

Abstract
Manduca sexta is a lepidopteran model widely used to study insect physiological processes, including innate immunity. In this study, we explored the proteomes of cell-free hemolymph from larvae injected with a sterile buffer (C for control) or a mixture of bacteria (I for induced). Of the 654 proteins identified, 70 showed 1.67 to >200-fold abundance increases after the immune challenge; 51 decreased to 0–60% of the control levels. While there was no strong parallel between plasma protein levels and their transcript levels in hemocytes or fat body, the mRNA level changes (i.e. I/C ratios of normalized read numbers) in the tissues concurred with their protein level changes (i.e. I/C ratios of normalized spectral counts) with correlation coefficients of 0.44 and 0.57, respectively. Better correlations support that fat body contributes a more significant portion of the plasma proteins involved in various aspects of innate immunity. Consistently, ratios of mRNA and protein levels were better correlated for immunity-related proteins than unrelated ones. There is a set of proteins whose apparent molecular masses differ considerably from the calculated Mr's, suggestive of posttranslational modifications. In addition, some low Mr proteins were detected in the range of 80 to >300 kDa on a reducing SDS-polyacrylamide gel, indicating the existence of high Mr covalent complexes. We identified 30 serine proteases and their homologs, 11 of which are known members of an extracellular immune signaling network. Along with our quantitative transcriptome data, the protein identification, inducibility, and association provide leads toward a focused exploration of humoral immunity in M. sexta.
Cell-free hemolymph (i.e. plasma) of insects serves as a medium that bathes tissues and cells, stores and transfers metabolites, and allows occurrence of physiological processes. Plasma protein concentrations in various insects range from 10 to 100 mg/ml, which fluctuate during development (1). Different groups of hemolymph proteins include hexamerins acting as amino acid sources for metamorphosis, lipophorins for lipid transport, vitellogenins for embryo development, enzymes (e.g. esterases, lipases) for lipid hydrolysis, cytokines for intercellular communications, peptide hormones for endocrine regulation, and carriers of lipid hormones. Additionally, a substantial body of literature is available on proteins involved in immune responses (2, 3). Hemolymph is also a battleground wherein plasma proteins and hemocytes attack invading organisms such as viruses, bacteria, fungi, and parasites (4, 5). Some proteins recognize pathogens and propagate the signals of wounding and microbe invasion, others either act as effectors to stop bleeding, kill the pathogens, or modulate potency and duration of the defense reaction (6–10). Fat body, analogous to vertebrate adipose tissues and liver, is the major source of insect plasma proteins. Several studies have described compositions of insect hemolymph proteomes of the fruit fly, honeybee, mosquito, and silkworm (11–15), but little is known about proteome–transcriptome correlations, posttranslational modifications, or protein complex formation during immune responses.
We have been studying the innate immune system of a biochemical model insect Manduca sexta, particularly serine proteases (SPs), serine protease homologs (SPHs), and serpins in larval hemolymph (2). Analogous to blood coagulation and complement activation in mammals, rapid responses around wounded tissues or invading pathogens are mediated by a plasma SP cascade in insects. Accumulating evidence indicates that pattern recognition receptors (PRRs), SPHs, serpins, phenoloxidases (POs), and antimicrobial peptides (AMPs) may form macromolecular complexes during an immune reaction (16–19). Gene sequences and expression profiles of these proteins are now available as a result of the M. sexta genome project and RNA-Seq analyses (6–8, 10). Immuno-transcriptome analyses (20–22) have elucidated sequences and levels of mRNAs that encode defense proteins in M. sexta fat body and hemocytes that are analogous to certain human leukocytes. Furusawa et al. (23) identified 58 nonredundant proteins in M. sexta larval plasma using one- and two-dimensional electrophoresis. We recently published an analysis of the plasma peptidome and identified 138 peptides (arbitrarily defined as Mr <25 kDa to include all AMPs) as well as 130 proteins that remained soluble after 50% acetonitrile precipitation (24). Here, we describe the identification of larger hemolymph proteins, their differential expression after a bacteria challenge, and correlations of induced changes between plasma proteins and the corresponding mRNAs in fat body and hemocytes. We also present evidence for immune-response-related posttranslational modifications and protein–protein interactions based on discrepancies between theoretical protein Mr's and mobility on an SDS-polyacrylamide gel.
EXPERIMENTAL PROCEDURES
Experimental Design and Statistical Rationale
Control (C) and induced (I) plasma samples were collected from M. sexta larvae injected with buffer or mixture of bacteria, respectively (24). The samples including biological replicates (n = 3, three larvae per replicate) were studied using a gel-LC approach (25) (Fig. 1). Proteins in the gel slices were identified by searching MS/MS data against a sequence database of M. sexta proteins. Their levels were quantified using normalized spectral counts (NSCs) and compared by Student's t test to reveal significant differences (p < 0.05) between the I and C. Correlations of plasma protein levels and corresponding mRNA levels in fat body (F) and hemocytes (H) were analyzed by ordinary least squares regression using NSCs and normalized RNA-Seq read numbers (NRNs) (22). Possible correlations between mRNA and protein level changes (i.e. I/C) were examined using log2(NRNI/NRNC) and corresponding log2(NSCI/NSCC) values. NSC distributions of proteins in different gel slices and their theoretical Mr values were examined to identify major posttranslational modifications in certain hemolymph proteins.
Fig. 1.
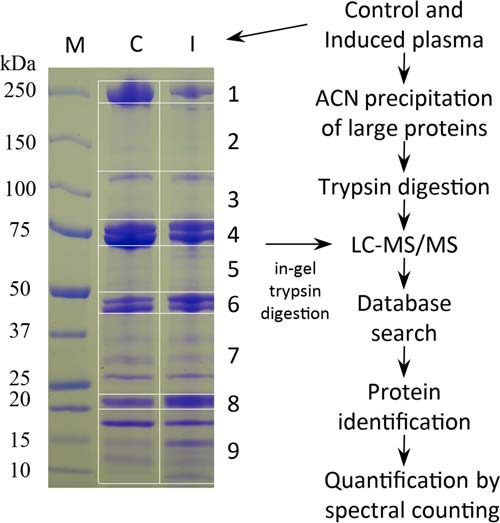
Schematic overview of sample preparation and data analysis. The control and induced plasma samples were separated by SDS-PAGE on a gradient gel that was later cut into nine pieces. Proteins in the gel slices were treated with trypsin and analyzed by nanoLC-MS/MS. As reported previously (24), the same plasma samples were treated with 50% ACN and proteins in the supernatants were analyzed. Protein identification and quantification were performed as described in the Materials and Methods.
Preparation of Cell-Free Hemolymph from Buffer- and Bacteria-Injected M. sexta Larvae
The same plasma samples used in our previous work (24) were analyzed using the gel-LC approach. Briefly, each of day 1, fifth instar larvae was injected with a mixture of Escherichia coli, Micrococcus luteus, and insoluble β-1,3-glucan from Alcaligenes faecalis or with sterile phosphate buffered saline as a negative control. At 24 h after the injection, prolegs of the insects were cut and hemolymph was collected using tubes containing a crystal of 1-phenyl-2-thiourea and 1 mm p-aminobenzamidine. After centrifugation, equal volumes of the plasma samples from three immune challenged insects were pooled as induced plasma-1 (IP1). Similarly, control plasma-1 (CP1) was prepared from three larvae injected with the saline. This experiment was repeated twice on different days to obtain CP2, CP3, IP2, and IP3.
SDS-PAGE Separation of Plasma Proteins, In-Gel Trypsinolysis, and Sample Preparation for MS Analysis
The pooled plasma samples (40 μl each) were separately mixed with 6×SDS sample buffer (8 μl) and heated at 95 °C for 5 min. Sixty μg of protein from each sample were loaded onto a 4–15% precast gradient polyacrylamide gel (Bio-Rad, Hercules, CA) and run at constant current of 30 mA for 45 min. After brief staining with Coomassie Brilliant Blue R-250, the gel was destained in 30% methanol and 10% acetic acid. Each of the six lanes was cut into nine gel slices (Fig. 1), and the 54 slices were individually cut into small pieces prior to extensive destaining by 50% acetonitrile (ACN)/50 mm NH4HCO3, pH 8.0. The gel pieces were then dehydrated with 100% ACN, dried briefly, and rehydrated with 10 mm tris (2-carboxyethyl) phosphine in 50 mm NH4HCO3 for 1 h at room temperature. The reducing solution was replaced with 55 mm iodoacetamide in 50 mm NH4HCO3 to alkylate Cys residues for 1 h at 25 °C in the dark. After further rinsing with 50 mm NH4HCO3, the gel pieces were dehydrated with ACN and then infiltrated with sequencing-grade trypsin (8 μg/ml) in 50 mm NH4HCO3. After digestion for 16 h at 37 °C, trypsinolytic peptides were extracted from the gel pieces with 1% trifluoroacetic acid for subsequent LC-MS/MS analysis.
LC-MS/MS Analysis
The 54 control and induced samples were individually analyzed on an LTQ-OrbitrapXL mass spectrometer (Thermo Fisher Scientific, Grand Island, NY) coupled to a New Objective PV-550 nanoelectrospray ion source and an Eksigent NanoLC-2D chromatography system (24). Briefly, peptides were separated using a 5–40% acetonitrile gradient in 0.1% formic acid performed over 40 min at a flow rate of 300 nl/min. During each 1 s full-range FT-MS scan, the six most intense ions were analyzed via MS/MS in the linear ion trap mode. MS/MS spectra were collected using a trigger threshold of 8,000 counts and monoisotopic precursor selection. Parental ions were rejected for MS/MS if their charge states were not assignable, if they were previously identified as contaminants on blank gradient runs, or if they had already been twice selected for MS/MS. Three to six technical replicates were performed for each biological sample in the LC-MS/MS analyses.
Protein Database, Identification, and Validation
The MS/MS spectra were searched against a high-quality, nonredundant database (denoted here as Msexta_060614.fasta, see supplemental text file) for protein identification. The database consisted of immunity-related protein sequences (6–10) and complete protein sequences deduced from the rectified open reading frames and confirmed by homology and domain search (26). Centroided ion masses were extracted by extract_msn.exe utility from Bioworks (v3.3.1) for database searching with Mascot (v2.2.04, Matrix Science, Boston, MA). The following parameters were used in the search: no charge state deconvolution or deisotoping, trypsin digestion, maximum missed cleavage of 1, parental ion mass tolerance of 5.0 ppm, fragment ion mass tolerance of 0.60 Da, formylation or acetylation of protein N termini, Met oxidization, cyclization of glutamate to pyroglutamate, iodoacetamide or acrylamide derivatization of Cys as variable modifications. Scaffold (v4.2.0, Proteome Software Inc., Portland, OR) was used to validate MS/MS-based peptide or protein identifications. Protein probabilities were assigned by Protein Prophet algorithm (27), and identifications were accepted if they could be established at greater than 98.0% probability and contained at least two identified peptides (Table S6). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principle of parsimony. Reversed protein sequences of the same database were used as decoys, demonstrating a project-wide false discovery rate of 0.5%. Average values of the unique spectral counts (USCs) in three biological replicates for individual proteins were used to analyze distributions of proteins in different gel slices.
Function Prediction, Statistical Analysis, and Ratio Calculation
The accepted protein identifications were used as queries to search the insect RefSeq collection at National Center for Biotechnology Information NCBI; by BLASTP under default conditions to identify their homologs in other species in order to predict function. To study changes in protein levels, NSCs for individual proteins in the CP1 or IP samples were calculated by multiplying their observed SCs by the ratio of the average SC of all matched peptides in all the six samples to the total SC of all matched peptides in the samples. As an example, since the total spectral counts for CP1–3 and IP1–3 proteins in gel were 63,886, 66,646, 64,540, 69,891, 70,899 and 70,538, respectively, the SC value for a protein identified in the IPgel2 sample was normalized according to: NSCIP2, gel = SCIP2, gel × (63,886 + 66,646 + 64,540 + 69,891 + 70,899 + 70,538)/(6 × 70,899). Peptidomic data from our previous study of the ACN-treated samples (24) were similarly reanalyzed using the improved database (Msexta_060614.fasta). NSCs were analyzed by Student's t test to reveal statistically significant differences (p < 0.05) between induced and control samples. Changes in protein levels (i.e. I/Cs) were calculated by dividing the average NSC values from the induced samples by the corresponding average NSC values from the control samples. In calculating I/C values, when NSCs in the denominator were all zero, their average (0) was replaced with 0.5 instead of 1 (24). Proteins were regarded as up-regulated if I/C ≥ 1.67, whereas proteins with I/C ≤ 0.60 as down-regulated.
Correlation of Protein and mRNA Levels as Well as Their Changes after the Immune Challenge
Amino acid sequences of the identified proteins were used to search the CIFH09 database (http://darwin.biochem.okstate.edu/blast/blast_links.html) by TBLASTN. CIFH09 is a collection of cDNA contigs assembled form RNA-Seq reads of control (C) and induced (I) fat body (F) and hemocytes (H) (21). Raw numbers of CF, CH, IF, and IH reads assembled into the cDNA contig with the highest sequence identity to a protein query were converted to normalized read numbers (NRNs) (22). To test if there is a direct correlation between mRNA and protein levels, the log2NRN and corresponding average log2NSC values were analyzed by ordinary least squares regression in the following pairs: CF-CP, CH-CP, IF-IP, and IH-IP (P for plasma). To test whether a correlation exists between mRNA and protein level changes (i.e. I/C), log2(NRNIF/NRNCF) and log2(NRNIH/NRNCH) and corresponding log2(NSCIP/NSCCP) values were examined similarly in terms of F-Pgel, F-PACN, H-Pgel, and H-PACN, respectively.
RESULTS
Proteomics Workflow and Protein Identification
To identify M. sexta plasma proteins, especially those involved in innate immunity, we collected hemolymph samples from the larvae injected with buffer or bacteria and analyzed them by the gel-LC-MS/MS approach (Fig. 1) (25, 28). A high-quality, nonredundant protein database (Msexta_060614.fasta) was searched with the MS/MS data for protein identification and spectral counting (Table S1). In the dataset, 217,791, or 13.7%, of the 1,594,513 spectra matched those of trypsinolytic peptides. The matching spectra corresponded to 654 independent proteins in the database, and 150 were known or predicted to be defense-related based on homology (Table S2). This represents a dramatic increase in coverage of the M. sexta hemolymph proteome, reflecting quality of the protein database, sensitivity increase due to prefractionation, as well as our selection of day 1, fifth instar larvae for injection. Insects at this stage are large enough to provide adequate hemolymph but not too much storage proteins that overwhelm the detection of less abundant proteins.
In this study, we identified a total of 401 proteins with a putative signal peptide, 222 of which were also detected in the peptidome analysis (Table S2) (24). Among the 401, 138 may be involved in defense, including PRRs, SPs (including hemolymph proteases or HPs), SPHs, serpins, and AMPs (6–10); the other 263 may participate in lipid/ion transport and other extracellular functions, based on sequence homology. To our surprise, 253 of the 654 proteins do not contain signal peptide for secretion. Some of them may be related to immunity, including galectin-3; serpin-2; serpin-17B; prophenoloxidase-1 (proPO1); proPO2; plasmatocyte spreading peptide (PSP) binding proteins 1, 6, 7; Eiger; Smt3; and Uev1A. While Eiger possesses a transmembrane region, the other cytosolic proteins (e.g. PSP binding proteins and proPOs) may enter plasma via nonconventional secretion pathway or cell rupture (29–31). Consistent with the latter possibility, we detected 44 ribosomal proteins, 7 translation factors, 4 histones, 8 proteasome subunits, and others. Noting that 90% of these “contaminants” had much lower normalized spectral counts (NSCs) in induced plasma (IP) than control plasma (CP) samples, decreased hemocyte lysis after immune challenge may be responsible for the bias.
Quantitative Analysis and Data Comparison of Gel-Fractionated and ACN-Treated Samples
The pairwise Pearson coefficients of the 654 proteins demonstrated excellent data consistency within the control (C, 0.97–0.99 for gel; 0.94–0.96 for ACN) or induced (I, 0.99 for gel; 0.94–0.96 for ACN) group (Table S3). The C-I coefficients were low (0.55–0.65) for ACN (24), indicating that levels of most ACN-stable proteins differ dramatically between the CP and IP samples. In contrast, the C-I coefficients were much higher (0.93–0.95) for gel, suggesting that most proteins identified in the gel did not change considerably in their abundances after the immune challenge.
We then examined the total number of proteins identified to compare the methods of sample preparation. A total of 654 proteins were identified in the gel-fractionated samples, over twice that (318) in the ACN-treated samples (i.e. supernatants of the hemolymph pools after solvent precipitation of larger proteins) (24) (Fig. 2A), confirming that ACN precipitated a large number of proteins. The smallest peptide was 6 kDa (cecropin-7) in ACN-treated and 7 kDa (cecropin-6) in gel-fractionated. The largest protein (Dumpy, 1,935 kDa) identified in one of the three ACN-treated samples was larger than that (apolipophorin-like, 475 kDa) in the gel-fractionated samples. In terms of numbers of proteins in different size ranges, the two methods detected 17 (gel) and 18 (ACN) below 10 kDa, 13 of which were identical (Fig. 2B). Most proteins fell into the Mr range of 10 to 80 kDa (239 or 75% in ACN; 553 or 85% in gel). In the range of >80 kDa, 60 and 84 proteins were identified in ACN and gel, respectively. The large proteins identified in ACN but not in gel were: C-type lectins X3, X5, and X6; Dumpy; papilin; SP56; PI3KC2A; ROS-like Tyr kinase; and seven others. Their low NSCs suggested that concentration of the ACN supernatants allowed their meagre detection (22). In terms of the ratios of NSCACN and NSCgel per protein, there was a monotonic decrease from 14.4 to 0.1 (Fig. 2C) as Mr increased from <10 to >200 kDa. This indicates that small proteins in the ACN-treated samples are better represented than large ones.
Fig. 2.
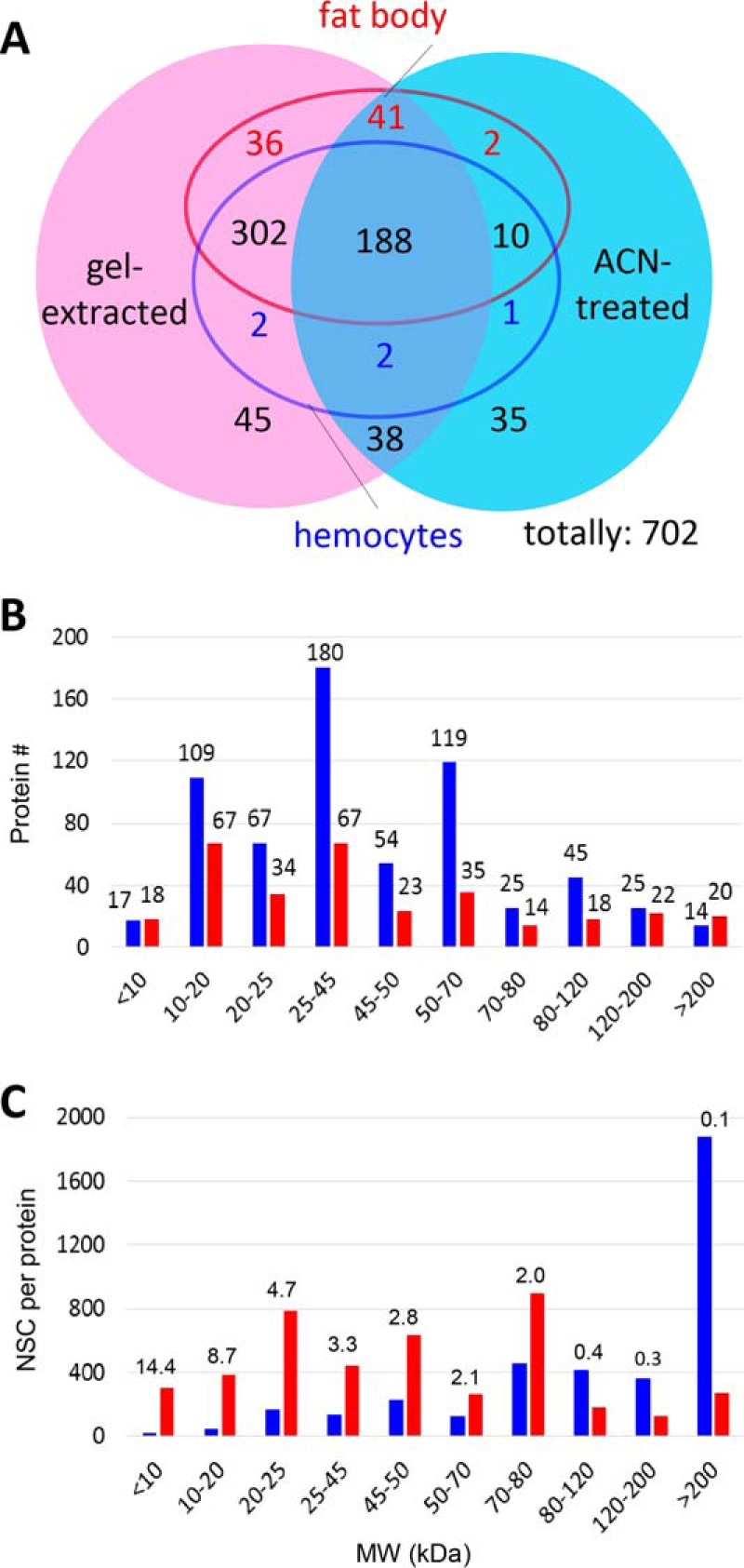
Comparison of proteins identified by different methods (A) and in various Mr ranges (B, C). (A) Among the 654 proteins identified in the gel-fractionated samples (shaded pink), 502 are expressed in hemocytes (blue circle) and 575 in fat body (red circle). For the 319 proteins in the ACN-treated samples (shaded cyan), 203 are expressed in hemocytes and 243 in fat body. (B, C) Size distribution of proteins identified in the gel slices (blue bars) and ACN-treated samples (red bars) with the numbers (B) or average NSCs (C) of proteins shown as bars. Protein numbers (#gel and #ACN) and ratios of the NSCs per protein (NSCACN/NSCgel) in various size ranges are listed on top of the bars in (B) and (C), respectively.
Up- and Down-Regulated Proteins
The levels of 77 proteins (including the gel and ACN data) increased significantly after the immune challenge (Table I). As anticipated, a majority (41, or 53%) of them are related to immune responses. These include eight PRRs: hemolin, immulectin-1, immulectin-12, peptidoglycan recognition protein-3 (PGRP3), PGRP5, Hdd1, hemicentin-2, and Reeler, whose mRNA levels were also up-regulated after the challenge. We detected seven SP-related proteins (HP5, HP17a, HP20, proPO-activating protease-2 (PAP2), PAP3, scolexin A, SPH4) and six protease inhibitors (serpin-2, 5, 11, protease inhibitor-6 (PI6), PI-like protein, dipetalogastin-like Kazal-type PI), which may mediate or modulate the extracellular immune signal transduction. The most dramatic increases occurred in the category of AMPs, including diapausins, attacins, lebocins, cecropins, gloverin, gallerimycin-1, and lysozyme-1. Most of the proteins increased >10-fold, especially the four attacins, which showed >200-fold changes in the ACN and gel samples. This is consistent with the fact that these effectors of the innate immune system are highly up-regulated at the transcription level after immune challenge. Other highly induced proteins include heat shock protein (HSP) 25.4's, lipases, esterases, peptidases, lipid-binding proteins, and some hypothetical proteins. While some of them mediate metabolic changes in response to the stress condition, functions for the others in immunity remain to be determined. In other words, the quantitative proteomic analysis provided significant new leads for investigation of insect immune responses.
Table I. A list of 77 proteins up-regulated in gel-fractionated and ACN-treated samples after immune challenge.
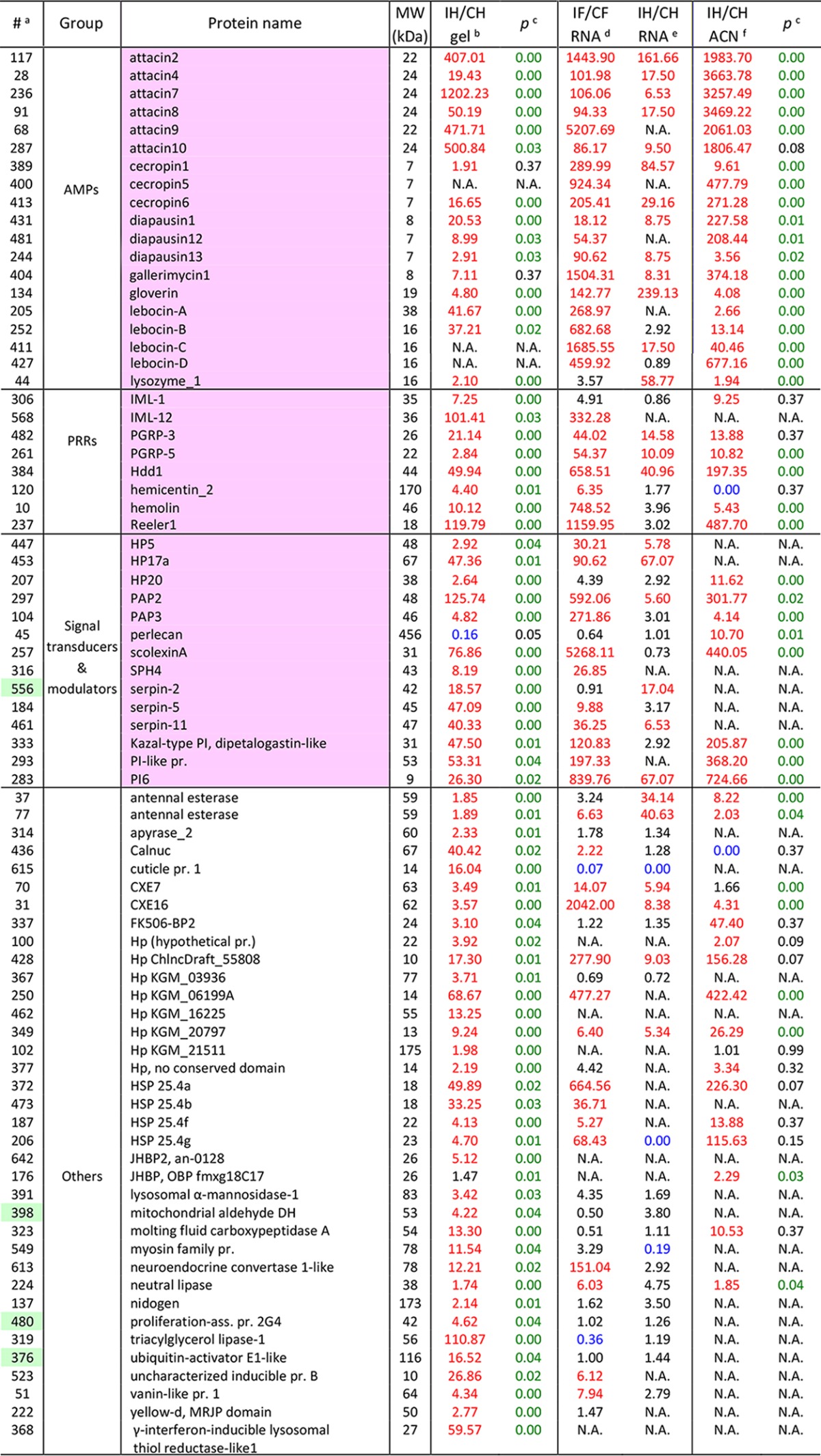
aProtein identification numbers with the intracellular ones shaded green.
bRatios of average normalized spectral counts (NSCs) of proteins in the induced (I) and control (C) samples separated by SDS-PAGE (I/C >1.67: red; I/C <0.60: blue).
cResults of the Student's t-tests conducted using NSCs of proteins in the I and C samples (p < 0.05: green).
d,eRelative abundances (RAs) or adjusted read numbers (ARNs) of mRNAs in fat body (IF/CF) and hemocytes (IH/CH) (RA or ARN >5, red; RA or ARN <0.2: blue; N.A.: not available) (21).
fRecalculated I/C ratios of average NSCs for proteins identified in the ACN-treated samples (I/C >1.67: red; I/C <0.60: blue). Detailed information of these proteins can be found in Table S2.
In contrast to the up-regulated proteins described above, the levels of 78 proteins significantly decreased after the challenge (Table S4). Eight proteins are related to defense responses, including diapausin-5, immulectin-9, extracellular leucine-rich repeat protein-8 (ELRRP8), peroxidasin, HP6, HP8, HP21, and metalloprotease inhibitor. The decreases in HP6, HP8, and HP21 (range: 0.69–0.94, average: 0.85) were small based on the gel data, while the other five reduced substantially (range: 0.00–0.56, average: 0.18). Most (35) of the 47 gel proteins were undetected in the ACN-treated samples, whereas 36 of the 37 ACN proteins were identified in the gel-fractionated samples. This anomaly may be due to their property differences and operational variances. The gel samples were obtained after electrophoresis, staining, destaining, and in-gel trypsin digestion; the simple ACN treatment had less operational variations but protein size, stability, concentration, and other properties affect their precipitation by ACN, especially those in the high Mr ranges (Fig. 2C) (24).
Proteins versus mRNAs
We compared the proteome results from gel and ACN to those obtained by transcriptome analyses of fat body and hemocytes. For this comparison, I/C ratios of the ACN data (24) were recalculated using the same method for the gel data. Based on RNA-Seq data, 491 of the 654 proteins in gel and 198 of the 317 in ACN were expressed in both fat body and hemocytes (Fig. 2A and Table S2). Adding those expressed in only one tissue, the 571 proteins in gel and 244 in ACN and their respective cDNA contigs were identified for retrieving read numbers from CF, IF, CH, and IH (24). Using normalized read numbers (NRNs) from the RNA-Seq analysis (22) and corresponding NSCs, we first compared the fat body mRNA and plasma protein levels after the immune challenge. As shown in Fig. 3A (upper panel), there was a correlation between CF and CPgel (0.43) and between IF and IPgel (0.41). The positive correlation remained when ACN data were analyzed, but the coefficients reduced to 0.41 (CF- CPACN) and 0.26 (IF-IPACN) (Fig. S1A, upper panel). The correlations were much lower between hemocyte transcripts and plasma proteins. The CH-CPgel, IH-IPgel, CH-CPACN, and IH-IPACN coefficients were 0.05, -0.19, 0.00, and -0.25, respectively (Fig. 3A and Fig. S1A, lower panel). The negative or low positive correlations between the transcript and protein levels seemed to result from relatively small contribution of hemocytes to plasma proteomes, as shown in the previous study (24). In contrast, due to the sheer volume of fat body and its high levels of protein synthesis, moderately positive correlations exist between fat body transcript and plasma protein abundances.
Fig. 3.

Correlation of the mRNA and protein abundances (A) and their changes (B) for gel-fractionated samples. (A) Levels. Upper panels: normalized read numbers (NRNs, y-axis) of specific transcripts in the control (CF, left) and induced (IF, right) fat body (red dot) and their corresponding normalized spectral counts (NSCs, x-axis) in the control (CP, left) and induced (IP, right) plasma are plotted in log2 scale, representing the relative mRNA and protein levels, respectively. Lower panels are the same as upper panels, except that the NRNs of mRNA in the control (CH, left) and induced (IH, right) hemocytes (blue dot) are shown. (B) Ratios. The mRNA and protein level changes are represented by NRNI/NRNC of fat body (red dot, upper panel) or hemocytes ((blue dot, lower panel) and NSCIP/NSCCP of plasma in log2 scale, respectively. The correlation coefficients for all the comparisons are indicated in the panels.
We then tested whether there was a stronger correlation between the challenge-induced changes (i.e. I/C ratios) in mRNA versus protein levels. The scatter plots (Fig. 3B and Fig. S1B) clearly demonstrated a positive correlation (0.44–0.69) in both samples: Most gel and ACN proteins showed the same tendency of changes as their mRNAs did, fat body mRNA in particular. Only a small number of proteins showed mRNA level increases but protein level decreases or vice versa. Similar to the relationships between protein and mRNA levels, the protein level changes showed a higher correlation to immune-induced mRNA level changes in fat body (gel: 0.57; ACN: 0.69) than in hemocytes (gel: 0.44; ACN: 0.52). These data support the long-standing notion that fat body makes greater contribution to the pool of plasma proteins than do hemocytes. Moreover, this is consistent with the functional disparity between fat body and hemocytes in humoral and cellular immunity (2–4).
Since the above correlations to certain degree reflect gene transcription and translation in fat body and hemocytes between the control and induced samples, we postulated that induced changes in levels of immunity-related mRNA versus defense proteins would be more tightly correlated than induced changes in levels of immunity-unrelated mRNA versus nondefense proteins. That is, the correlation of changes in immune-related mRNAs versus proteins would improve upon removal of nonimmune proteins and their mRNAs from the population. Similarly, we postulated that separating extracellular from intracellular proteins and their mRNAs would lead to similar segregation of correlation coefficients. The correlations for immunity-related genes did increase to 0.75 (versus 0.57 for all genes) for fat body, whereas a slight decrease was seen for hemocytes (from 0.44 to 0.41; Fig. 4A); those for immunity-unrelated genes decrease to 0.34 (fat body) and 0.30 (hemocytes). This is consistent with the first prediction. On the other hand, the correlations for extracellular proteins slightly increased from 0.57 to 0.63 (fat body) and from 0.44 to 0.46 (hemocytes) (Fig. 4B). Those for intracellular proteins dramatically decreased to 0.15 (fat body) and 0.21 (hemocytes). The smaller increase (0.63 versus 0.75) is consistent with the fact that extracellular proteins include not only immunity-related (e.g. PRRs, SPs, serpins, AMPs) but also immunity-unrelated (e.g. lipophorins, storage proteins). While some PRRs and AMPs showed large increases in mRNA and protein levels, their good correlation is partly masked by those unrelated (i.e. noises). In contrast, a few intracellular, immunity-related proteins were detected in the plasma samples (Table S2). Our previous studies on M. sexta immune signal transducers showed their gene expression changed less dramatically after the challenge (9, 22). The weak correlation is largely covered by the noises of other proteins (e.g. ribosomal proteins).
Fig. 4.

Comparison of the mRNA level changes in fat body (red dot, upper) or hemocytes (blue dot, lower) and their protein level changes in the gel samples. (A) immunity-related and -unrelated; (B) extra- and intracellular proteins. See Fig. 3 for the definitions of x- and y-axes.
Predicted versus Observed Protein Sizes
In theory, proteins should migrate to positions corresponding to their calculated Mr's after SDS-PAGE and major deviations often suggest posttranslational modifications. By comparing the calculated Mr's and size ranges of the gel slices in which the proteins were detected, we noticed marked discrepancies—some proteins are detected a few slices away from the expected locations (Table S5). This can be an artifact since protein levels have a major impact on their apparent distributions in gel and hypothetical Mr is not a perfect predictor of electrophoretic migration (Fig. S2). Ten highly abundant proteins (ΣUSC: 6000–500) were detected in nearly all slices, suggestive of diffusion among gel fractions during electrophoresis or staining. Thirty-nine abundant proteins (ΣUSC: 500–150) were mostly located in 5–7 gel slices. A majority of the 105 (ΣUSC: 150–50), 212 (ΣUSC: 50–15), and 289 (ΣUSC: 15–0) proteins were identified in 3–4, 1–2, and 1 gel slice, respectively. Therefore, we reexamined the protein distribution patterns in gel slices and found that abnormal migration of 130 proteins still cannot be explained by diffusion.
Proteolytic activation of SP, SPH, and other protein precursors and its regulation by serpins account for some of the discrepancies, which actually represent the immune signaling, modulation, and execution mechanisms in insect hemolymph (2, 32). Due to their relatively low abundances, the narrow spreads in the gel in many cases reflect proteolytic cleavage or serpin-protease complex formation rather than diffusion. We identified 22 SPs (HP1a, 1b, 2, 5, 6, 8, 9, 14a, 15, 16a, 17a, 19, 20, 21, 22, 25, GP57, GP59, SP34, PAP1–3), 8 SPHs (SPH1b, 2–4, 33, 101, Scolexins A&B), and 17 serpins (1A, 1E, 1K, 1N, 1Z, 2–6, 9, 11–13, 15b, 17b, and 30) in the gel slices (Table S5). Most intact serpins are 45–50 kDa SP inhibitors, and their N-terminal most part (40–45 kDa) form covalent complexes with the catalytic domains (30–35 kDa) of their cognate SPs and then slowly dissociate from the latter (32). Likewise, each hemolymph protease may be detected as a precursor, N- and C-terminal fragments of the active SP, and a 75 kDa complex with the serpin fragment. Among five of the M. sexta serpin-1 splicing variants, serpin-1E forms SDS-stable complexes with HP1 and HP8 (33). Serpin-3 through -6 are also known to form complex with HPs, including PAP1, PAP3, HP1, HP6, HP8, and HP21 (34, 35). These findings are consistent with the detection of serpin-1E, -3, -4, and -12 in the range of 25–80 kDa in induced hemolymph (Table II). HP8 and PAP3 identified at 70–80 kDa may represent their catalytic domains in complex with serpin-6 (19). Since most serpin-HP complexes resided in gel slice-4 (70–80 kDa), we were unable to distinguish them or identify their components in this analysis. Serpin-9 displayed interesting changes: its level was higher in naïve plasma and became lower after the challenge (Table II). It existed as an intact protein, with some in a cleaved form and some spectral counts in the higher Mr slices, possibly representing a complex with an unknown HP. In addition, 18 other proteins may be cleaved by proteases, including Cys protease inhibitor (36), lebocin-A (37), cathepsin 26–29 kDa like-2 (38), furin, Eiger, and lacunin (Table S5).
Table II. Distribution of selected immunity-related proteins in different gel slices.
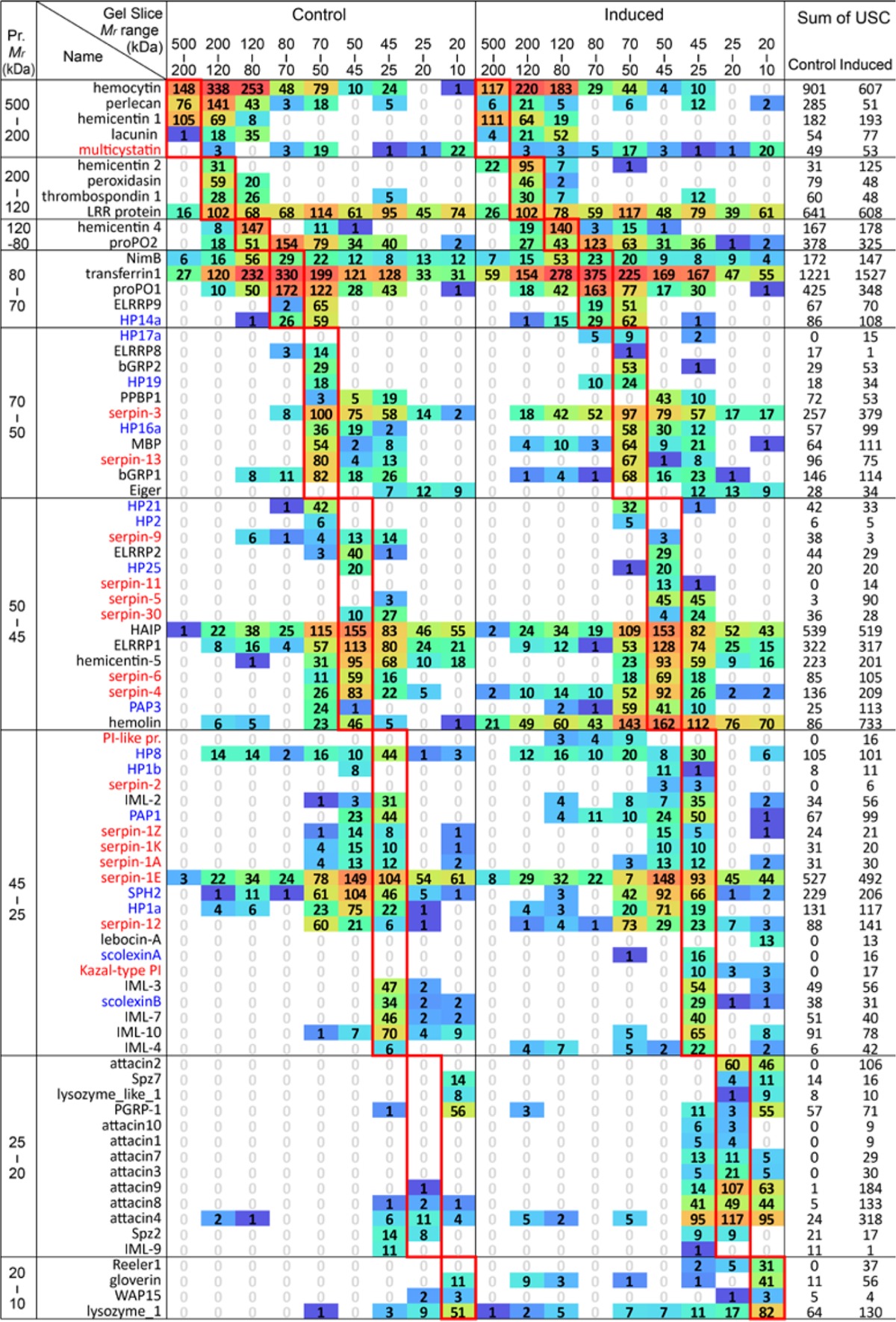
Proteins with unique spectral counts (USCs) in two or more gel slices (Table S5) are tabulated. So are the ones with USCs in one gel slice but their theoretical Mr values do not match the Mr range of that slice. Each cell, labeled with average USC of the three biological replicates, is colored with a gradient from blue to green and then to red based on its log2(USC+1). Red rectangles enclose the slices in which theoretical sizes of the proteins correspond; USCs outside are considered abnormal. SPs/SPHs (including HPs) and protease inhibitors (including serpins) are colored blue and red, respectively.
There is a special type of Mr discrepancies unrelated to diffusion, proteolysis, or formation of serpin-protease complexes. Low but considerable spectral counts in gel slices 1–3 (80 to >200 kDa) were detected for HP1a; HP8; HP14a; PAP1; PAP3; SPH2; SPH101; and serpin-1E, -3, -4, -9, and -12. Most of them are involved in the generation of active POs that produce quinone intermediates to crosslink nucleophiles in proteins and polysaccharides (5, 17). We have observed 54 other proteins migrate to gel slices 1–3 (80 to >400 kDa), which are a lot greater than their theoretical Mr's. Among them, β-1,3-glucan recognition protein-1; ELRRP1; hemocyte aggregation inhibitor protein; Hdd1; hemolin; immulectin-2, -4; LRR transmembrane protein-3; apolipophorins III; HSP25.4d; microbe-binding protein; Nimrod B; PGRP1; proPO1; proPO2; proPSP; ML2; attacin-4; gloverin; lysozyme-1; vanin-like-1; and PI-like protein (totally 22) are known or predicted to play roles in the humoral and cellular immune responses. Although it is unclear whether or not the other 32 proteins participate in defense, carboxylesterase (CXE)-7, CXE16, and antennal esterase-1 and -2 were up-regulated considerably. We interpret these 66 proteins as components of high Mr immune complexes crosslinked by covalent bonds, which allow them to sustain the reducing and denaturing conditions of SDS-PAGE.
DISCUSSION
A Dynamic Proteome of the Larval Hemolymph
Here, we used orthogonal fractionation by SDS-PAGE and LC-MS/MS to perform a deep analysis of the plasma proteome in M. sexta larvae. By combining results from the gel-free approach (i.e. ACN precipitation) (24), we identified a total of 702 proteins (Fig. S2), some with known functions and others with functions predicted based on sequence homology. These findings provide an overview of larval hemolymph proteins in the lepidopteran insect. Among them, 171 (24%) are related to immunity, 77 up-regulated (I/C >1.67), and 430 (61%) extracellular. Additionally, hemocyte lysis seems to contribute 272 intracellular proteins to the plasma.
Hemolymph SPs and SPHs mediate extracellular signal transduction and pathogen killing (2). Understanding their functions and inhibitory regulation in M. sexta is useful for investigating similar systems in insect vectors of human diseases. However, these systems are formidably large based on genome information. For instance, there are 193 SP/SPH and 32 serpin genes in the M. sexta genome ((8) and Kanost et al., unpublished data). Identification of the 30 SPs (eight in the ACN supernatants), 8 SPHs, and 13 serpins substantially narrows the scope of our exploration to those detected in plasma. A focused research is expected to facilitate the elucidation and reconstitution of the SP-SPH system and its regulation by serpins in M. sexta.
Another proteome-level finding is that 272 (39%) of the 702 plasma proteins do not contain a signal peptide, and their abundances decreased considerably after the immune challenge. Since the CP and IP samples were prepared under the same conditions, this is unlikely to be a technical artifact. Rather, we propose three mechanisms to explain this phenomenon: 1) control and induced hemocytes attach to and are stabilized by tissues to different extents, 2) induced hemocytes as a whole or in a subpopulation are more resilient to centrifugal force that causes cell rupture, and 3) unconventional secretory mechanisms (29) assist the release of some cytosolic proteins, which may differ between the control and induced states. Based on the number of identified proteins and their abundances, we consider cell lysis to be an important source of plasma proteins, such as proPOs.
Differential Protein Expression in Response to the Immune Challenge
We observed 155 changes in protein expression associated with immune challenge, and posit that most, if not all, of these changes represent the Manduca defense response. Of the 77 up-regulated proteins (Table I), over half are known to be related to immunity. These proteins recognize microbial surface components, transduce or modulate extracellular signals (HPs, SPHs, serpins), or act as effectors (AMPs, PIs). We also detected two putative intracellular immune signal transducers (Smt3, Uev1A) (Table S2). Additionally, some of the up-regulated proteins (e.g. HSP25.4's, hypothetical proteins, lipid-binding proteins) may participate in defense in unknown ways. HSP25.4 isoforms a–c, f, and g were up-regulated 49.9, 33.3, 2.1, 4.1, and 4.7-fold, and their functions are worth exploring. HPs KGM_06199A and _06199B, each containing a von Willebrand factor type C domain stabilized by four disulfide bonds, may act as PIs or AMPs. Uncharacterized inducible proteins A and B, 59- and 62-residue peptides rich in certain residues (e.g. Gly), could also be novel AMPs. Others (e.g. apyrase-2, CXEs, esterases, lipases, neuroendocrine convertase-1, lysosomal carboxypeptidases) are probably responsible for changes in metabolism and protein processing after immune challenge.
Correlations of mRNA and Protein Abundances
Discrepancies between mRNA and protein levels have been well documented in mammalian systems (39, 40). Here, we demonstrated the same phenomenon in a systematic way in an insect for the first time. As shown in Fig. 3A and Fig. S1A, the correlation coefficients between plasma and fat body or hemocytes were 0.43CF-CP, gel, 0.41IF-IP, gel, 0.41CF-CP, ACN, 0.26IF-IP, ACN, 0.05CH-CP, gel, -0.19IH-IP, gel, 0.00CH-CP, ACN, and -0.25IH-IP, ACN. These results, supported by a well-established quantitative method with biological replicates, reinforce concerns regarding the overreliance of transcriptome or PCR data to interpret biological functions.
Despite the poor correlations in levels, we performed additional comparisons, such as mRNA and protein level changes (i.e. I/C), major versus minor contributors of plasma proteins (F–H), gel versus ACN data, immunity-related versus -unrelated proteins, and extra- versus intra-cellular proteins (Figs. 3B and 4 and Fig. S1B). The gel data (I/C) were more extensive than the ACN data, but the latter showed better parallel, likely due to strong induction of AMP genes (0.69ACN > 0.57gel for fat body; 0.52ACN > 0.44gel for hemocytes). Extracellular protein level changes were better correlated than intracellular ones (0.63 > 0.15 for fat body and 0.46 > 0.21 for hemocytes). Immunity-related protein level changes had the highest correlation coefficient (0.75F) with the corresponding mRNA level changes in fat body, much higher than those in hemocytes (0.41H) and immunity-unrelated (0.34F and 0.30H) (Fig. 4). Therefore, although direct correspondence of mRNA and protein levels was poor, choosing a set of process-related genes expressed in a major contributing tissue allowed a relatively good correlation uncovered based on the level changes.
Posttranslational Modification and Immune Complex Formation
While prefractionation of the protein mixtures by SDS-PAGE was performed primarily to enhance protein discoveries (41, 42), we also gained important insights into the changes in hemolymph proteome, one being posttranslational modifications and the other immune complex formation.
For 162 of the 654 proteins identified, we noted major discrepancies between their theoretical Mr values and their mobility of gel electrophoresis (Table II and Table S5). Most (130) of the inconsistencies remained after the effect of diffusion (Fig. S2) were considered. Glycosylation and proteolytic processing can be used to explain the Mr differences, which are neglected in most proteomic studies. Fortunately, proteolytic activation of the precursors of SPs (including HPs, PAPs), SPHs, POs, PSP, and spätzle-1 has been well documented in M. sexta (2, 16, 32, 43). It is also known that serpins and other protease inhibitors are involved in the control of immune SPs (19, 33–35, 44). While the current analysis lacks the resolution needed to reveal components of the serpin-protease complexes, it did provide consistent results and new leads (e.g. serpin-9 and -12) for future studies.
One important result of this study is the detection of high Mr immune complexes in the plasma proteome, which are stable in the presence of SDS and β-mercaptoethanol. By identifying 66 proteins (10–80 kDa) in gel slices 1–3 (80 to >200 kDa) (Table S5), we validated and extended the previous observations of such complexes (16, 19, 43, 45, 46). Nearly half (32) of them (e.g. PRRs, SPs, SPHs, proPOs, AMPs) may take part in immune responses. In contrast, only 171 or 24% of the 702 plasma proteins are related to immunity, indicating that defense proteins are more frequently captured in the high Mr complexes. Their coexistence with POs led us to propose that PO-catalyzed covalent crosslinking is responsible for their stability. Consistent with this working model, we have also seen similar components in >500 kDa complexes eluted from a gel filtration column (data not shown). It is possible that the complexes observed in the gel slices 1–3 were dissociated from higher Mr ones that are partially crosslinked since complete crosslinking may prevent them from entering the gel. Consequently, the nature and extent of these covalent modifications remain to be fully defined.
Supplementary Material
Acknowledgments
We thank Dr. Jack Dillwith for his critical comments on the manuscript.
Footnotes
Author contributions: H.J and S.H. designed the research; S.Z. and J.R. performed the experiment; Y.H., X.C., and S.Z. analyzed data; Y.H., S.Z., S.H., and H.J. wrote the paper.
* This work was supported by National Institutes of Health Grant GM58634 (to H. Jiang). Mass spectrometry analyses were performed in the DNA/Protein Resource Facility at Oklahoma State University, using resources supported by the NSF MRI and EPSCoR programs (DBI/0722494). This article was approved for publication by the Director of the Oklahoma Agricultural Experiment Station and supported in part under project OKL02450. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
The mass spectrometry data (PXD003267 and 10.6019/PXD003267) have been deposited in the PRIDE repository (http://www.ebi.ac.uk/pride) using tools provided by the ProteomeXchange Consortium.
1 The abbreviations used are:
- CP
- control plasma from larvae injected with buffer
- IP
- induced plasma from larvae injected with bacteria
- CF
- IF, CH, and IH, control (C) and induced (I) fat body (F) and hemocytes (H)
- NRN
- normalized cDNA read number
- AMP
- antimicrobial peptide
- CXE
- carboxylesterase
- Hp
- hypothetical protein
- HP
- hemolymph protease
- PAP
- proPO-activating protease
- PGRP
- peptidoglycan recognition protein
- PO and proPO
- phenoloxidase and its precursor
- PI
- protease inhibitor
- PRR
- pattern recognition receptors
- PSP
- plasmatocyte spreading peptide
- SP and SPH
- serine protease and its homolog.
REFERENCES
- 1.Kanost M. R. (2003) Hemolymph. Encyclopedia of Insects, Resh V. H., and Carde R., eds. Academic Press, New York, 505–508 [Google Scholar]
- 2.Jiang H., Vilcinskas A., and Kanost M. R. (2010) Immunity in lepidopteran insects. Adv. Exp. Med. Biol. 708, 181–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lemaitre B., and Hoffmann J. (2007) The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 25, 697–743 [DOI] [PubMed] [Google Scholar]
- 4.Strand M. R. (2008) The insect cellular immune response. Insect Sci. 15, 1–14 [Google Scholar]
- 5.Zhao P., Lu Z., Strand M. R., and Jiang H. (2011) Antiviral, anti-parasitic, and cytotoxic effects of 5,6-dihydroxyindole (DHI), a reactive compound generated by phenoloxidase during insect immune response. Insect Biochem. Mol. Biol. 41, 645–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang X., He Y., Cao X., Gunaratna R. T., Chen Y. R., Blissard G., Kanost M. R., and Jiang H. (2015) Phylogenetic analysis and expression profiling of the pattern recognition receptors: Insights into molecular recognition of invading pathogens in Manduca sexta. Insect Biochem. Mol. Biol. 62, 38–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rao X. J., Cao X., He Y., Hu Y., Zhang X., Chen Y. R., Blissard G., Kanost M. R., Yu X. Q., and Jiang H. (2015) Structural features, evolutionary relationships, and transcriptional regulation of C-type lectin-domain proteins in Manduca sexta. Insect Biochem. Mol. Biol. 62, 75–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao X., He Y., Hu Y., Zhang X., Wang Y., Zou Z., Chen Y., Blissard G. W., Kanost M. R., and Jiang H. (2015) Sequence conservation, phylogenetic relationships, and expression profiles of nondigestive serine proteases and serine protease homologs in Manduca sexta. Insect Biochem. Mol. Biol. 62, 51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao X., He Y., Hu Y., Wang Y., Chen Y. R., Bryant B., Clem R. J., Schwartz L. M., Blissard G., and Jiang H. (2015) The immune signaling pathways of Manduca sexta. Insect Biochem. Mol. Biol. 62, 64–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He Y., Cao X., Li K., Hu Y., Chen Y. R., Blissard G., Kanost M. R., and Jiang H. (2015) A genome-wide analysis of antimicrobial effector genes and their transcription patterns in Manduca sexta. Insect Biochem. Mol. Biol. 62, 23–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y., Dong Z., Wang D., Wu Y., Song Q., Gu P., Zhao P., and Xia Q. (2014) Proteomics of larval hemolymph in Bombyx mori reveals various nutrient-storage and immunity-related proteins. Amino Acids 46, 1021–1031 [DOI] [PubMed] [Google Scholar]
- 12.Woltedji D., Fang Y., Han B., Feng M., Li R., Lu X., and Li J. (2013) Proteome analysis of hemolymph changes during the larval to pupal development stages of honeybee workers (Apis mellifera ligustica). J. Proteome Res. 12, 5189–5198 [DOI] [PubMed] [Google Scholar]
- 13.Karlsson C., Korayem A. M., Scherfer C., Loseva O., Dushay M. S., and Theopold U. (2004) Proteomic analysis of the Drosophila larval hemolymph clot. J. Biol. Chem. 279, 52033–52041 [DOI] [PubMed] [Google Scholar]
- 14.Handke B., Poernbacher I., Goetze S., Ahrens C. H., Omasits U., Marty F., Simigdala N., Meyer I., Wollscheid B., Brunner E., Hafen E., and Lehner C. F. (2013) The hemolymph proteome of fed and starved Drosophila larvae. PLoS ONE 8, e67208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dwivedi S. B., Muthusamy B., Kumar P., Kim M. S., Nirujogi R. S., Getnet D., Ahiakonu P., De G., Nair B., Gowda H., Prasad T. S., Kumar N., Pandey A., and Okulate M. (2014) Brain proteomics of Anopheles gambiae. OMICS 18, 421–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu X. Q., Jiang H., Wang Y., and Kanost M. R. (2003) Nonproteolytic serine proteinase homologs are involved in prophenoloxidase activation in the tobacco hornworm, Manduca sexta. Insect Biochem. Mol. Biol. 33, 197–208 [DOI] [PubMed] [Google Scholar]
- 17.Nappi A. J., and Christensen B. M. (2005) Melanogenesis and associated cytotoxic reactions: Applications to insect innate immunity. Insect Biochem. Mol. Biol. 35, 443–459 [DOI] [PubMed] [Google Scholar]
- 18.Wang Y., and Jiang H. (2004) Prophenoloxidase (proPO) activation in Manduca sexta: an analysis of molecular interactions among proPO, proPO-activating proteinase-3, and a cofactor. Insect Biochem. Mol. Biol. 34, 731–742 [DOI] [PubMed] [Google Scholar]
- 19.Zou Z., and Jiang H. (2005) Manduca sexta serpin-6 regulates immune serine proteinases PAP-3 and HP8. cDNA cloning, protein expression, inhibition kinetics, and function elucidation. J. Biol. Chem. 280, 14341–14348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou Z., Najar F., Wang Y., Roe B., and Jiang H. (2008) Pyrosequence analysis of expressed sequence tags for Manduca sexta hemolymph proteins involved in immune responses. Insect Biochem. Mol. Biol. 38, 677–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang S., Gunaratna R. T., Zhang X., Najar F., Wang Y., Roe B., and Jiang H. (2011) Pyrosequencing-based expression profiling and identification of differentially regulated genes from Manduca sexta, a lepidopteran model insect. Insect Biochem. Mol. Biol. 41, 733–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gunaratna R. T., and Jiang H. (2013) A comprehensive analysis of the Manduca sexta immunotranscriptome. Dev. Comp. Immunol. 39, 388–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furusawa T., Rakwal R., Nam H. W., Hirano M., Shibato J., Kim Y. S., Ogawa Y., Yoshida Y., Kramer K. J., Kouzuma Y., Agrawal G. K., and Yonekura M. (2008) Systematic investigation of the hemolymph proteome of Manduca sexta at the fifth instar larvae stage using one- and two-dimensional proteomics platforms. J. Proteome Res. 7, 938–959 [DOI] [PubMed] [Google Scholar]
- 24.Zhang S., Cao X., He Y., Hartson S., and Jiang H. (2014) Semi-quantitative analysis of changes in the plasma peptidome of Manduca sexta larvae and their correlation with the transcriptome variations upon immune challenge. Insect Biochem. Mol. Biol. 47, 46–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blagoev B., Ong S. E., Kratchmarova I., and Mann M. (2004) Temporal analysis of phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat. Biotechnol. 22, 1139–1145 [DOI] [PubMed] [Google Scholar]
- 26.Cao X., and Jiang H. (2015) Integrated modeling of protein-coding genes in the Manduca sexta genome using RNA-Seq data from the biochemical model insect. Insect Biochem. Mol. Biol. 62, 2–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nesvizhskii A. I., Keller A., Kolker E., and Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 28.Hubner N. C., Ren S., and Mann M. (2008) Peptide separation with immobilized pI strips is an attractive alternative to in-gel protein digestion for proteome analysis. Proteomics 8, 4862–4872 [DOI] [PubMed] [Google Scholar]
- 29.Nickel W. (2010) Pathways of unconventional protein secretion. Curr. Opin. Biotechnol. 21, 621–626 [DOI] [PubMed] [Google Scholar]
- 30.Matsumoto Y., Oda Y., Uryu M., and Hayakawa Y. (2003) Insect cytokine growth-blocking peptide triggers a termination system of cellular immunity by inducing its binding protein. J. Biol. Chem. 278, 38579–38585 [DOI] [PubMed] [Google Scholar]
- 31.Kanost M. R., Gorman M. J. (2008) Phenoloxidases in insect immunity. In Insect Immunology, Beckage N. E., ed. Elsevier, San Diego, 30 [Google Scholar]
- 32.Jiang H., and Kanost M. R. (2000) The clip-domain family of serine proteinases in arthropods. Insect Biochem. Mol. Biol. 30, 95–105 [DOI] [PubMed] [Google Scholar]
- 33.Ragan E. J., An C., Yang C. T., and Kanost M. R. (2010) Analysis of mutually exclusive alternatively spliced serpin-1 isoforms and identification of serpin-1 proteinase complexes in Manduca sexta hemolymph. J. Biol. Chem. 285, 29642–29650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tong Y., Jiang H., and Kanost M. R. (2005) Identification of plasma proteases inhibited by Manduca sexta serpin-4 and serpin-5 and their association with components of the prophenol oxidase activation pathway. J. Biol. Chem. 280, 14932–14942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christen J. M., Hiromasa Y., An C., and Kanost M. R. (2012) Identification of plasma proteinase complexes with serpin-3 in Manduca sexta. Insect Biochem. Mol. Biol. 42, 946–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyaji T., Murayama S., Kouzuma Y., Kimura N., Kanost M. R., Kramer K. J., and Yonekura M. (2010) Molecular cloning of a multidomain cysteine protease and protease inhibitor precursor gene from the tobacco hornworm (Manduca sexta) and functional expression of the cathepsin F-like cysteine protease domain. Insect Biochem. Mol. Biol. 40, 835–846 [DOI] [PubMed] [Google Scholar]
- 37.Rayaprolu S., Wang Y., Kanost M. R., Hartson S., and Jiang H. (2010) Functional analysis of four processing products from multiple precursors encoded by a lebocin-related gene from Manduca sexta. Dev. Comp. Immunol. 34, 638–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nägler D. K., and Ménard R. (1998) Human cathepsin X: A novel cysteine protease of the papain family with a very short proregion and unique insertions. FEBS Lett. 434, 135–139 [DOI] [PubMed] [Google Scholar]
- 39.Chen G., Gharib T. G., Huang C. C., Taylor J. M., Misek D. E., Kardia S. L., Giordano T. J., Iannettoni M. D., Orringer M. B., Hanash S. M., and Beer D. G. (2002) Discordant protein and mRNA expression in lung adenocarcinomas. Mol. Cell. Proteomics 1, 304–313 [DOI] [PubMed] [Google Scholar]
- 40.Kendrick N. (2014) A gene's mRNA level does not usually predict its protein level. Kendrick Laboratories, Inc., Madison, WI [Google Scholar]
- 41.Gautier V., Mouton-Barbosa E., Bouyssié D., Delcourt N., Beau M., Girard J. P., Cayrol C., Burlet-Schiltz O., Monsarrat B., and Gonzalez de Peredo A. (2012) Label-free quantification and shotgun analysis of complex proteomes by one-dimensional SDS-PAGE/NanoLC-MS: Evaluation for the large scale analysis of inflammatory human endothelial cells. Mol. Cell. Proteomics 11, 527–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao B. B., Stuart L., and Feener E. P. (2008) Label-free quantitative analysis of one-dimensional PAGE LC/MS/MS proteome: Application on angiotensin II-stimulated smooth muscle cells secretome. Mol. Cell. Proteomics 7, 2399–2409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang H., Wang Y., Yu X. Q., and Kanost M. R. (2003) Prophenoloxidase-activating proteinase-2 from hemolymph of Manduca sexta. A bacteria-inducible serine proteinase containing two clip domains. J. Biol. Chem. 278, 3552–3561 [DOI] [PubMed] [Google Scholar]
- 44.Suwanchaichinda C., Ochieng R., Zhuang S., and Kanost M. R. (2013) Manduca sexta serpin-7, a putative regulator of hemolymph prophenoloxidase activation. Insect Biochem. Mol. Biol. 43, 555–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clark K. D., and Strand M. R. (2013) Hemolymph melanization in the silkmoth Bombyx mori involves formation of a high molecular mass complex that metabolizes tyrosine. J. Biol. Chem. 288, 14476–14487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clark K. D. (2015) Altered tyrosine metabolism and melanization complex formation underlie the developmental regulation of melanization in Manduca sexta. Insect Biochem. Mol. Biol. 58, 66–75 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.