

Abstract
Preclinical modelling studies are beginning to aid development of therapies targeted against key regulators of pancreatic cancer progression. Pancreatic cancer is an aggressive, stromally-rich tumor, from which few people survive. Within the tumor microenvironment cellular and extracellular components exist, shielding tumor cells from immune cell clearance, and chemotherapy, enhancing progression of the disease. The cellular component of this microenvironment consists mainly of stellate cells and inflammatory cells. New findings suggest that manipulation of the cellular component of the tumor microenvironment is possible to promote immune cell killing of tumor cells. Here we explore possible immunogenic therapeutic strategies. Additionally extracellular stromal elements play a key role in protecting tumor cells from chemotherapies targeted at the pancreas. We describe the experimental findings and the pitfalls associated with translation of stromally targeted therapies to clinical trial. Finally, we discuss the key inflammatory signal transducers activated subsequent to driver mutations in oncogenic Kras in pancreatic cancer. We present the preclinical findings that have led to successful early trials of STAT3 inhibitors in pancreatic adenocarcinoma.
Keywords: Pancreatic cancer, Inflammation, Stroma, Microenvironment
Core tip: Many advances have been made in preclinical assessment of therapies in pancreatic cancer. Here we review the successes and failures of translation to clinical trial of therapies targeting the pancreatic cancer microenvironment. Using data from preclinical trials we expose opportunities for further clinical trial within pancreatic cancer. We focus on therapies that modulate the immune response to pancreatic cancer, stromally active therapies and therapies targeting inflammatory signal transduction that are key in pancreatic cancer progression. We provide experimental results that have led to clinical trial and those findings that may be exploited in future. We attempt to rationalize the failure of certain therapies to translate to clinical practice and provide a realistic overview of why at present tumor microenvironment targeted therapies are not licensed in pancreatic cancer.
INTRODUCTION
Rationale for targeting inflammation in pancreatic cancer
Inflammation is a hallmark of cancer[1]. For over 100 years scientists have been interested in the relationship between inflammation and cancer. Researchers within Glasgow Royal Infirmary have for some time been interested in the relationship between cachexia, inflammation and poor prognosis in cancers of different origins. The modified Glasgow Prognostic Score (mGPS) that assesses blood albumin in combination with inflammation, C-reactive protein (CRP), has for over a decade been used to accurately predict outcome across a range of tumor types. Raised mGPS correlates with poor patient prognosis in colorectal, renal and pancreatic cancers[2]. Additionally, large observational studies have analyzed both cancer incidence and outcome based on daily aspirin use during previously performed randomized controlled trials. The long-term use of aspirin, a non-selective COX inhibitor, improves survival from cancer as a result of reduction in cancer incidence and metastatic burden[3-5]. These findings demonstrate a clear link between inflammation and cancer initiation and behaviour. Thus, there is observational evidence that inflammation promotes incidence, enhances progression and impacts on prognosis in patients with cancer.
Pancreatic adenocarcinoma (PDAC) presents at a late stage of progression and is associated with very poor outcomes (www.cancerresearchuk.org/cancer-info/cancerstats/). Surgery remains the only potentially curative treatment, though as few as 15% of patients have disease amenable to surgical intervention, and despite surgery the majority of these patients will succumb to recurrent disease. Therefore, new therapies and methods of instituting these therapies are required if survival is to improve in PDAC.
In addition to standard clinicopathological features, presence of systemic inflammation, as assessed by CRP, is a poor prognostic factor in patients undergoing surgical resection for PDAC. In a cohort of 135 patients who underwent potentially curative Whipple’s resection for PDAC an elevated mGPS was independently associated with lower overall survival[6]. Furthermore a high neutrophil to lymphocyte ratio (NLR), a further index of host innate response, has been categorically shown to confer poor prognosis in PDAC[7]. Interestingly, in this study of 74 patients, NLR had improved utility at predicting disease recurrence than CRP. This phenomenon was not confined to resectable cases of PDAC, inoperable cases of PDAC appear to respond poorly to chemotherapy in the presence of a raised NLR[8]. Indeed in the randomized controlled clinical trial of nab-paclitaxel in PDAC an elevated NLR conferred poor prognosis in both treatment arms[9]. Therefore, assessment of host inflammation at the time of diagnosis of PDAC, has clinical implications for patient survival regardless of therapeutic modality.
The treating physician should consider the inflammatory insults they are subjecting patients to during their treatment course. Over the last decade minimally invasive surgery of the pancreas has increased significantly. When 65033 resections of liver and pancreas were assessed, patients who had minimally invasive pancreatic resections had reduced morbidity, mortality, and length of stay in hospital compared with those having open resections. Traditionally inflammatory insults generated by minimally invasive surgery are smaller than open procedures, however, at present, studies show no oncological benefit[10]. Intra-operative blood transfusion is associated with loss of immune surveillance in cancer patients, with associated increases in morbidity and mortality following surgery. These data suggest transfusion should be avoided in the peri-operative period if possible[11]. Furthermore, a profoundly elevated systemic inflammatory response in the post-operative period has been associated with increasing rates of infectious complications following a number of operations including pancreatectomy[12]. To our knowledge no randomized controlled data exist to confirm the findings of this meta-analysis, however, the study raises the question of the benefits of use of anti-inflammatories in the post-operative setting. Such benefits would have to be offset against potential increases in the risk of anastomotic leak. Thus, when dealing with the small percentage of patients who have PDAC suitable for operative management, surgeons must consider the implications of their treatments. Limiting inflammatory insults involved seems sensible but requires clarification via clinical trial.
In vivo models of PDAC
Preclinical studies in PDAC have improved greatly in the past decade with the development of murine models that genetically and histologically recapitulate the human disease. Murine models use pancreas specific promoters to drive oncogenic Kras and tumor suppressor gene mutations including mutant Tp53 to create experimental PDAC murine models with an active microenvironment. These models permit preclinical interrogation of targeted therapies in the hope of translation to patients via clinical trial.
Importantly, progression of murine models of PDAC based on initiating oncogenic Kras mutations are greatly accelerated in the presence of pancreatic specific inflammation[13]. Further work by the same authors revealed this was due to the requirement of pancreatitis to overcome oncogene-induced senescence, which can be blocked by anti-inflammatory medication[14]. Lee et al[15] found that when Kras was mutated within the pancreas, pancreatic inflammation led to reduction in Ink4a expression. Low levels of Ink4a allowed tumor cells to escape senescence and progress to form tumors. In the presence of Kras mutations, pancreatic inflammation is sufficient to induce tumor formation.
Following initiation of PDAC, inflammation promotes tumor progression. Within the tumor microenvironment there are many pro and anti-tumoral interactions[16]. Immune cells present have plasticity that permits differing both pro or anti-tumorigenic actions based on received stimulus. Ultimately, during PDAC evolution, a myriad of stromal elements, immune cells and key transducers of inflammatory signals cooperate to permit disease progression. Improvements in understanding the tumor microenvironment are permitting trial of novel therapeutics against key disease progression mediators. Ongoing research in this area will elucidate more completely the complex interactions within the tumor microenvironment and help future development and assessment of multi-target drug regimens.
DISCUSSION
Inflammatory targets identified by in vivo modeling studies in PDAC
Possible inflammatory therapeutic targets in PDAC can be classified into one of three categories: (1) immune modulation to target tumors; (2) targeting tumor stroma; (3) targeting signal transduction (Table 1).
Table 1.
Preclinical assessment of inflammatory targets in pancreatic adenocarcinoma
| Target | Drug | PMID | Year | Authors summaries |
| Hedgehog acyltransferase (Hhat) | RU-SKI 43 | 24469057 | 2015 | In vivo mouse study targeting Hedgehog acyltransferase (Hhat). A lentivirally delivered hairpin RNA impeded the proliferation of pancreatic cancer in vitro and in vivo |
| Hedgehog | GDC-0449 | 25679326 | 2015 | Combination therapy with GDC-0449 or miR-let7b vs single agent therapy effectively inhibited tumor growth when injected to athymic nude mice bearing ectopic tumors generated using MIA PaCa-2 cells |
| Sonic hedgehog pathways | Ormeloxifene | 25840985 | 2015 | Ormeloxifene caused potent inhibition of the SHH signaling pathway via downregulation of SHH and its related important downstream targets. Ormeloxifene potentiated the antitumorigenic effect of gemcitabine by 75% in PDAC xenograft mice |
| Hedgehog pathway | MEDI-5304 | 24344235 | 2014 | MEDI-5304 displayed robust pharmacodynamic effects in stromal cells that translated to antitumor efficacy as a single agent in an HT-29/MEF coimplantation model of paracrine hedgehog signaling. MEDI-5304 also improved responses to carboplatin in the HT-29/MEF model. The antibody, however, had no effect as a single agent or in combination with gemcitabine on the CSC frequency or growth of several primary pancreatic cancer explant models |
| Hedgehog | GDC-0449 | 25278454 | 2014 | GDC-0449 for 3 wk leads to downmodulation of GLI1 and PTCH1, without significant changes in CSCs compared with baseline. GDC-0449 and gemcitabine were not superior to gemcitabine alone in the treatment of metastatic pancreatic cancer |
| Hedgehog pathway | Metformin | 24692708 | 2014 | In vitro, BxPC3 human pancreatic cancer cells were treated with metformin, and Sonic hedgehog (Shh) mRNA and protein levels were examined. Metformin reduces the expression of Shh in several cancer cell lines including pancreatic cancer cells |
| Hedgehog | Curcumin | 23563640 | 2013 | Curcumin can inhibit the proliferation of TGF-β1-stimulated PANC-1 cells, it can induce apoptosis, and reverse the EMT. The possible underlying molecular mechanisms are through inhibition of the Shh-GLI1 signaling pathway |
| COX 5-lipoxygenase (5-LOX) | Dietary licofelone | 25906749 | 2015 | In vivo mouse study of licofelone, an agent that targets both COX-2 and 5-LOX |
| LOX | Zileuton | 25483364 | 2014 | Zileuton suppressed the proliferation of SW1990 cells in a concentration- and time-dependent manner. In addition, zileuton induced SW1990 cells to undergo apoptosis and significantly decreased 5-LOX expression |
| STAT3 | Thiosemicarbazones | 25561562 | 2015 | In vitro and in vivo iron-binding ligands inhibit constitutive and interleukin 6-induced activation of STAT3 signaling DFO, Dp44mT, and DpC significantly decreased constitutive phosphorylation of the STAT3 transcription factor at Tyr705 in the pancreatic cancer cell lines and when injected in vivo |
| STAT3 | Aspirin metformin | 26056043 | 2015 | Metformin combined with aspirin significantly inhibited the phosphorylation of mTOR and STAT3, and induced apoptosis as measured by caspase-3 and PARP cleavage Taken together, the combination of metformin ad aspirin significantly inhibited pancreatic cancer cell growth in vitro and in vivo |
| JAK2 STAT3 | MicroRNA (miR)-216a | 25220761 | 2014 | MiR-216a overexpression markedly inhibited the JAK2/STAT3 signaling pathway and xenograft tumor growth in vivo |
| ALK pathway including STAT3 | Crizotinib | 25193856 | 2014 | Crizotinib strongly suppressed the growth and proliferation of pancreatic cancer cells in a dose-dependent manner. Crizotinib strongly inhibited the expression of activated ALK in pancreatic cancer cells, modulating its downstream mediators such as STAT3, AKT, and ERK |
| STAT3 NF-κB COX-2 EP4 | Nexrutine | 24520096 | 2014 | Nexrutine treatment inhibited growth of pancreatic cancer cells through induction of apoptosis Reduced levels and activity of STAT3, NF-κB, and their crosstalk led to transcriptional suppression of COX-2 and subsequent decreased levels of prostaglandin E2 (PGE2) and PGF2. Nexrutine intervention reduced the levels of NF-κB, STAT3, and fibrosis in vivo. Expression of prostaglandin receptor EP4 that is known to play a role in fibrosis was significantly elevated in human pancreatic tumors. Dual inhibition of STAT3-NF-κB by Nexrutine may overcome problems associated with inhibition of either pathway |
| JAK/STAT Src/FAK | Guggulsterone | 23920124 | 2013 | In vitro, guggulsterone treatment decreased mucin MUC4 expression in Capan1 and CD18/HPAF cells through transcriptional regulation by inhibiting Jak/STAT pathway |
| Notch JAK2 | GSI IX and AG-490 | 24293409 | 2014 | Combinational treatment with anti-NOTCH and JAK/STAT drugs significantly attenuates tumor progression in vivo and suppresses conversion from acinar-ductal-metaplasia to PDAC |
Outlines the significant interest shown by preclinical researchers in targeting inflammation in PDAC. Using the search criteria pancreatic cancer/pancreatic adenocarcinoma + hedgehog, JAK/STAT, LOX we identified preclinical studies that have attempted to assess therapeutics targeted against these important inflammatory mediators of PDAC progression in the past 2 years. We have included those published in journals with an impact factor > 5. PDAC: Pancreatic adenocarcinoma; PARP: Poly-ADP-ribose polymerase; JAK: Janus kinase; STAT: Signal transducer and activator of transcription.
This review will consider the progress made by preclinical studies in each of these three areas and how better understanding of the PDAC microenvironment has potential to translate to the clinical arena. Figure 1 provides a summary of potential inflammatory targets for therapy in PDAC.
Figure 1.
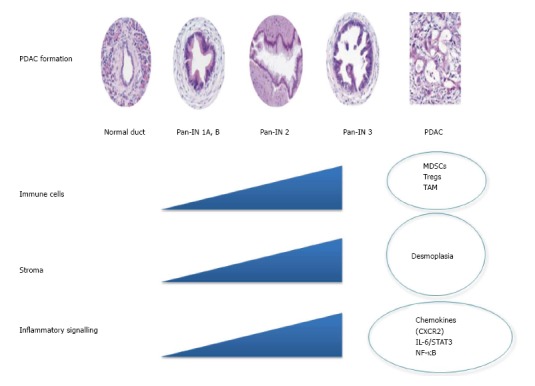
Changes in the pancreatic adenocarcinoma microenvironment during tumor formation. Pancreatic cancer forms from normal tissue via progression through pre-invasive pancreatic intra-epithelial neoplasia (Pan-IN) to invasive PDAC. Changes in immune cell components, stroma, and inflammatory signaling pathways all contribute to PDAC progression. Here we identify possible targets for therapy in PDAC. PDAC: Pancreatic adenocarcinoma; NF-κB: Nuclear facter kappa B; STAT: Signal transducer and sctivator of transcription; IL: Interleukin; MDSCs: Myeloid derived suppressor cells.
Immune modulation
Tumor immunosurveillance is a term that refers to identification and clearance of tumor cells in the early stages of tumorigenesis by the adaptive immune system. It is the role of CD8+ T cells to provide cytotoxic protection against “foreign” tumor cells, and hence the development of tumor immunogenicity. Different facets of immunosurveillance are now being interrogated to establish how PDAC so effectively evades detection.
Dendritic cells are a good example of an immune cell capable of adopting dual roles within the PDAC microenvironment. Dendritic cells can engage both CD8+ and CD4+ T cell responses dependent on stimulus[17]. Chemokine CXCL17 may be important for migration of dendritic cells to tumor sites while ICAM 2 upregulation was necessary for activation of a CD8+ cytotoxic response against tumor cells. Downregulation of CXCL17 and ICAM2 by tumor cells during evolution from precursor lesions to PDAC allowed tumors to develop immune tolerance[18]. In contrast Ochi et al[19] have demonstrated that blockade of TLR4 signaling promotes CD4+ T helper cell activity which has a positive effect on pancreatic tumourigenesis through mediation of pro-tumorigenic inflammatory responses. The plasticity of the immune system was highlighted by the findings of Beatty et al[20]. In patients with metastatic PDAC, targeting CD40 with monoclonal antibodies led to tumor regression. Authors anticipated CD40 ligation would result in enhanced anti-tumoral T cell responses, however in fact resulted in anti-tumoral effects through macrophage infiltration. This phase 1 trial holds promise for trials of similar agents to activate an anti-tumoral immune response.
Tumor-associated macrophages (TAMs) are ever present from pre-invasive Pan-Ins to established PDAC[21]. TAMs exhibit an M2 phenotype that is pro-tumorigenic while suppressing adaptive immunity[22]. In cancer, signals received by macrophages from tumor cells including interleukin (IL)-10 and transforming growth factor (TGF)-β lead to adoption of an M2 phenotype[22]. Macrophages are attracted to the tumor microenvironment via production of chemokines by tumor cells[23]. CSF1 and CCL2 are crucial mediators of this chemo-attraction. CCL2 overexpression mediates migration of M2 macrophages to PDAC and is thought to play a key role in recruiting pro-tumourigenic macrophages to metastatic sites in development of the metastatic niche[24,25]. In vivo studies of anti-CCL2 drugs were effective in enhancing tumor immunity and impacting on metastasis in PDAC[26]. In addition, patients with high CCL2 expression and low CD8 T cell infiltration suffer poor outcomes following tumor resection.
Direct depletion of TAMs may also be a therapeutic option. Trabectedin has recently been licenced for study in PDAC and is currently in phase 2 trials in advanced disease (NCT01339754). Trabectedin can actively target macrophages via caspase-8 dependent apoptosis with selectivity to TAMs achieved through differential expression of TRAIL receptors by macrophages[27].
Promotion of anti-tumoral cell mediated responses has been successful, particularly in metastatic melanoma. These strategies focus on engaging T cell responses. CTLA4 inhibitor, ipilimumab, was the first drug shown to improve outcome in patients with metastatic melanoma. Following this development and success of Programmed cell death 1 (PD1)/Programmed cell death ligand (PDL1) T cell checkpoint inhibitors has led to great excitement in the field of oncology. These drugs have made a significant impact on survival of patients with metastatic melanoma. CTLA4 is a cell surface protein that suppresses T cell function. When drugs such as Ipilimumab bind CTLA4, T cell function is activated. Likewise, PD1 inhibits T cell function. Production of PD1s major ligand PDL1 by tumor cells and pro-tumorigenic immune cells permits tumors to escape T cell mediated adaptive immunosurveillance[28]. Hence PD1 is an ideal target when attempting to generate anti-tumoral immune responses.
Concerns exist that PDAC may not respond to such T cell interference because they are extremely fibrotic and desmoplastic. As a result PDACs have a relative paucity of anti-tumoral T lymphocytes seen at histology compared with other epithelial tumors. Preliminary studies assessing ipilimumab have proven unsuccessful as a single agent[29]. Feig et al[30] have recently shown that immunosurveillance can be overcome in PDAC in vivo by expression of fibroblast activating protein (FAP) and production of CXCL12 by cancer associated fibroblasts (CAFs). These CAFs were able to prevent T cell infiltration to the tumor microenvironment. Interestingly when these CAFs were depleted genetically, or indeed CXCL12 was inhibited, tumors were sensitised to T cell checkpoint inhibition. Furthermore, myeloid derived suppressor cells (MDSCs) are orchestrated by PDAC to suppress proliferation and induce apoptosis in activated T cells[31]. Selective depletion of this granulocytic subset of MDSCs led to enhanced CD8+ T cell responses promoting immunosurveillance. These data suggest that greater understanding of the processes of evasion of tumor immunosurveillance by PDAC will open up therapeutic opportunities via combination with therapies that enhance the effectiveness of immunogenics.
Tumor vaccines are designed to target tumor specific antigens, activating adaptive immunity, eradicating tumor cells. Studies have assessed PDAC specific antigens including MUC-1 that is expressed by over 90% of PDAC cells. Vaccine PANVAC-VF was developed to be active against cells expressing MUC-1, oncofetal protein carcinoembryonic antigen (CEA) and 3 co-stimulatory molecules. Unfortunately no survival benefit was seen with the addition of PANVAC-VF to standard therapy in a phase III trial in palliative PDAC patients[32]. Ribonucleoprotein enzyme Telomerase, which maintains telomeric stability, has been assessed unsuccessfully as a potential vaccine target in the Telovac trial[33]. 1062 palliative patients were randomised to standard chemotherapy, sequential chemotherapy with Telovac or concurrent chemotherapy and Telovac. Neither Telovac groups showed any survival advantage. Smaller trials have been established against other targets including KRAS, although none has proven successful in clinical trial as yet showing how difficult it is to raise a successful adaptive immune response against PDAC.
Targeting tumor stroma
The majority of the tumor bulk of PDAC is composed of stromal cells. This dynamic network of immune cells, stellate cells and extracellular matrix is now believed to play a crucial role in sustenance and support for invasive tumor cells[34]. Stellate cells are key coordinators of fibrosis as a result of received signals from tumor cells in PDAC[35]. SPARC is one such factor present in high levels in the tumor microenvironment. SPARC functions normally to promote wound healing, however its role in PDAC is less certain. High expression of SPARC is associated with poor patient survival in resected cohorts of PDAC patients[36]. Albumin-bound Paclitaxel (nab-paclitaxel) binds SPARC-expressing fibroblasts, allowing therapeutic targeting of this cell type. When nab-paclitaxel was combined with gemcitabine patients with metastatic PDAC survived significantly longer compared with standard gemcitabine chemotherapy[37]. Surprisingly no appreciable histological changes to the stroma were evident in the tumors of mice treated with nab-paclitaxel raising the possibility that targeting SPARC improved outcome via a different biological mechanism than predicted[38].
PDAC is desmoplastic, avascular and relatively acellular. These attributes are believed to be responsible for failure of chemotherapies to adequately access tumor cells. Recently, high tissue pressures within the PDAC stroma have been suggested to prevent chemotherapy delivery to tumor cells[31]. Preclinical model work has suggested relieving such pressures will enhance chemotherapy delivery and subsequent tumor cell death. Unfortunately, these findings have not translated to patients with drugs including anti-MMP and VEGF inhibitors failing to have a therapeutic effect in clinical trials[35]. Sonic hedgehog paracrine signaling through smoothened has been implicated in the coordination of stromal elements by tumor cells in PDAC. Despite this, a phase II trial based on preclinical data assessing smoothened inhibition in PDAC was stopped early as patients receiving gemcitabine alone survived longer than those receiving the smoothened inhibitor in addition to gemcitabine[39] (clinicaltrials.gov, Infinity Pharmaceuticals, Cambridge, MA). In addition Catenacci et al[40] have recently found that Vismodegib, a Sonic hedgehog antagonist, when combined with gemcitabine provided no survival benefit in advanced PDAC patients than gemcitabine alone in a multicentre randomised controlled phase II trial. Hyaluronan is a prominent element within the stroma of PDAC and when targeted in preclinical studies experimenters have found significant improvements in tumor vasculature and lowering of tissue pressures permitting access by chemotherapeutics[41,42]. This agent is currently the subject of phase II clinical trials in combination with best current chemotherapeutic regimens FOLFIRINOX and gemcitabine/nab-paclitaxel (clinicaltrials.gov: NCT01959139 and NCT01839487).
At present no stromal agent is licenced for therapeutic use in PDAC, with sonic hedgehog in particular representing a cautionary tale of “bench to bedside” medicine. Recent studies, contrary to the findings of Olive et al[39], have proven that deletion of Shh specifically within the pancreas of in vivo PDAC models led to development of more aggressive tumors[43]. Furthermore, Özdemir et al[44] found eliminating CAFs from the tumor microenvironment led conversely to suppression of immunosurveillance, increasing numbers of T regulatory cells infiltrating the microenvironment, leading to tumor progression. What is clear from this work is that the PDAC stroma exists in a state of flux, with an interdependent network of stromal components which when manipulated therapeutically do not always produce expected results.
Targeting inflammatory signal transduction
Mutant KRAS is the major oncogenic driver of PDAC in more than 90% of cases[45]. Development of temporally controlled, inducible models of PDAC has recently permitted interrogation of signaling mechanisms required for PDAC tumorigenesis and progression. Use of inducible and reversible Kras alleles has demonstrated the requirement of ongoing stimulus from Kras for precursor lesions to progress to PDAC. Removal of Kras stimuli prevented progression from pan-INs to PDAC. However, when mutant Kras expression remained switched on, striking stimulation of the hedgehog signaling pathway was observed in addition to upregulation of inflammatory mediators IL-6, STAT3, and COX2. Kras inactivation resulted in decreased expression of these inflammatory mediators and resultant pan-IN regression[46], providing clear evidence of the relationship between Kras mutation and coordination of the inflammatory response in PDAC. KRAS activates RAF phosphorylation resulting in production of chemokines including CXCL1 and CXCL8[47]. CXCR2 a G-protein coupled receptor is crucial for MDSC migration to the tumor microenvironment and metastatic sites in breast and colon cancer and is activated by CXCL1, 2, 5, 7 and 8[48,49]. CXCR2 inhibition in preclinical models of PDAC successfully delayed tumor progression, suggesting it merits further study[50].
Ochi et al[51] recently reported high expression of TLR7 in PDAC. Activation of TLR7 promotes PDAC formation via downstream signalling through inflammatory signalling pathways including STAT3 and nuclear facter kappa B (NF-κB). When TLR7 was knocked out of immune cells within a murine model of PDAC and exposed to the same pro-tumorigenic conditions animals were completely protected from pancreatic carcinogenesis. Pharmacological TLR7 inhibition is yet to be assessed.
Transcription factor STAT3 represents a key-signaling node in PDAC[52,53]. When mouse models expressing endogenous mutant Kras combined with experimentally induced pancreatitis were assessed, STAT3 activation was significantly increased. Absence of STAT3 from the pancreata of these mice led to a block in acinar to ductal metaplasia and pan-IN formation, while reduced immune cell infiltration and IL-6 expression was also observed. Infiltrating macrophages were identified as producing IL-6 leading to STAT3 upregulation. Corcoran et al[54] have proposed that patients could be selected to trial based on phosphoSTAT3 levels as they predict PDAC cell sensitivity to JAK/STAT inhibitors. As the Jak/STAT signaling cascade has been recognised to impact on survival following resection for PDAC, investigators are now beginning to target it in randomized controlled trials[55].
Ruxolitinib targets the IL6/JAK/STAT signaling cascade. Assessment via double blind randomised controlled trial in advanced PDAC with capecitabine vs capecitabine/placebo showed a marginal survival advantage in the ruxolitinib group[56]. Intriguingly, those patients that benefited most were those with high mGPS scores. This work and that in preclinical studies by Corcoran et al[54] suggests trial of anti-inflammatory agents requires careful patient selection to optimise outcome in PDAC. Two ongoing phase III trials of JAK/STAT inhibition in PDAC, JANUS 1 and 2, basing patient selection on high systemic inflammatory scores, will test the hypothesis of the need for better patient selection in PDAC trials of inflammatory targeted agents.
NF-κB is also important in PDAC progression downstream of Kras mutation, specifically IKK2/β releases NF-κB from inhibition leading to progression of pancreatitis, ductal metaplasia, PanIN formation and eventually PDAC formation[57]. Genetic inactivation of IKK2/β in preclinical PDAC models led to failure of mice to develop tumors, while IKK2/β deficient animals showed reductions in pancreatic cell proliferation rates and reduced inflammatory cell infiltrate. These observations support a critical role for NF-κB in PDAC tumorigenesis. Unfortunately NF-κB is difficult to pharmacologically target effectively due to the complexity of regulation within the signaling cascade. Inhibitors of IKK2/β have so far failed to reach clinical trial.
SUMMARY
Preclinical trials are beginning to inform us as to tumor generated and tumor associated inflammation, how these factors help progress PDAC, and how they may be countered therapeutically.
Robust murine models of human disease now exist to allow preclinical trial of therapeutic agents. From these models researchers have established MDSCs, CAFs and TAMs are key cellular mediators of immunosuppression. Targeting these cells may sensitise tumors to immunotherapies such as anti-PD1 and CTLA4 antibodies. Immunotherapies have been extremely successful in diseases such as metastatic melanoma and if tumors can be “unmasked” from immunosuppressive elements this strategy is an exciting prospect in PDAC. IL-6/STAT3 and NF-κB represent established inflammatory signaling nodes that progress PDAC. Trial of JAK/STAT inhibition shows early promise in clinical trial. However, NF-κB remains an extremely difficult target to develop drugs against.
Early trials with trabectedin (immune cells), targeting hyaluronan (tumor stroma), and JAK/STAT inhibitors (inflammatory signaling), are extremely promising, however, as yet no large randomised controlled phase III trials have been published.
In future, we envisage combination trials targeting all three aspects of the pro-tumoral PDAC microenvironment will lead to better results in carefully selected patients. Pre-clinical assessment of such strategies is already under trial in robust mouse models of PDAC. A number of inflammatory targets, as outlined in this commentary, have been identified for trial, therefore the next decade of randomised controlled clinical trial data will determine the effectiveness of agents against these key inflammatory mediators in PDAC.
It is important to note the relative lack of success of translation of stromal/inflammation targeting therapies to clinical trial from the laboratory at present. Only those therapies that demonstrate the most robust preclinical data should be taken forward. Patient selection for tumor microenvironment targeted therapies is a key issue, as is identification of biomarkers of response to such therapies. While those patients presenting with high levels of inflammation are easy to identify clinically, objective monitoring of response to therapy is more difficult due to the lack of robust biomarkers and paucity of available tissue to assess response. Strategies that incorporate pre and post-treatment endoscopic ultrasound biopsies must be considered to help develop techniques required to run robust clinical trials. Immune cell profiling could be employed to stratify subgroups of resected PDACs, potentially enabling individualized targeted immunotherapeutic strategies.
In PDAC the multitude of pathways and factors that determine progression remains the main obstacle in combating this aggressive disease. It is probable that to generate durable responses, in such a plastic disease as PDAC, carefully selected combination therapies will be required. Such strategies are likely to evolve to incorporate chemotherapeutics, immunogenics and therapies targeted against tumor stroma and signal transduction. The recent steady progression made in advanced PDAC with FOLFIRINOX and nab-paclitaxel will hopefully progress further with complementary therapeutics targeted against these different components of the tumor microenvironment.
Footnotes
Conflict-of-interest statement: We have no conflicts of interest to disclose.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: November 30, 2015
First decision: December 28, 2015
Article in press: February 16, 2016
P- Reviewer: Bilir C, Kang CM, Kleeff J, Nagahara H, Sperti C S- Editor: Qiu S L- Editor: A E- Editor: Lu YJ
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.McMillan DC. The systemic inflammation-based Glasgow Prognostic Score: a decade of experience in patients with cancer. Cancer Treat Rev. 2013;39:534–540. doi: 10.1016/j.ctrv.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 3.Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012;379:1591–1601. doi: 10.1016/S0140-6736(12)60209-8. [DOI] [PubMed] [Google Scholar]
- 4.Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011;377:31–41. doi: 10.1016/S0140-6736(10)62110-1. [DOI] [PubMed] [Google Scholar]
- 5.Rothwell PM, Price JF, Fowkes FG, Zanchetti A, Roncaglioni MC, Tognoni G, Lee R, Belch JF, Wilson M, Mehta Z, et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet. 2012;379:1602–1612. doi: 10.1016/S0140-6736(11)61720-0. [DOI] [PubMed] [Google Scholar]
- 6.Jamieson NB, Denley SM, Logue J, MacKenzie DJ, Foulis AK, Dickson EJ, Imrie CW, Carter R, McKay CJ, McMillan DC. A prospective comparison of the prognostic value of tumor- and patient-related factors in patients undergoing potentially curative surgery for pancreatic ductal adenocarcinoma. Ann Surg Oncol. 2011;18:2318–2328. doi: 10.1245/s10434-011-1560-3. [DOI] [PubMed] [Google Scholar]
- 7.Garcea G, Ladwa N, Neal CP, Metcalfe MS, Dennison AR, Berry DP. Preoperative neutrophil-to-lymphocyte ratio (NLR) is associated with reduced disease-free survival following curative resection of pancreatic adenocarcinoma. World J Surg. 2011;35:868–872. doi: 10.1007/s00268-011-0984-z. [DOI] [PubMed] [Google Scholar]
- 8.Stotz M, Gerger A, Eisner F, Szkandera J, Loibner H, Ress AL, Kornprat P, AlZoughbi W, Seggewies FS, Lackner C, et al. Increased neutrophil-lymphocyte ratio is a poor prognostic factor in patients with primary operable and inoperable pancreatic cancer. Br J Cancer. 2013;109:416–421. doi: 10.1038/bjc.2013.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldstein D, El-Maraghi RH, Hammel P, Heinemann V, Kunzmann V, Sastre J, Scheithauer W, Siena S, Tabernero J, Teixeira L, et al. nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: long-term survival from a phase III trial. J Natl Cancer Inst. 2015;107:pii: dju413. doi: 10.1093/jnci/dju413. [DOI] [PubMed] [Google Scholar]
- 10.Mehrabi A, Hafezi M, Arvin J, Esmaeilzadeh M, Garoussi C, Emami G, Kössler-Ebs J, Müller-Stich BP, Büchler MW, Hackert T, et al. A systematic review and meta-analysis of laparoscopic versus open distal pancreatectomy for benign and malignant lesions of the pancreas: it’s time to randomize. Surgery. 2015;157:45–55. doi: 10.1016/j.surg.2014.06.081. [DOI] [PubMed] [Google Scholar]
- 11.Al-Refaie WB, Parsons HM, Markin A, Abrams J, Habermann EB. Blood transfusion and cancer surgery outcomes: a continued reason for concern. Surgery. 2012;152:344–354. doi: 10.1016/j.surg.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 12.Adamina M, Steffen T, Tarantino I, Beutner U, Schmied BM, Warschkow R. Meta-analysis of the predictive value of C-reactive protein for infectious complications in abdominal surgery. Br J Surg. 2015;102:590–598. doi: 10.1002/bjs.9756. [DOI] [PubMed] [Google Scholar]
- 13.Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 14.Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernández-Porras I, Cañamero M, Rodriguez-Justo M, Serrano M, Barbacid M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell. 2011;19:728–739. doi: 10.1016/j.ccr.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee KE, Bar-Sagi D. Oncogenic KRas suppresses inflammation-associated senescence of pancreatic ductal cells. Cancer Cell. 2010;18:448–458. doi: 10.1016/j.ccr.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baumgart S, Ellenrieder V, Fernandez-Zapico ME. Oncogenic transcription factors: cornerstones of inflammation-linked pancreatic carcinogenesis. Gut. 2013;62:310–316. doi: 10.1136/gutjnl-2011-301008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finn OJ. Cancer immunology. N Engl J Med. 2008;358:2704–2715. doi: 10.1056/NEJMra072739. [DOI] [PubMed] [Google Scholar]
- 18.Hiraoka N, Yamazaki-Itoh R, Ino Y, Mizuguchi Y, Yamada T, Hirohashi S, Kanai Y. CXCL17 and ICAM2 are associated with a potential anti-tumor immune response in early intraepithelial stages of human pancreatic carcinogenesis. Gastroenterology. 2011;140:310–321. doi: 10.1053/j.gastro.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 19.Ochi A, Nguyen AH, Bedrosian AS, Mushlin HM, Zarbakhsh S, Barilla R, Zambirinis CP, Fallon NC, Rehman A, Pylayeva-Gupta Y, et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J Exp Med. 2012;209:1671–1687. doi: 10.1084/jem.20111706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 22.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamilton JA, Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol. 2013;34:81–89. doi: 10.1016/j.it.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, Mitchem JB, Plambeck-Suess SM, Worley LA, Goetz BD, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. 2013;19:3404–3415. doi: 10.1158/1078-0432.CCR-13-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, Erba E, Uboldi S, Zucchetti M, Pasqualini F, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23:249–262. doi: 10.1016/j.ccr.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 28.Sullivan RJ, Lorusso PM, Flaherty KT. The intersection of immune-directed and molecularly targeted therapy in advanced melanoma: where we have been, are, and will be. Clin Cancer Res. 2013;19:5283–5291. doi: 10.1158/1078-0432.CCR-13-2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I, Rosenberg SA. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33:828–833. doi: 10.1097/CJI.0b013e3181eec14c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. 2013;110:20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stromnes IM, DelGiorno KE, Greenberg PD, Hingorani SR. Stromal reengineering to treat pancreas cancer. Carcinogenesis. 2014;35:1451–1460. doi: 10.1093/carcin/bgu115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Choi M. Immune Therapy in Pancreatic Cancer: Now and the Future? Rev Recent Clin Trials. 2015;10:317–325. doi: 10.2174/1574887110666150916142537. [DOI] [PubMed] [Google Scholar]
- 33.Middleton G, Silcocks P, Cox T, Valle J, Wadsley J, Propper D, Coxon F, Ross P, Madhusudan S, Roques T, et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): an open-label, randomised, phase 3 trial. Lancet Oncol. 2014;15:829–840. doi: 10.1016/S1470-2045(14)70236-0. [DOI] [PubMed] [Google Scholar]
- 34.Neesse A, Algül H, Tuveson DA, Gress TM. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut. 2015;64:1476–1484. doi: 10.1136/gutjnl-2015-309304. [DOI] [PubMed] [Google Scholar]
- 35.Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM, et al. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60:861–868. doi: 10.1136/gut.2010.226092. [DOI] [PubMed] [Google Scholar]
- 36.Infante JR, Matsubayashi H, Sato N, Tonascia J, Klein AP, Riall TA, Yeo C, Iacobuzio-Donahue C, Goggins M. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J Clin Oncol. 2007;25:319–325. doi: 10.1200/JCO.2006.07.8824. [DOI] [PubMed] [Google Scholar]
- 37.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frese KK, Neesse A, Cook N, Bapiro TE, Lolkema MP, Jodrell DI, Tuveson DA. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012;2:260–269. doi: 10.1158/2159-8290.CD-11-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, Marsh R, Wallace J, Kozloff M, Rajdev L, et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J Clin Oncol. 2015;33:4284–4292. doi: 10.1200/JCO.2015.62.8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112–120. doi: 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6:2969–2972. [PubMed] [Google Scholar]
- 46.Collins MA, Bednar F, Zhang Y, Brisset JC, Galbán S, Galbán CJ, Rakshit S, Flannagan KS, Adsay NV, Pasca di Magliano M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639–653. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Balkwill FR. The chemokine system and cancer. J Pathol. 2012;226:148–157. doi: 10.1002/path.3029. [DOI] [PubMed] [Google Scholar]
- 48.Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, Manova-Todorova K, Leversha M, Hogg N, Seshan VE, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150:165–178. doi: 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jamieson T, Clarke M, Steele CW, Samuel MS, Neumann J, Jung A, Huels D, Olson MF, Das S, Nibbs RJ, et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J Clin Invest. 2012;122:3127–3144. doi: 10.1172/JCI61067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ijichi H, Chytil A, Gorska AE, Aakre ME, Bierie B, Tada M, Mohri D, Miyabayashi K, Asaoka Y, Maeda S, et al. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J Clin Invest. 2011;121:4106–4117. doi: 10.1172/JCI42754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ochi A, Graffeo CS, Zambirinis CP, Rehman A, Hackman M, Fallon N, Barilla RM, Henning JR, Jamal M, Rao R, et al. Toll-like receptor 7 regulates pancreatic carcinogenesis in mice and humans. J Clin Invest. 2012;122:4118–4129. doi: 10.1172/JCI63606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fukuda A, Wang SC, Morris JP, Folias AE, Liou A, Kim GE, Akira S, Boucher KM, Firpo MA, Mulvihill SJ, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell. 2011;19:441–455. doi: 10.1016/j.ccr.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Klöppel G, Yoshimura A, Reindl W, Sipos B, Akira S, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–469. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 54.Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA, Bardeesy N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011;71:5020–5029. doi: 10.1158/0008-5472.CAN-11-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Denley SM, Jamieson NB, McCall P, Oien KA, Morton JP, Carter CR, Edwards J, McKay CJ. Activation of the IL-6R/Jak/stat pathway is associated with a poor outcome in resected pancreatic ductal adenocarcinoma. J Gastrointest Surg. 2013;17:887–898. doi: 10.1007/s11605-013-2168-7. [DOI] [PubMed] [Google Scholar]
- 56.Hurwitz HI, Uppal N, Wagner SA, Bendell JC, Beck JT, Wade SM, Nemunaitis JJ, Stella PJ, Pipas JM, Wainberg ZA, et al. Randomized, Double-Blind, Phase II Study of Ruxolitinib or Placebo in Combination With Capecitabine in Patients With Metastatic Pancreatic Cancer for Whom Therapy With Gemcitabine Has Failed. J Clin Oncol. 2015;33:4039–4047. doi: 10.1200/JCO.2015.61.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, Li J, Peng B, Fleming JB, Wang H, et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105–120. doi: 10.1016/j.ccr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]