMC-specific autophagy deficient mice show several features of senescence, we cannot exclude that other pathways, besides senescence, are engaged in response to defective VSMC autophagy and involved in the accelerated plaque formation. Overall, our study uncovers for the first time the role of VSMC autophagy in postinjury neointima formation and diet-induced atherosclerosis and reveals SQSTM1 as a key player in the induction of VSMC senescence, supporting the growing body of evidence that autophagic dysfunction plays a major role in cardiovascular disease.
Materials and Methods
Mouse studies
Mice homozygous for a vector that disrupts Atg7 in exon 14 by Cre-loxP technology (Atg7F/F mice)25 were crossbred for >10 generations with Tagln-Cre+ mice (C57BL/6, Jackson Laboratory, 004746), which express Cre recombinase under the control of the mouse Tagln/Sm22α promoter, to obtain VSMC-specific atg7 knockout mice (referred to as Atg7F/FTagln-Cre+ mice or simply atg7−/− mice). To induce neointimal lesions, blood flow was disrupted in the left common carotid artery of Atg7+/+ and atg7−/− mice for 5 d (n = 4/group) or 5 wk (n = 14/group) by ligating the vessel near the bifurcation. During the surgical procedure, 0.1 mg/g BW ketamine i.p. (Anesketin, 100mg/ml, Eurovet) and 0.01 mg/g BW xylazine i.p. (Rompun, 20 mg/ml, Bayer Health Care) were used as anesthetics and 0.1 mg/kg BW brupenorfine s.c. (Temgesic, 0.3 mg/ml, Schering-Plough) as analgesic agent. For atherosclerosis studies, Atg7+/+ and atg7−/− mice were crossbred with apoe−/− mice (C57BL/6, Jackson Laboratory, 002052) and fed a western-type diet (Harlan Teklad, TD88137) for 10 (n = 16/group) or 14 wk (n = 16/group). At the end of the experiment, mice were fasted overnight and blood was collected by puncture of the retro-orbital plexus. Plasma lipoprotein profiles were determined by fast performance liquid chromatography gel filtration on a Superose 6 column. Cholesterol levels were assessed by a commercially available kit (Randox, CH200). Tissues were fixed in formalin 4% for 24 h before paraffin imbedding. In some experiments, tissues were imbedded in OCT (Thermo Scientific, Neg-50) and stored at −80°C. Macrophage-specific atg7 knockouts were obtained by crossbreeding Atg7F/F mice for > 10 generations with Lysm-Cre+ mice (C57BL/6, Jackson Laboratory, 004781) which express Cre recombinase under the control of the mouse lysozyme M promoter. All experiments were approved by the Ethics Committee of the University of Antwerp.
Cell culture
VSMCs were isolated from mouse aorta as previously described.62,63 Briefly, aortas were preincubated in HBSS (Gibco Life Technologies, 14025-050) containing 1 mg/ml collagenase (Worthington, type II CLS, 4176), 1 mg/ml soybean trypsin inhibitor (Worthington, 3571) and 0.74 units/ml elastase (Worthington, 2279) for 15 min at 37°C, in 95% air/5% CO2. Next, the adventitia was stripped off and the aorta was opened longitudinally to remove blood clots and endothelial cells. Subsequently, aortas were placed in fresh enzyme solution and incubated for 1 h (37°C, in 95% air/5% CO2). Isolated cells were centrifuged, washed and resuspended in DMEM/F-12 medium (Gibco Life Technologies, 11320-074) supplemented with 20% fetal bovine serum (FBS; Sigma Aldrich, F6178). Cells were used from passage 4 till 10 and cultured in DMEM/F-12 medium supplemented with 10% FBS. Cells were isolated from 2 or 3 mice. In each individual experiment, cells from the same passage number were used. Bone marrow-derived macrophages (BMDM) were harvested by flushing bone marrow from the hind limbs of mice. Cells were cultured for 7 d in RPMI medium (Gibco Life Technologies, 61870-010) supplemented with 15% L-cell conditioned medium (LCCM). LCCM is produced by L929 cells cultured in RPMI medium. Once cells are confluent, medium is changed and cells are left alone for 7 d to secrete M-CSF (monocyte colony-stimulating factor). Then, the medium (=LCCM) is collected and stored at −80°C. To induce oxidative stress, cells were treated with 25 µmol/l tert-butyl hydroperoxide (tBHP; Sigma-Aldrich, 458139) or 50 µg/ml oxidized LDL (oxLDL) for 24 h. oxLDL was prepared as previously described.64 In some experiments, cells were treated with 10 µmol/l puromycin (Sigma Aldrich, P8833) for 12 h or exposed to UV-irradiation for 10 min. Cell viability was evaluated by a neutral red assay.65 ROS production was determined using the fluorogenic marker carboxy-H2DCFDA (Image-iT™ LIVE Green Reactive Oxygen Species Detection Kit, Molecular Probes, I36007). The DCFDA-positive cells were immediately visualized by fluorescence microscopy and then quantified using ImageJ software. To measure cellular size, VSMCs were labeled with 1 µmol/l calcein AM (Molecular probes, C3099) and visualized with an inverted microscope attached to a microlens-enhanced dual spinning disk confocal system (UltraVIEW VoX, PerkinElmer, Seer Green, UK). Images were analyzed with Volocity software (PerkinElmer). Cellular protein quantity was determined using the BCA method (Pierce BCA Protein Assay Kit, Thermo Scientific, 23225). To induce starvation, cells were incubated in Earle's balanced salt solution (EBSS; Gibco Life Technologies, 14155-048) for 48 h.
Transmission electron microscopy
Samples were prepared for transmission electron microscopy (TEM) as previously described.66 Sections were examined with a Tecnai microscope (FEI, Eindhoven, The Netherlands) at 120 kV.
Analysis of proliferation, collagen amount and migration
Cellular proliferation was assessed using a BrdU incorporation test. Once 80% confluent, BMDM and VSMCs were treated with 10 µmol/l BrdU (Sigma-Aldrich, B5002) for 2 h or 6 h respectively, followed by an anti-BrdU staining (rabbit anti-BrdU antibody; Abcam, ab6326). The BrdU-positive nuclei were quantified using ImageJ software. Collagen synthesis in VSMCs was stimulated by 10 ng/ml TGFB (Peprotech, 100-21) for 48 h in serum-free medium. Total collagen amount was assayed using Sirius red staining as previously described.67 COL1A1/2 and COL3A1 expression was analyzed by immunofluorescence with rabbit anti-COL1A1/2 antibody (Abcam, ab21286) and anti-rabbit Alexa Fluor 555 secondary antibody (Molecular Probes, A21428), and goat anti-COL3A1 antibody (Biodesign, T33330G) and anti-goat Alexa Fluor 488 secondary antibody (Molecular Probes, A11078). The migratory capacity was evaluated, independently of cell size and chemotactic effects, using an Oris™ Cell Migration Assay (Platypus Technologies, CMA1.101). Briefly, 2.5×104 cells/well were added to a 96-well plate with Cell Seeding Inserts and incubated overnight to permit cell attachment. Subsequently, the stoppers were removed to allow cell migration into the empty zone; 24 h later, cells were fixed and stained with 0.1% crystal violet (Sigma Aldrich, C3886) in 20% methanol for 3 min to visualize and quantify the migrated cells using an inverted microscope. The migratory capacity of the cells was evaluated by measuring the percentage closure area: 100 × ((premigration)area – (migration)area) / (premigration)area.
Western blotting
Cells were lysed in Laemmli sample buffer (Bio-Rad, 161-0737) containing β-mercaptoethanol (Sigma Aldrich, M3148) and boiled for 4 min. Protein samples were loaded on NuPAGE 4-12% Bis-Tris gels (Life Technologies, BG04120) and after electrophoresis transferred to Immobilon-P membranes (Millipore, IPVH304F0). Membranes were probed with the following primary antibodies: goat anti-GSTA (ab53940), rabbit anti-NFE2L2 (ab137550), mouse anti-CDKN2A/p16 (ab54210) and rabbit anti-CDKN1A/p21 (ab7960) from Abcam; rabbit anti-CXCL12 (Bioss, bs-4938); rabbit anti-GAPDH (14C10), rabbit anti-TGFB (3711) and rabbit anti-phospho RB (8516) from Cell Signaling Technology; mouse anti-MAP1LC3B (Nanotools, clone 5F10, 0231-100); rabbit anti-NQO1 (Novus Biologicals, NBP1-40663); rabbit anti-PARP1 (sc-7150), rabbit anti-CDKN2A/p16 (sc-1207) and rabbit anti-total RB (sc-50) from Santa Cruz Biotechnology; mouse anti-ACTB (clone AC-15, A5441), rabbit anti-ATG7 (A2856), rabbit anti-ATG5 (A0856), rabbit anti-SQSTM1/p62 (P0067) and rabbit anti-acetyl-TP53 (SAB4503014) from Sigma-Aldrich. Thereafter, membranes were incubated with HRP-conjugated secondary antibodies (Dako, anti-rabbit P0399, anti-mouse P0260, anti-goat P0160) to allow chemiluminescent detection. In some experiments, cytoplasmic and nuclear fractions were isolated from cultured VSMCs and BMDM using the NE-PER® Nuclear and Cytoplasmic Extraction kit (Thermo Scientific, 78833) prior to western blot analysis. Tissue samples were homogenized (see section gelatin zymography) before western blot analysis.
Gelatin zymography
Ligated left common carotid arteries were collected, gently flushed to remove all blood clots and immediately snap frozen. Tissues were homogenized in RIPA buffer (Sigma Aldrich, R0278) and protein content was determined using the BCA method (Pierce BCA Protein Assay Kit, Thermo Scientific, 23225). Samples were mixed with Laemmli sample buffer without β-mercaptoethanol before loading on a 10% Zymogram Gelatin gel (Life Technologies, EC6175). After electrophoresis in Tris-Glycine SDS Running Buffer (Life Technologies, LC2675), proteins were renaturated in Zymogram Renaturing Buffer (Life Technologies, LC2670) and incubated with Zymogram Developing Buffer (Life Technologies, LC2671) overnight. Gels were stained with Coomassie brilliant blue (Merck, 115444) for 3 h and subsequently destained twice for 1 h. Finally, gels were scanned to visualize the gelatinolytic activity of MMP9 and MMP2 in the left common carotid artery.
MicroArray
Total RNA was prepared using the Absolutely RNA Miniprep Kit (Agilent, 400800) and treated with RNase-free DNase I. RNA quality was verified on an Agilent 2100 Bioanalyzer System (Agilent Technologies, Waldbronn, Germany) using the RNA 6000 Nano LabChip kit (Agilent Technologies, 5067-1511). Samples were then analyzed by the Microarray Facility of the Flanders Interuniversity Institute for Biotechnology using a Whole Mouse Genome Oligo Microarray Kit (Agilent Technologies, G4122F) representing over 41,000 mouse genes and transcripts.
Real time RT-PCR
TaqMan gene expression assays (Applied Biosystems) for Nfe2l2, Gsta and Nqo1 were performed in duplicate on an ABI Prism 7300 sequence detector system (Applied Biosystems, Foster City, CA, USA) in 25-µl reaction volumes containing Universal PCR Master Mix (Applied Biosystems, 4324018). The parameters for PCR amplification were 95°C for 10 min followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. Relative expression of mRNA was calculated using the comparative threshold cycle method. All data were normalized for quantity of cDNA input by performing measurements on the endogenous reference gene Actb.
Nfe2l2 silencing and CDKN2A and SQSTM1 overexpression
VSMCs were transfected with 100 nmol/l Nfe2l2-specific siRNA (ON-TARGETplus® SMART Pool, Mouse NFE2L2, Dharmacon, L-040766-00-0005) or siRNA control (ON-TARGETplus® Control Pool, nontargeting pool, Dharmacon, D-001810-10-05) via nucleofection using the Human AoSMC Nucleofector™ Kit (Amaxa, VPC-1001). Silencing efficiency was assessed by real time RT-PCR and western blotting.
To overexpress CDKN2A or SQSTM1 in VSMCs, full-length cDNA encoding mouse CDKN2A or SQSTM1 protein was amplified by PCR from VSMCs using Platinum Pfx DNA polymerase (Invitrogen, 11708-013). The following primers were used: 5′-CCAAGCTTAGCAGCATGGAGTCCGCTGCAGACAG-3′ (Cdkn2a FW), 5′-CCTCTAGATTAGCTCTGCTCTTGGGATTGGCC-3′ (Cdkn2a RV), 5′-CAGAATTCGTTATGGCGTCGTTCACGGTGAAGGC-3′ (Sqstm1 FW) and 5′-CAGCGGCCGCTATCACAATGGTGGAGGGTGCTTCG-3′ (Sqstm1 RV) from Sigma Aldrich. The resultant PCR products were HindIII/XbaI (Cdkn2a) or EcoRI/NotI (Sqstm1) digested and cloned in the similarly opened plasmid pEGFP-N3 (Clontech, 6080-1), thereby replacing the eGFP coding sequence. After sequencing, VSMCs were transfected with 5 µg plasmid DNA via nucleofection using the Human AoSMC Nucleofector™ kit (Amaxa, VPC-1001).
Analysis of cellular senescence
Senescence was determined using the Senescence Cells Histochemical Staining Kit (Sigma-Aldrich, CS0030). Briefly, cells were incubated with 1x Fixation Buffer for 7 min at RT. Next, cells were washed and incubated with staining mixture (containing X-gal) at 37°C for 24 h. Subsequently, cells were counterstained with Nuclear Fast Red (BDH, 34209). The percentage of senescence-associated GLB1-positive cells was quantified using ImageJ software. Cell cycle analysis was performed by flow cytometry using BD FACScan (BD, Franklin Lakes, NJ, USA) as previously described.68 Results were analyzed with FCS Express 4 Flow software. To detect DNA damage, a comet assay was performed as previously described.69
Histological analysis
The percentage neointima formation after ligation [(neointima area/lumen area) × 100] as well as the plaque area and necrotic core in the brachiocephalic artery and aortic root were measured on H&E sections. Atherosclerotic plaques located in the aortic root were analyzed in 5 different sections sliced at equally spaced intervals (every 50 µm). A 3000 μm2 minimum threshold was implemented in order to avoid counting of regions that likely do not represent substantial areas of necrosis.70 Lesions were further analyzed by immunohistochemistry with the following primary antibodies: rabbit anti-LAMP2 (BD PharMingen, 553322), rabbit anti-cleaved CASP3 (Cell Signaling Technology, 9661), rabbit anti-phospho RB (Cell Signaling Technology, 8516), rabbit anti-CDKN2A/p16 (Santa Cruz Biotechnology, sc-1207), goat anti-TAGLN (Abcam, ab10135), mouse anti-ACTA2 (FITC labeled; Sigma-Aldrich, A2547) and rabbit anti-SQSTM1/p62 (Sigma Aldrich, P0067). Thereafter, tissue sections were incubated with species-appropriate HRP-conjugated secondary antibodies followed by 60 min of reactive ABC (Vector Laboratories, pk4001). 3,3′-diaminobenzidine (Sigma Aldrich, D5637) or 3-amino-9-ethyl-carbazole (Sigma Aldrich, A5754) were used as a chromogen. A Sirius red (Sigma Aldrich, 365548) staining was used for detection of total collagen and COL1A1/2 under polarized light. COL3A1 was detected using goat anti-COL3A1 antibody (Biodesign, T33330G). The Senescence Cells Histochemical Staining Kit (Sigma Aldrich, CS0030) was applied on the left common carotid artery (LCCA) and aortic root. Frozen sections of the LCCA were counterstained with periodic acid-Schiff (PAS) to visualize the basal lamina. Sections of the aortic root were counterstained with Nuclear Fast Red (BDH, 34209). Consecutive sections of the aortic root were double stained with rabbit anti-phospho-RB antibody (Cell Signaling Technology, 8516) and anti-rabbit Alexa Fluor 555 secondary antibody (Molecular Probes, A21428), and mouse anti-ACTA2-FITC (Sigma Aldrich, F3777) antibody and visualized by fluorescence microscopy. All other images were acquired with Universal Grab 6.1 software using an Olympus BX40 microscope (Tokyo, Japan) and quantified with ImageJ software.
Statistical analysis
All data were analyzed with SPSS 22.0 software (SPSS Inc.) and presented as mean ± SEM. All in vitro experiments were analyzed by the Student t test or two-way ANOVA with genotype and treatment as category factors, followed by a Dunnett Post Hoc test if applicable. The n-value represents the number of independently performed experiments unless mentioned otherwise. The in vivo data were analyzed by a Student t test or a Mann Whitney test in case of nonparametric analysis, and the n-value represents the number of mice. In some cases, a repeated measure or univariate analysis was applied whereas the n-value represents the number of measurements per mouse. Differences were considered significant at P < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors acknowledge Dr Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science, Japan) for providing Atg7F/F mice and Rita Van den Bossche, Hermine Fret, Anne-Elise Van Hoydonck, Lieve Svensson, Dominique De Rijck, Ken Op de Beeck, Ridha Limame, Hadis Shakeri and Lynn Roth for excellent technical support. The UltraVIEW VoX spinning disk confocal microscope and TEM were purchased with support of the Hercules Foundation. The authors also thank Erik Biessen and Carlie De Vries for fruitful discussions.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was supported by the Fund for Scientific Research (FWO)-Flanders (grant G044312N) and the University of Antwerp (BOF). Mandy O.J. Grootaert is a fellow of the Agency for Innovation by Science and Technology (IWT).
References
- 1.Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 2014; 24:24-41; PMID:24366339; http://dx.doi.org/ 10.1038/cr.2013.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; http://dx.doi.org/ 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- 3.Martinet W, De Bie M, Schrijvers DM, De Meyer GRY, Herman AG, Kockx MM. 7-ketocholesterol induces protein ubiquitination, myelin figure formation, and light chain 3 processing in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2004; 24:2296-301; PMID:15458974; http://dx.doi.org/ 10.1161/01.ATV.0000146266.65820.a1 [DOI] [PubMed] [Google Scholar]
- 4.Jia G, Cheng G, Gangahar DM, Agrawal DK. Insulin-like growth factor-1 and TNF-α regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol Cell Biol 2006; 84:448-54; PMID:16942488; http://dx.doi.org/ 10.1111/j.1440-1711.2006.01454.x [DOI] [PubMed] [Google Scholar]
- 5.Hill BG, Haberzettl P, Ahmed Y, Srivastava S, Bhatnagar A. Unsaturated lipid peroxidation-derived aldehydes activate autophagy in vascular smooth-muscle cells. Biochem J 2008; 410:525-34; PMID:18052926; http://dx.doi.org/ 10.1042/BJ20071063 [DOI] [PubMed] [Google Scholar]
- 6.Xu K, Yang Y, Yan M, Zhan J, Fu X, Zheng X. Autophagy plays a protective role in free cholesterol overload-induced death of smooth muscle cells. J Lipid Res 2010; 51:2581-90; PMID:20484746; http://dx.doi.org/ 10.1194/jlr.M005702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding Z, Wang X, Schnackenberg L, Khaidakov M, Liu S, Singla S, Dai Y, Mehta JL. Regulation of autophagy and apoptosis in response to ox-LDL in vascular smooth muscle cells, and the modulatory effects of the microRNA hsa-let-7 g. Int J Cardiol 2013; 168:1378-85; PMID:23305858; http://dx.doi.org/ 10.1016/j.ijcard.2012.12.045 [DOI] [PubMed] [Google Scholar]
- 8.Salabei JK, Cummins TD, Singh M, Jones SP, Bhatnagar A, Hill BG. PDGF-mediated autophagy regulates vascular smooth muscle cell phenotype and resistance to oxidative stress. Biochem J 2013; 451:375-88; PMID:23421427; http://dx.doi.org/ 10.1042/BJ20121344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He C, Zhu H, Zhang W, Okon I, Wang Q, Li H, Le YZ, Xie Z. 7-Ketocholesterol induces autophagy in vascular smooth muscle cells through Nox4 and Atg4B. Am J Pathol 2013; 183:626-37; PMID:23770348; http://dx.doi.org/ 10.1016/j.ajpath.2013.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salabei JK, Hill BG. Implications of autophagy for vascular smooth muscle cell function and plasticity. Free Radic Biol Med 2013; 65:693-703; PMID:23938401; http://dx.doi.org/ 10.1016/j.freeradbiomed.2013.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grube E, Buellesfeld L. Everolimus for stent-based intracoronary applications. Rev Cardiovasc Med 2004; 5 Suppl 2:S3-8 [PubMed] [Google Scholar]
- 12.Zhao HQ, Nikanorov A, Virmani R, Schwartz LB. Inhibition of experimental neointimal hyperplasia and neoatherosclerosis by local, stent-mediated delivery of everolimus. J Vasc Surg 2012; 56:1680-8; PMID:22841285; http://dx.doi.org/ 10.1016/j.jvs.2012.04.022 [DOI] [PubMed] [Google Scholar]
- 13.Martinet W, De Meyer GRY. Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res 2009; 104:304-17; PMID:19213965; http://dx.doi.org/ 10.1161/CIRCRESAHA.108.188318 [DOI] [PubMed] [Google Scholar]
- 14.Perrotta I. The use of electron microscopy for the detection of autophagy in human atherosclerosis. Micron 2013; 50:7-13 [DOI] [PubMed] [Google Scholar]
- 15.Bennett MR. Reactive oxygen species and death: oxidative DNA damage in atherosclerosis. Circ Res 2001; 88:648-50; PMID:11304484; http://dx.doi.org/ 10.1161/hh0701.089955 [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Tabas I. Emerging roles of mitochondria ROS in atherosclerotic lesions: causation or association? J Atheroscler Thromb 2014; 21:381-90; PMID:24717761; http://dx.doi.org/ 10.5551/jat.23929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 2015; 22:377-88; PMID:25257172; http://dx.doi.org/ 10.1038/cdd.2014.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perrotta I, Aquila S. The role of oxidative stress and autophagy in atherosclerosis. Oxid Med Cell Longev 2015; 2015:130315; PMID:25866599; http://dx.doi.org/ 10.1155/2015/130315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI et al.. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 2010; 12:213-23; PMID:20173742 [DOI] [PubMed] [Google Scholar]
- 20.Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8:729-40; PMID:17667954; http://dx.doi.org/ 10.1038/nrm2233 [DOI] [PubMed] [Google Scholar]
- 21.Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res 2007; 100:15-26; PMID:17204661; http://dx.doi.org/ 10.1161/01.RES.0000256837.40544.4a [DOI] [PubMed] [Google Scholar]
- 22.Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 2012; 15:545-53; PMID:22445600; http://dx.doi.org/ 10.1016/j.cmet.2012.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al.. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149-63; PMID:18083104; http://dx.doi.org/ 10.1016/j.cell.2007.10.035 [DOI] [PubMed] [Google Scholar]
- 24.Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 2010; 30:3275-85; PMID:20421418; http://dx.doi.org/ 10.1128/MCB.00248-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y et al.. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425-34; PMID:15866887; http://dx.doi.org/ 10.1083/jcb.200412022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al.. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880-4; PMID:16625205; http://dx.doi.org/ 10.1038/nature04723 [DOI] [PubMed] [Google Scholar]
- 27.Jung HS, Chung KW, Won KJ, Kim J, Komatsu M, Tanaka K, Nguyen YH, Kang TM, Yoon KH, Kim JW et al.. Loss of autophagy diminishes pancreatic β cell mass and function with resultant hyperglycemia. Cell Metab 2008; 8:318-24; PMID:18840362; http://dx.doi.org/ 10.1016/j.cmet.2008.08.013 [DOI] [PubMed] [Google Scholar]
- 28.Blagosklonny MV. Cell senescence: hypertrophic arrest beyond the restriction point. J Cell Physiol 2006; 209:592-97; PMID:17001692; http://dx.doi.org/ 10.1002/jcp.20750 [DOI] [PubMed] [Google Scholar]
- 29.Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol 2013; 75:685-705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 2013; 123:966-72; PMID:23454759; http://dx.doi.org/ 10.1172/JCI64098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res 2012; 111:245-59; PMID:22773427; http://dx.doi.org/ 10.1161/CIRCRESAHA.111.261388 [DOI] [PubMed] [Google Scholar]
- 32.Erusalimsky JD, Kurz DJ. Cellular senescence in vivo: its relevance in ageing and cardiovascular disease. Exp Gerontol 2005; 40:634-42; PMID:15970413; http://dx.doi.org/ 10.1016/j.exger.2005.04.010 [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez-Menocal L, Pham SM, Mateu D, St-Pierre M, Wei Y, Pestana I, Aitouche A, Vazquez-Padron RI. Aging increases p16 INK4a expression in vascular smooth-muscle cells. Biosci Rep 2010; 30:11-18; http://dx.doi.org/ 10.1042/BSR20080128 [DOI] [PubMed] [Google Scholar]
- 34.Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest 2004; 114:1299-307; PMID:15520862; http://dx.doi.org/ 10.1172/JCI22475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell 2011; 146:682-95; PMID:21884931; http://dx.doi.org/ 10.1016/j.cell.2011.07.030 [DOI] [PubMed] [Google Scholar]
- 36.Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle 2009; 8:1888-95; PMID:19471117; http://dx.doi.org/ 10.4161/cc.8.12.8606 [DOI] [PubMed] [Google Scholar]
- 37.Demidenko ZN, Blagosklonny MV. At concentrations that inhibit mTOR, resveratrol suppresses cellular senescence. Cell Cycle 2009; 8:1901-4; PMID:19471118; http://dx.doi.org/ 10.4161/cc.8.12.8810 [DOI] [PubMed] [Google Scholar]
- 38.Kang HT, Lee KB, Kim SY, Choi HR, Park SC. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS One 2011; 6:e23367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. A possible involvement of p62/sequestosome-1 in the process of biliary epithelial autophagy and senescence in primary biliary cirrhosis. Liver Int 2012; 32:487-99; PMID:22098537 [DOI] [PubMed] [Google Scholar]
- 40.Fujii S, Hara H, Araya J, Takasaka N, Kojima J, Ito S, Minagawa S, Yumino Y, Ishikawa T, Numata T, et al.. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology 2012; 1:630-41; PMID:22934255; http://dx.doi.org/ 10.4161/onci.20297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gewirtz DA. Autophagy and senescence: a partnership in search of definition. Autophagy 2013; 9:808-12; PMID:23422284; http://dx.doi.org/ 10.4161/auto.23922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takasaka N, Araya J, Hara H, Ito S, Kobayashi K, Kurita Y, Wakui H, Yoshii Y, Yumino Y, Fujii S, et al.. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J Immunol 2014; 192:958-68; PMID:24367027; http://dx.doi.org/ 10.4049/jimmunol.1302341 [DOI] [PubMed] [Google Scholar]
- 43.Carmeliet P, Moons L, Collen D. Mouse models of angiogenesis, arterial stenosis, atherosclerosis and hemostasis. Cardiovasc Res 1998; 39:8-33; PMID:9764187; http://dx.doi.org/ 10.1016/S0008-6363(98)00108-4 [DOI] [PubMed] [Google Scholar]
- 44.Newby AC. Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates. Cardiovasc Res 2006; 69:614-24 [DOI] [PubMed] [Google Scholar]
- 45.Louis SF, Zahradka P. Vascular smooth muscle cell motility: From migration to invasion. Exp Clin Cardiol 2010; 15:e75-85; PMID:21264073 [PMC free article] [PubMed] [Google Scholar]
- 46.Godin D, Ivan E, Johnson C, Magid R, Galis ZS. Remodeling of carotid artery is associated with increased expression of matrix metalloproteinases in mouse blood flow cessation model. Circulation 2000; 102:2861-6; PMID:11104745; http://dx.doi.org/ 10.1161/01.CIR.102.23.2861 [DOI] [PubMed] [Google Scholar]
- 47.Khan R, Agrotis A, Bobik A. Understanding the role of transforming growth factor-beta1 in intimal thickening after vascular injury. Cardiovasc Res 2007; 74:223-34; PMID:17349984; http://dx.doi.org/ 10.1016/j.cardiores.2007.02.012 [DOI] [PubMed] [Google Scholar]
- 48.Rekhter MD. Collagen synthesis in atherosclerosis: too much and not enough. Cardiovasc Res 1999; 41:376-84; PMID:10341837; http://dx.doi.org/ 10.1016/S0008-6363(98)00321-6 [DOI] [PubMed] [Google Scholar]
- 49.Herrera RE, Makela TP, Weinberg RA. TGF β-induced growth inhibition in primary fibroblasts requires the retinoblastoma protein. Mol Biol Cell 1996; 7:1335-42; PMID:8885230; http://dx.doi.org/ 10.1091/mbc.7.9.1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Debacq-Chainiaux F, Pascal T, Boilan E, Bastin C, Bauwens E, Toussaint O. Screening of senescence-associated genes with specific DNA array reveals the role of IGFBP-3 in premature senescence of human diploid fibroblasts. Free Radic Biol Med 2008; 44:1817-32; PMID:18329388; http://dx.doi.org/ 10.1016/j.freeradbiomed.2008.02.001 [DOI] [PubMed] [Google Scholar]
- 51.Schober A, Knarren S, Lietz M, Lin EA, Weber C. Crucial role of stromal cell-derived factor-1alpha in neointima formation after vascular injury in apolipoprotein E-deficient mice. Circulation 2003; 108:2491-7; PMID:14581398; http://dx.doi.org/ 10.1161/01.CIR.0000099508.76665.9A [DOI] [PubMed] [Google Scholar]
- 52.Zernecke A, Schober A, Bot I, von Hundelshausen P, Liehn EA, Mopps B, Mericskay M, Gierschik P, Biessen EA, Weber C. SDF-1alpha/CXCR4 axis is instrumental in neointimal hyperplasia and recruitment of smooth muscle progenitor cells. Circ Res 2005; 96:784-91; PMID:15761195; http://dx.doi.org/ 10.1161/01.RES.0000162100.52009.38 [DOI] [PubMed] [Google Scholar]
- 53.Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, Goddard M, Bennett M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res 2006; 99:156-64; PMID:16794190; http://dx.doi.org/ 10.1161/01.RES.0000233315.38086.bc [DOI] [PubMed] [Google Scholar]
- 54.Gorenne I, Kavurma M, Scott S, Bennett M. Vascular smooth muscle cell senescence in atherosclerosis. Cardiovasc Res 2006; 72:9-17; PMID:16824498; http://dx.doi.org/ 10.1016/j.cardiores.2006.06.004 [DOI] [PubMed] [Google Scholar]
- 55.Johnson JL. Emerging regulators of vascular smooth muscle cell function in the development and progression of atherosclerosis. Cardiovasc Res 2014; 103:452-60 [DOI] [PubMed] [Google Scholar]
- 56.Schrijvers DM, De Meyer GRY, Martinet W. Autophagy in atherosclerosis: a potential drug target for plaque stabilization. Arterioscler Thromb Vasc Biol 2011; 31:2787-91; PMID:22096098; http://dx.doi.org/ 10.1161/ATVBAHA.111.224899 [DOI] [PubMed] [Google Scholar]
- 57.De Meyer GRY, Grootaert MOJ, Michiels CF, Kurdi A, Schrijvers DM, Martinet W. Autophagy in vascular disease. Circ Res 2015; 116:468-79; PMID:25634970; http://dx.doi.org/ 10.1161/CIRCRESAHA.116.303804 [DOI] [PubMed] [Google Scholar]
- 58.Lavandero S, Chiong M, Rothermel BA, Hill JA. Autophagy in cardiovascular biology. J Clin Invest 2015; 125:55-64; PMID:25654551; http://dx.doi.org/ 10.1172/JCI73943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo Z, Mitchell-Raymundo F, Yang H, Ikeno Y, Nelson J, Diaz V, Richardson A, Reddick R. Dietary restriction reduces atherosclerosis and oxidative stress in the aorta of apolipoprotein E-deficient mice. Mech Ageing Dev 2002; 123:1121-31; PMID:12044962; http://dx.doi.org/ 10.1016/S0047-6374(02)00008-8 [DOI] [PubMed] [Google Scholar]
- 60.Gorenne I, Kumar S, Gray K, Figg N, Yu H, Mercer J, Bennett M. Vascular smooth muscle cell sirtuin 1 protects against DNA damage and inhibits atherosclerosis. Circulation 2013; 127:386-96; PMID:23224247; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.112.124404 [DOI] [PubMed] [Google Scholar]
- 61.Martinet W, De Loof H, De Meyer GRY. mTOR inhibition: a promising strategy for stabilization of atherosclerotic plaques. Atherosclerosis 2014; 233:601-7; PMID:24534455; http://dx.doi.org/ 10.1016/j.atherosclerosis.2014.01.040 [DOI] [PubMed] [Google Scholar]
- 62.Owens GK, Loeb A, Gordon D, Thompson MM. Expression of smooth muscle-specific α-isoactin in cultured vascular smooth muscle cells: relationship between growth and cytodifferentiation. J Cell Biol 1986; 102:343-52; PMID:3944187; http://dx.doi.org/ 10.1083/jcb.102.2.343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Geisterfer AA, Peach MJ, Owens GK. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ Res 1988; 62:749-56; PMID:3280155; http://dx.doi.org/ 10.1161/01.RES.62.4.749 [DOI] [PubMed] [Google Scholar]
- 64.Schrijvers DM, De Meyer GRY, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol 2005; 25:1256-61; PMID:15831805; http://dx.doi.org/ 10.1161/01.ATV.0000166517.18801.a7 [DOI] [PubMed] [Google Scholar]
- 65.Lowik CW, Alblas MJ, van de Ruit M, Papapoulos SE, van der Pluijm G. Quantification of adherent and nonadherent cells cultured in 96-well plates using the supravital stain neutral red. Anal Biochem 1993; 213:426-33; PMID:7694524; http://dx.doi.org/ 10.1006/abio.1993.1442 [DOI] [PubMed] [Google Scholar]
- 66.Martinet W, Timmermans JP, De Meyer GRY. Methods to assess autophagy in situ - Transmission electron microscopy vs. immunohistochemistry. In: Galluzzi L, Kroemer G, editors. Methods in Enzymology: cell-wide metabolic alterations associated with malignancy. Waltham, Massachusetts: Academic Press; 2014:89-114; PMID:24924129 [DOI] [PubMed] [Google Scholar]
- 67.Yu H, Stoneman V, Clarke M, Figg N, Xin HB, Kotlikoff M, Littlewood T, Bennett M. Bone marrow-derived smooth muscle-like cells are infrequent in advanced primary atherosclerotic plaques but promote atherosclerosis. Arterioscler Thromb Vasc Biol 2011; 31:1291-9; PMID:21372299; http://dx.doi.org/ 10.1161/ATVBAHA.110.218578 [DOI] [PubMed] [Google Scholar]
- 68.Vindelov LL, Christensen IJ. Detergent and proteolytic enzyme-based techniques for nuclear isolation and DNA content analysis. Methods Cell Biol 1994; 41:219-29; PMID:7532263; http://dx.doi.org/ 10.1016/S0091-679X(08)61720-3 [DOI] [PubMed] [Google Scholar]
- 69.Martinet W, Knaapen MW, De Meyer GRY, Herman AG, Kockx MM. Oxidative DNA damage and repair in experimental atherosclerosis are reversed by dietary lipid lowering. Circ Res 2001; 88:733-9; PMID:11304497; http://dx.doi.org/ 10.1161/hh0701.088684 [DOI] [PubMed] [Google Scholar]
- 70.Seimon TA, Wang Y, Han S, Senokuchi T, Schrijvers DM, Kuriakose G, Tall AR, Tabas IA. Macrophage deficiency of p38alpha MAPK promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. J Clin Invest 2009; 119:886-98; PMID:19287091 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.