

Abstract
The KEAP1/Nrf2 pathway senses and responds to changes in intracellular oxidative stress. Mutations that result in constitutive activation of Nrf2 are present in several human tumors, especially non-small cell lung cancer. Therefore, compounds that inhibit Nrf2 activity might be beneficial in treating patients whose tumors show activation of this pathway. Recent reports suggest that the natural product brusatol can potently and selectively inhibit Nrf2 activity, resulting in cell cytotoxicity, and can be effectively combined with chemotherapeutic agents. Here, we analyzed the effects of brusatol on the cellular proteome in the KEAP1 mutant non-small cell lung cancer cell line A549. Brusatol was found to rapidly and potently decrease the expression of the majority of detected proteins, including Nrf2. The most dramatically decreased proteins are those that display a short half-life, like Nrf2. This effect was confirmed by restricting the analysis to newly synthesized proteins using a labeled methionine analogue. Moreover, brusatol increased the expression of multiple components of the ribosome, suggesting that it regulates the function of this macromolecular complex. Finally, we show that brusatol induces its potent cellular cytotoxicity effects on multiple cancer cell lines in a manner independent of KEAP1/Nrf2 activity and with a profile similar to the protein translation inhibitor silvestrol. In conclusion, our data show that the activity of brusatol is not restricted to Nrf2 but, rather, functions as a global protein synthesis inhibitor.
The KEAP11/Nrf2 pathway is one of the most important cellular mechanisms to react and respond to oxidative stresses. Under normal physiological conditions, Nrf2 protein is maintained at low levels due to constitutive ubiquitination and degradation. This is achieved through recruitment to the Cul3 ubiquitin ligase complex by the substrate adaptor protein KEAP1. KEAP1 exists as a dimer, which binds a single Nrf2 molecule through two peptide motifs at its N terminus, a high affinity GluThrGlyGLU motif and a lower affinity AspLeuGly motif. This positions Nrf2 for effective ubiquitin transfer. Polyubiquitinated Nrf2 is subsequently degraded by the proteasome. In response to oxidative stress or electrophiles, key cysteine residues in KEAP1 become oxidized, thereby impairing its ability to recruit and ubiquitinate Nrf2 (1). This results in Nrf2 stabilization, allowing it to translocate to and accumulate in the nucleus (reviewed in (2)). Nrf2 binds specific DNA sequences in complex with the small Maf proteins, which were originally identified as viral oncogenes containing leucine zipper motifs (3). Hundreds of target genes have been identified using ChIP-seq and RNAseq experiments (4–6), highlighting the importance of Nrf2 in reducing levels of oxidative stress by increasing glutathione and NADPH synthesis, and up-regulating drug efflux pumps. Therefore, this pathway senses and responds to oxidative stress to maintain cellular redox homeostasis.
When dysregulated, this pathway also contributes to many human pathologies including cardiovascular and neurodegenerative diseases, inflammation and cancer. Large-scale genome sequencing efforts have identified mutations in both KEAP1 and Nrf2 in several human tumor indications, but especially in non-small cell lung cancers (NSCLC). Mutations in KEAP1 are distributed across the gene and occur in ∼20 and ∼12% of the adenomatous tumors and squamous NSCLC, respectively (7). In addition to KEAP1, ∼15% of squamous NSCLC cases present with mutations in Nrf2 (8), supporting the hypothesis that Nrf2 degradation is frequently dysregulated in this indication (9). Nrf2 mutations cluster in two hotspot regions, ∼10 amino acids each, near the DLG and ETGE KEAP1 interacting regions, resulting in constitutive Nrf2 activation. A reasonable expectation is that these tumors become addicted to the high levels of Nrf2 activity, and some data have been presented that supports this hypothesis (10). Identification of a selective inhibitor of this pathway would be of great interest for potential therapeutic applications, which led us to explore the use of brusatol as an Nrf2 inhibitor. Brusatol is a representative compound from a family of natural product quassinoids. Recent publications have suggested that it can decrease the levels of Nrf2 and sensitize cells to various chemotherapy agents (11, 12).
To identify and characterize the cellular targets of brusatol, we chose to employ a mass-spectrometry-based strategy. The binding of small molecules to their target proteins can induce conformational changes and alter their thermal stability compared with the apo form. This fact has been recently exploited to demonstrate engagement of known protein targets by small molecules in cells using Western blotting in an approach termed the cellular thermal shift assay (CETSA) (13). More recently, this assay was coupled with multiplexed mass spectrometry to yield an assay capable of surveying small molecule–protein interactions across a large fraction of the proteome (14). Our work using this approach, and follow up studies, suggest that brusatol regulates Nrf2 through an indirect mechanism involving global inhibition of protein synthesis.
EXPERIMENTAL PROCEDURES
Cell Culture
A549 cells (obtained from ATCC) were maintained in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS, Sigma Aldrich) and 10 mm l-glutamine. Cells were maintained in a humidified incubator at 37 °C/5% CO2.
Brusatol Extraction
The ground, air-dried seeds of Brucea sumatrana (200 g) were exhaustively extracted with n-hexane (1 l). The residue, after the removal of hexane, was further extracted with methanol (1 l). The total methanol extract evaporated to a viscous residue that was dark green in color and extremely bitter. The syrupy material was mixed with an equal volume of water and the filtered solution extracted with hexane until the extracts were colorless, then with methylene chloride. The methylene chloride extract was dried and evaporated to yield a dark yellow resinous material (500 mg). TLC (DCM/MeOH = 50:1) of the crude material showed two main components that gave green colors with a ferric chloride spray. This crude material was purified via column chromatography on silica gel (DCM/MeOH = 100:1∼40:1) to give crude brusatol, which was recrystallized from DCM/ether to give pure brusatol (30 mg) as a white solid (20 mg was used for analytical studies). Analytical nmr spectra match literature reports (15, 16).
CETSA Assays
For lysate assays, A549 cells were seeded in 10 cm plates and grown to 80% confluence. The cells were trypsinized, pelleted, and washed two times with PBS. The cell pellet was resuspended in 4.4 ml KB buffer (25 mm Tris, pH 7.5, 1 mm DTT, 10 mm MgCl2, complete protease inhibitor (Roche) and phosphatase inhibitor mixture II (Sigma)). The cell suspension was subjected to three cycles of freezing in liquid nitrogen and thawing at 37 °C, vortexed, and centrifuged at 20,000 × g for 20 min at 4 °C. The supernatant (lysate) was divided into two aliquots and treated with 500 nm brusatol or DMSO for 30 min at 25 °C. Following treatment, the lysates were aliquoted into PCR tubes (50 μl per tube) and heated in a thermal cycler at 38 °C, 42 °C, 46 °C, 50 °C, 54 °C, 58 °C, 62 °C, 66 °C, or 70 °C for 3 min, followed by a cool-down at room temperature for an additional 3 min. Lysates were pooled by treatment and temperature into 1.5 ml Eppendorf tubes and centrifuged at 20,000 × g for 20 min at 4 °C. The supernatants were isolated and submitted for MS analysis.
For intact cell assays, A549 cells were seeded in 10 cm plates and grown to 80% confluence. Cells were treated with either 500 nm brusatol or DMSO for 4 h (five plates per treatment). Following treatment, the cells were trypsinized, pelleted, and washed 2× with PBS. Cells from each treatment arm resuspended in 2.2 ml PBS supplemented with protease and phosphatase inhibitors. Cells were aliquoted into PCR tubes (50 μl per tube) and pelleted, removing all but ∼10 μl of remaining PBS. The PCR tubes were then heated as previously described and subjected to three cycles of freeze/thaw as before. Cells were then resuspended in KB buffer, pooled by treatment and temperature, vortexed, and centrifuged at 20,000 × g for 20 min at 4 °C. The supernatants were isolated and submitted for MS analysis. These datasets were acquired once for potential target discovery, and hits were subsequently validated by Western blots.
Western Blots
Lysates were prepared as described above. For dose/time-course experiments, cells were treated with brusatol or cyclohexamide (EMD Millipore) at the dose and time points indicated, followed by lysate harvest in KB buffer using the snap-freeze method described above. Proteins were separated on 4–20% Tris-glycine gels and transferred to nitrocellulose membranes via iBlot (Invitrogen). Membranes were blotted with the following primary antibodies: Nrf2 (Abcam ab62352), cystatin-C (Abcam ab133495), IGFBP4 (Thermo PA5–25925), PAF1 (Bethyl A304–374A), NRCAM (ProteinTech 21608–1-AP), MCL-1 (Cell Signaling 5453), p21 (Cell Signaling 2947), Gemin-4 (Abcam ab31581), FAM96A (Acris AP51504PU-N), RL22 (Santa Cruz sc-136413), GCN1L1 (Abcam ab86139), RPL11 (Abcam ab79352), RL9 (Santa Cruz sc-292593), survivin (sc-17779), cyclin A (sc-751), and β-actin (Cell Signaling 12620). Western blotting was performed a minimum of two times using biological replicates, and a representative example is shown.
Mass Spectrometry
For each sample, 8 m urea was added in 1:1 ratio by volume to aid in denaturation of proteins before digestion. Proteins were reduced with 5 mm dithiothreitol (DTT) at 65 °C for 25 min and then alkylated with 11 mm iodoacetamide at room temperature, in the dark for 20min. Samples were further diluted to 2 m urea with 20 mm HEPES (pH 8). Trypsin (Promega) was then added at a ratio of 1:100 (enzyme:substrate by weight)(17) and digestion carried out overnight at 37 °C. The reaction was quenched with 1% trifluoroacetic acid, and samples were desalted using C-18 Sep-Pak (Waters). Eluents were dried to completion, resuspended with 100 μl of 100 mm HEPES, pH 8.0, and labeled with TMT10plex labeling reagent (Thermo) according to the manufacturer's protocol. TMT-labeling reactions were quenched with 5% hydroxylamine for 15 min before combining into the two sets (Set 1: DMSO and brusatol at 38 °C, 42 °C, 46 °C, 50 °C, and 54 °C; Set 2: DMSO and brusatol at 38 °C, 58 °C, 62 °C, 66 °C, and 70 °C). Both sets were acidified with trifluoroacetic acid and dried to completion using a speedvac. Each set was fractionated by high pH reverse phase using an Agilent 1100 series offline HPLC system. Peptides were loaded onto an Agilent Zorbax 300 Extend C-18 analytical column with 2.1 × 150 mm dimensions and 3.5 μm particle size. Solvent A consisted of 25 mm ammonium formate (pH 9.7) while solvent B was 100% ACN. Peptide fractions were collected in 45 s intervals for a total of 63 min with a linear gradient of 15% to 60% B. Ninety-six fractions were collected, and every 16th fraction was combined to form a final set of 16 distinct groups: fraction 1, 17, 33, 49, 65, 81 into one fraction, etc. Samples were lyophilized, subjected to STAGEtip for desalting, and injected for LC-MS/MS analysis.
TMT-labeled samples were analyzed on an Orbitrap-Fusion mass spectrometer (Thermo Fisher Scientific, San Jose, CA USA) connected to a NanoAcquity UPLC system (Waters, Milford, MA) via an EASY-Spray ion source (Thermo Scientific). Peptide separation was achieved over a PepMap RSLC EASY-Spray column (75 μm inner diameter, 15 cm length, 2 μm C18 beads from Thermo Scientific) flowing at 800 nl/min. A linear gradient of 2% Solvent B to 25% Solvent B was performed over 85 min with a total method run time of 120 min. Solvent B consisted of 98% ACN/2% water/0.1% formic acid, and Solvent A was composed of 98% water/2% ACN/0.1% formic acid. The mass spectrometer was operated with a spray voltage of 1.9kV. Full MS scans were collected in the Orbitrap at 120,000 resolution across a mass range of 375 to 1600 m/z. The MS1 automatic gain control target was set to of 2 × 105 with a maximum injection time of 50 ms. MS2 ions (automatic gain control target 1 × 104, maximum injection time of 50 ms) were selected using a top speed data-dependent mode and fragmented with normalized collision energy of 30 and analyzed in the linear ion trap. Utilizing the Orbitrap Fusion's SPS mode, the top 10 precursor ions previously analyzed in MS2 were selected again for MS3 analysis (isolation width of 2 Th, automatic gain control target 1 × 105, maximum injection time 120 ms). Fragment ions were generated via higher collision dissociation (normalized collision energy 65) and analyzed in the Orbitrap at a resolution of 60,000.
Raw files were converted to mzXML using ReAdW (version 4.3.1 with minor modification to enable parsing rapid scan and SPS scan headers). MS/MS spectra were searched using Mascot (v.2.3.02) against target sequences from the human proteome database (UniProt Dec. 2011) and known contaminants concatenated with a decoy database containing reverse sequences of each entry (total entries are 238,494, including both forward and decoy hits). Search parameters included tryptic specificity with allowance of up to two missed cleavage events, a precursor ion tolerance of 50 ppm, and a fragment ion tolerance of 0.8 Da. Searches permitted variable modifications of methionine oxidation (+15.9949 Da) and static modifications of cysteine (+57.0215) for carbamidomethylation as well as TMT10plex tags on lysine and peptide N termini (+229.1629). Reporter ions produced by the TMT tags were quantified with an in-house software package known as Mojave. In brief, TMT reporter ions were extracted by isolating peaks within 20 ppm of theoretical TMT reporter mass. The intensity of the highest peak within the window is reported. Peptide spectra matches were filtered with a false discovery rate of 5% on the peptide level and subsequently at 2% on the protein level using linear discrimination (17). Additional filtering of quantified peptide spectra matches incorporated an isolation specificity of 0.7 or greater and a combined TMT intensity greater than 3000. The resulting dataset was then subjected to an R script for melting curve analysis. Briefly, two TMT sets were combined based on the shared 38 °C temperature point samples. A sigmoid curve was fitted to the temperature series for each treatment group using R nonlinear least-squares estimates function nls() based on logistic model “SSlogis” (package “stats,” version 3.2.2). Parameters for these functions were illustrated below, where “data” is an R data frame containing temperature series and the corresponding protein intensity and “Asym,” “xmid,” “scal” are all computed by SSlogis function internally.
 |
The melting point was calculated at 50% of initial protein abundance level. All mass spectrometry raw datasets have been deposited to the MASSIVE database (http://massive.ucsd.edu/) and can be downloaded by the identifier MSV000079314. A complete list of peptide-spectrum-matches passing abovementioned filters (except for TMT intensity threshold) can be found in Supplemental Table 1 for the cell lysate CETSA and intact cell CETSA experiments. A complete list of proteins with normalized abundance in each condition and melting points can be found in the Supplemental Table 2. Annotated MS/MS for all one hit wonders can be found in the Supplemental Fig. 2.
Taqman Assays
RNA was harvested via RNEasy Mini kits (Qiagen), cDNA synthesized with High Capacity cDNA reverse transcription kit (Life Technologies, Foster City, CA), and Taqman assays run using the following primer/probe sets (Life Technologies): Nrf2 (Hs00975961_g1), cystatin C (Hs00264679_m1), PAF1 (Hs00219496_m1), NRCAM (Hs01031598_m1), GCN1L (Hs00412445_m1), RPL11 (Hs00831112_s1), and RL22 (Hs01865331_s1). Endogenous control assays multiplexed with the target assays using GAPDH-Vic primers/probe (Hs03929097_g1). Taqman assays were performed twice, and technical replicates from one experiment are shown in the figure.
Click-IT Assays
Cells were seeded in six-well plates and grown to 80% confluence, then treated with brusatol or cyclohexamide for 15 min. The medium was removed and cells washed three times with PBS. Methionine-free medium was then added (with brusatol/cyclohexamide) for 30 min. Labeling compound l-azidohomoalanine (Invitrogen, Grand Island, NY) was added at a final concentration of 50 μm for 1 h. Cells were then washed three times with PBS, and lysates harvested using 200 μl lysis buffer per well (1% SDS in 50 mm Tris-HCl, pH 8.0, protease/phosphatase inhibitors). Click reactions were carried out using 90 μg of input lysate, biotinylated alkyne reagent (B10185, Invitrogen), and protein reaction buffer kit (C10276, Invitrogen) as per manufacturer's protocol. Biotinylated proteins were precipitated/cleaned up via methanol/chloroform extraction, and analyzed by Western blotting. Membranes were blotted with anti-streptavidin-HRP antibody (Thermo N504). Unprocessed lysates were also run on Western blot and probed for Nrf2 and β-actin. These experiments were performed twice, Western blots from one experiment are shown.
Cell Viability Following Brusatol or Silvestrol Treatment
Cells were grown in RPMI 1640 containing 2 mm glutamine and 10% fetal bovine serum (heat-inactivated) for suspension lines and 5% fetal bovine serum for adherent lines and maintained in a humidified incubator (37 °C, 5% CO2). A Multidrop Combi Reagent Dispenser (Thermo Scientific; Waltham, MA) was used for plating cells into barcoded Falcon 384-well, black, clear-bottom plates (Corning; Tewksbury, MA; Catalogue No. 353962). On the following day, the compound was added using an Oasis integrated Liquid Handling Robot Platform (Dynamic Devices; Wilmington, DE) with LiCONiC incubators (LiCONiC Instruments; Woburn, MA) with automated barcode tracking. After 72 h, 25 μl CellTiter-Glo reagent was added using a MultiFlo Microplate Dispenser (BioTek; Winooski, VT). Cell lysis was induced by mixing for 30 min on an orbital shaker; plates were then incubated at room temperature for 10 min to stabilize the luminescent signal. Luminescence readout was done via a 2104 EnVision Multilabel Plate Reader (PerkinElmer; Waltham, MA). Experiments were performed as biological triplicates, and average IC50 and viability values used for analysis.
Average doubling times were calculated based on 3–5 seeding densities. Cells were harvested at 80% confluency and doubling times calculated using the following formula, where Δ h = time of harvest – time of seeding in hours, C1 = initial cell seeding, C2 = total cell number at harvest = LOG10(2)/(LOG10(C2/C1)/Δ h).
Generation of Cell Lines Expressing dox-Inducible Nrf2 shRNAs
Nrf2-targeting and nontargeting control shRNA lentiviral vectors were constructed in the pINDUCER10 vector (18) by annealing and ligation of DNA oligos into the XhoI-EcoRI sites of the vector. shRNA lentiviral vectors were cotransfected with pCMV-VSVG and pCMV-dR8.9 plasmids into 293T cells using the Lipofectamine 2000 reagent (Life Technologies). Forty-eight hours after transfection, viral supernatants were collected and used to infect A549 H1048 H441 and H460 cells (ATCC, Manassas, VA) in the presence of 8 μg/ml polybrene (EMD Millipore Corp, Billerica, MA). Cells with stable integrants were selected with puromycin for 10 days. Stably selected pools were maintained in puromycin selective media.
Correlation of Cell Molecular Features with Brusatol Sensitivity
In preparation for predictive modeling, several cell line features were gathered: tissue of origin, growth rate, and doubling time. All sample descriptors were combined into one matrix for predictive modeling. Expression, absolute DNA copy number and loss-of-heterozygosity, seeding density and growth rate were expressed as continuous values. Mutation status for individual hotspots, for gene hotspot and pathway metafeatures, and for the three categories of function-altering mutation were each individually coded as binary variables. Categorical variables, such as tissue type, were coded with one binary dummy variable per factor level. All features, including the binary features, were standardized prior to imputation and predictive modeling.
Missing data, due to a lack of genomic data or insufficient sequencing coverage, were imputed using glmnet (19). Missing values were initially replaced with the average value for their column (feature). Missing data were then imputed, one feature at a time, in a serial fashion, by treating one column as the response variable and all other feature columns as dependent variables. Training was performed using all samples with data for the response variable, and values were then predicted for the samples previously missing data.
Classification and feature selection were performed using glmnet with binarized or continuous valued mean viability as the response variable. We selected an alpha value of 0.5 to increase the richness of the selected features and to increase interpretability. We selected the minimum lambda value within one standard deviation of the absolute minimum value under cross validation. This cross validation was performed with 25 randomly selected hold-out groups consisting of 20% of the samples. Hold-out groups were selected using the caret (20) tool to select sample groups with sensitive/resistant proportions or continuous response variable distributions similar to that of the full set of samples. Following cross validation and lambda selection, a glmnet run with all samples was performed to select potential biomarkers. All features with a nonzero coefficient under the selected lambda were used for further analyses. Ranked absolute coefficient values were used to assess the relative importance of features.
RESULTS
Recent publications have suggested that the natural product brusatol reduces Nrf2 protein levels (11, 12). To better understand the underlying mechanism, we first attempted to confirm this observation in the KEAP1 mutant A549 lung cancer cell line. As previously reported, brusatol was able to rapidly and potently inhibit Nrf2 protein levels (Fig. 1A) without altering the overall pattern of abundant cellular proteins as observed by Coomassie Blue staining (Fig. 1B). To determine whether brusatol directly engages Nrf2, cellular thermal shift (CETSA) assays were performed both in lysates and cells (13, 21). Brusatol was added directly to cell lysates for 30 min (Fig. 1C) and to intact cells for 4 h (Fig. 1D) before subjecting aliquots of lysate or cells to increasing temperatures. Insoluble and aggregated proteins were removed by centrifugation prior to SDS-PAGE and Western blotting. In cell lysates, Nrf2 signal was diminished at 50 °C and the protein fully insoluble by 58 °C (Fig. 2A); however, the addition of brusatol to cell lysates did not alter the thermostability of Nrf2, suggesting that Nrf2 may not be the direct target of this compound. As expected, addition of brusatol to cells for 4 h resulted in complete loss of Nrf2 protein at all temperatures. Nrf2 becomes thermally insoluble in cells at a slightly lower temperature than in lysates, similarly to what was previously seen for most proteins (21) and likely reflecting a stabilizing effect of the lysis buffers used.
Fig. 1.

Brusatol inhibits Nrf2 protein expression. (A) Increasing doses of brusatol were added to A549 cells. 3 h later, cells were lysed in RIPA buffer and analyzed by Western blotting using antibodies against Nrf2 and actin. (B) The same extracts were separately separated by SDS-PAGE and proteins were visualized by Coomassie blue staining. (C) A549 cell lysates (4.5 mg/ml) were treated with 0.1% DMSO or 500 nm brusatol for 30 min at room temperature (25 °C), then a total of 200 μl lysate per temperature point (400 μl for 38 °C) was heated in 200 μl thin-walled PCR tubes at the indicated temperature for 3 min and allowed to cool to room temperature for an additional 3 min. Lysates were then spun to remove insoluble proteins, and proteins remaining in solution analyzed by Western blotting using Nrf2 antibodies. (D) A549 cells were treated with 0.1% DMSO or 500 nm brusatol for 4 h, and then cells were removed from plates by trypsinization. 4.8 × 106 cells per temperature point were then pipetted in 200 μl thin-walled PCR tubes, pelleted, and heated at the indicated temperature for 3 min. Cells were then lysed and solubilized protein analyzed by Western blotting.
Fig. 2.
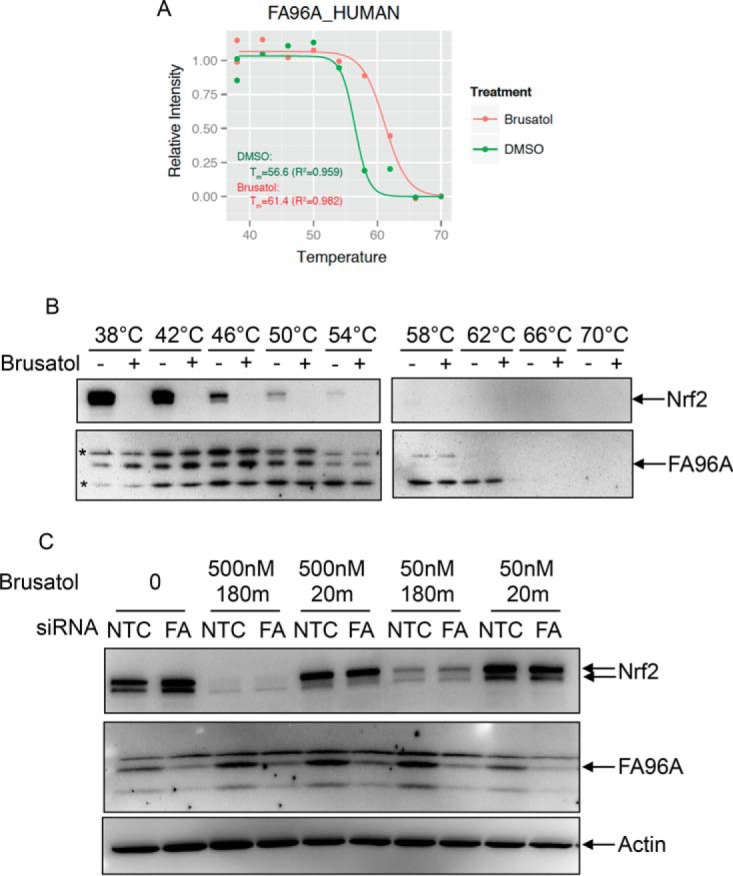
FA96A is not involved in the cellular response to brusatol. (A) Melting curve of FA96A from lysates of cells treated with DMSO or 500 nm brusatol for 30 min. Green: DMSO treatment; red: brusatol treatment. (B) A549 lysates were treated with 500 nm brusatol for 30 min and soluble proteins from the indicated temperatures fractionated by SDS-PAGE and Western blotted using the indicated antibodies. *denotes nonspecific bands. (C) A549 cells were treated with either nontargeting (42) or siRNA oligos targeting FA96A for 3 days. Lysates were then treated with the indicated amounts of brusatol for 30 min, and indicated proteins detected by Western blotting.
Next, we attempted to identify proteins that are direct targets of brusatol by performing thermal proteomic profiling using 10plex Tandem Mass Tags (TMT). Protein digests were labeled with TMT10plex as two sets and subjected to MS analysis to determine the melting curves for each protein in DMSO and brusatol-treated intact cells and lysates. The decay of each protein as a function of temperature was fitted to a logistic curve for the DMSO or brusatol treatment using “nls” function based on a self-start model “SSlogis” in the R package “stats” (version 3.2.2). A minimum coefficient of determination (R2) of 0.8 was applied to establish minimum quality of curve fitting. Melting temperature (Tm) was determined by calculating the temperature from the fitted curve at 50% protein abundance. After filtering, a total of 2851 and 2656 proteins were fitted into melting curves for the CETSA in lysate and intact cells, respectively. The full dataset is shown in Supplemental Tables 1 and 2 and Supplemental Figs. 1A (lysate) and 1B (intact cells). While individual proteins were identified in both lysate and cells that showed differential thermostability upon brusatol treatment (e.g. FA96A and RABL5 from cell lysates, and HPCL1 and PYRG2 from intact cells, Supplemental Fig. 1), these small changes could not be validated by Western blotting, and siRNA knockdown did not affect brusatol-induced changes in Nrf2 levels as expected for physiologically relevant targets of brusatol (see, for example, FA96A, Figs. 2A-2C). In fact, while there was good consistency in the melting curves across the proteome comparing lysates to cells, there were few/no proteins showing consistent brusatol-induced Tm shifts between the lysate and cell-based CETSA MS experiments (Supplemental Table 2). Interestingly, we also found that proteins showed greater thermal stability in lysates compared with cells (Fig. 3A), similar to previous reports (14).
Fig. 3.

Effects of brusatol on total protein synthesis. (A) Scatterplot showing the melting temperatures of all proteins detected in both lysates and intact cells under the DMSO condition. The solid line shows the best fit with an R2 concordance of 0.43, while the red dashed line indicates a hypothetical unity response, to demonstrate that most proteins are more thermostable when heated in lysis buffer compared with intact cells. (B) The ratios of protein abundance of 3499 proteins at the 38 °C temperature comparing brusatol-treated to DMSO-treated A549 cells. Protein abundance was quantified from summed TMT reporter ion intensities of peptide-spectrum-matchings. The scatter plot shows data from two replicate measurements indicating that the majority of proteins show reduced abundance following brusatol treatment. cystatin C (red), IGFBP4 (blue), PAF (purple), and NRCAM (brown) are indicated. (C) MS abundance data for cystatin C, IGFBP4, and PAF over the temperature ranges used are shown. X axis: temperature points to heat intact cells in the CETSA assay. Y axis: protein abundance at each temperature point normalized to the average of DMSO and brusatol-treated protein abundance at 38 °C. Green: DMSO treatment; red: brusatol treatment. (D) The same lysates used for MS analysis were used for Western blotting using antibodies specific for cystatin C, IGFBP4, and PAF.
Examining data from the soluble fraction at the lowest temperature analyzed (38 °C), the majority of identified proteins showed lower abundance in brusatol-treated cells, (median = -0.38, standard deviation = 0.3), with only 37 showing up-regulation by more than 20% (log2 ratio ≥ 0.26) (Fig. 3B). Three decreased proteins that were validated by Western blotting (cystatin C, IGFBP4, and PAF), are shown in Figs. 3C and 3D. Analysis of the 37 up-regulated proteins by gene set enrichment analysis showed more than half to be involved in ribosome assembly and protein translation (Table I and Supplemental Table 3). Although the fold increase in individual proteins was quite modest (∼1.2–1.5-fold), the concerted up-regulation of the ribosomal and translational machinery was dramatic and unexpected. Three examples (RL11, RL22, and GCN1L) of melting curves and Western blots of proteins increased by brusatol treatment are shown in Figs. 4A and 4B, respectively.
Table I. Top gene sets over-representing proteins up-regulated by brusatol treatment.
| Gene set name [# Genes (K)] | Description | # Genes in overlap (k) | p value | FDR q-value |
|---|---|---|---|---|
| KEGG_RIBOSOME [88] | Ribosome | 21 | 7.93 e−49 | 8.21 e−45 |
| REACTOME_PEPTIDE_CHAIN_ELONGATION [153]α | Genes involved in peptide chain elongation | 20 | 1.16 e−40 | 6.02 e−37 |
| REACTOME_INFLUENZA_VIRAL_RNA_TRANSCRIPTION_AND_REPLICATION [169] | Genes involved in influenza viral RNA transcription and replication | 20 | 9.62 e−40 | 2.73 e−36 |
| REACTOME_TRANSLATION [222] | Genes involved in translation | 21 | 1.05 e−39 | 2.73 e−36 |
| REACTOME_3_UTR_MEDIATED_TRANSLATIONAL_REGULATION [176] | Genes involved in 3′-UTR-mediated translational regulation | 20 | 2.27 e−39 | 3.91 e−36 |
Fig. 4.

Ribosomal proteins are increased following brusatol treatment. (A) MS abundance data for RL22, GCN1L, and RL11 over the temperature ranges used are shown. (B) The same lysates used for MS analysis were used for Western blotting using antibodies specific for RL22, GCN1L, and RL11.
The combination of most proteins showing decreased expression and ribosomal proteins showing increased expression suggested that modulation of protein translation could be the mechanism whereby brusatol decreases Nrf2 protein levels. We, therefore, compared the ability of brusatol and cycloheximide, a well-characterized protein synthesis inhibitor, to decrease the levels of Nrf2 and additional proteins. Figure 5A shows that brusatol and cycloheximide decreased the levels of Nrf2, PAF, cystatin C, and NRCAM (an additional protein down-regulated by brusatol) to a similar extent and with similar kinetics. The effects of brusatol and cycloheximide on protein abundance were independent of effects on mRNA levels, as the expression of these genes did not decrease following treatment compared with untreated control (Fig. 5B). In fact, Nrf2 and PAF mRNA levels increased following brusatol treatment. We also analyzed the effects of brusatol and cycloheximide on levels of p21 and Mcl-1, short half-life proteins (22, 23), and found that both were similarly decreased by brusatol and cycloheximide treatments. Brusatol and cycloheximide increased the levels of GCN1L and ribosomal proteins RL9 and RL22 with similar kinetics and to a similar extent (Fig. 5A), without affecting their mRNA levels (Fig. 5B). In comparison to a previously reported global protein dataset showing abundance change after cycloheximide treatment (24), good agreement was seen between proteins changing more than 2σ (i.e. log2 fold change ≤ -2σ or ≥ 2σ) after brusatol treatment and those proteins differentially expressed upon cycloheximide (Fig. 5C, Pearson correlation = 0.367). The effect was most notable among proteins down-regulated by more than 3σ (log2ratio<-0.9) (highlighted proteins in Fig. 5C). We also analyzed the abundance of two proteins that had previously been shown to be unaffected following treatment with brusatol (cyclin A and survivin) (12) and confirmed that neither brusatol nor cycloheximide conferred strong effects on their corresponding protein levels (Fig. 5D).
Fig. 5.

Brusatol-induced effects on protein abundance are independent of changes in mRNA. (A) A549 cells were treated with 0.1% DMSO, 50 nm or 500 nm brusatol or 5 or 50 μg/ml cycloheximide for the indicated times before harvesting the cells. Lysates were separated by SDS-PAGE and Western blotted using the indicated antibodies. (B) mRNA levels of Nrf2, cystatin C, PAF, NRCAM, GCN1L, RL22, and RL11 were analyzed at the 30 min and 4 h time points of brusatol and cycloheximide treated cells compared with no treatment control. NTC = nontargeted control siRNA. Error bars show standard deviation (n = 2). (C) Protein abundance change quantified after 4-h brusatol treatment or 6-h cycloheximide treatment (data from (24)). To filter out noise and look at more profound changes, a minimum protein level change of 0.6 (2σ) was required for the brusatol dataset. Proteins showing greater than 3σ changes were highlighted. Pearson correlation was calculated. (D) A549 cells were treated with 0.1% DMSO, 50 nm or 500 nm brusatol or 5 or 50 μg/ml cycloheximide for the indicated times before harvesting the cells. Lysates were separated by SDS-PAGE and Western blotted using the indicated antibodies.
These data are consistent with the hypothesis that brusatol, like cycloheximide, functions as a general inhibitor of protein synthesis. To test this more directly, we utilized a methionine analogue that allows for biotinylation via a “click” alkyne reaction (25). A 1-h pulse of the methionine analogue followed by biotinylation allows visualization of newly synthesized proteins using HRP-coupled streptavidin. Pretreatment of cells with either brusatol or cycloheximide completely abolished new protein synthesis under the same conditions that it inhibited Nrf2 expression (Fig. 6). In contrast, knockdown of Nrf2 had no effect on nascent protein synthesis under these conditions (data not shown). Consistent with its activity as a protein synthesis inhibitor, brusatol also showed broad cytotoxicity across a large (475) cell line panel encompassing several cancer indications at very low (nm) concentrations (Fig. 7A, Supplemental Table 4). The response of cells to brusatol was highly correlated with that seen with silvestrol (Fig. 7B, Supplemental Table 4), a known inhibitor of protein translation (26). There was no selective effect in KEAP1 mutant NSCLC lines compared with WT KEAP1 lines (Fig. 7C) nor did Nrf2 depletion significantly affect the sensitivity of cells to brusatol toxicity (Figs. 7D and 7E) as might have been expected from an agent that was a selective inhibitor of Nrf2 expression. In contrast, cell doubling time was the most predictive of brusatol sensitivity from among over >70,000 cellular features considered (Figs. 7F and 7G and Supplemental Table 4). Interestingly, the RNA and DNA levels of translation factor eIF6 also showed a correlation with brusatol sensitivity (Figs. 7F and 7H and Supplemental Table 4), in a manner independent of doubling time (Supplemental Table 4 and data not shown). This is consistent with the notion that faster-growing cells show a stronger requirement for protein synthesis compared with slower-growing cells.
Fig. 6.

Brusatol inhibits nascent protein synthesis. A549 cells were pretreated with brusatol or cycloheximide at the indicated concentrations for 15 min, then washed three times with PBS and the medium replaced with methionine-free medium (plus brusatol or cycloheximide) for 30 min. The methionine analog l-azidohomoalanine was then added to a final concentration of 50 μm for a further 1 h. l-azidohomoalanine was withheld from a control well to assess background signal. Cells were then harvested into lysis buffer, and 90 μg used for labeling with 20 μm biotin alkyne for 20 min. Biotin-labeled newly synthesized proteins were then separated by SDS-PAGE, and analyzed by Western blotting using HRP-coupled streptavidin. The lower blots show lysates analyzed by Western blotting using Nrf2 and actin antibodies.
Fig. 7.

Brusatol inhibits the proliferation of many cancer cell lines. (A) 475 cell lines were profiled using brusatol in a nine-point dilution curve, with 10 μm set as the highest concentration. The IC50s against the cell lines of different indications are plotted. Supplemental Table 4 provides the IC50 values for this figure. (B) 468 of these cell lines were also profiled against silvestrol in a nine-point dilution curve with 0.3 μm set as the highest concentration. The area under the curve viability over the entire dose–response for brusatol and silvestrol is shown as a scatterplot. R2 and p value calculated using linear regression. (C) The data from the 103 NSCLC cell lines where KEAP1 genotype information is known are shown. p value was calculated using a two-sided, two-sample Wilcoxon rank sum test. (D) The indicated cell lines expressing a doxycycline-inducible nontargeting control (42) or Nrf2 shRNA were treated with 500 ng/ml doxycycline for 48 h. Cells were lysed and proteins separated by SDS-PAGE and Western blotted using Nrf2 and β-actin antibodies. *denotes a nonspecific protein running very close to Nrf2 present in some cell lines. (E) The cells lines in (D) were treated with a brusatol dose response (highest concentration 10 μm), following 0.1% DMSO or 500 ng/ml doxycycline treatment for 48 h. Viability was assessed using Celltiter-Glo 72 h following brusatol treatment. (F) The Elastic Net algorithm was used to fit a linear model relating brusatol mean viability in cell lines to more than 70,000 genomic and phenotypic features of those lines. The largest 20 absolute values of the 61 nonzero coefficients are plotted to represent the features most strongly associated with brusatol mean viability. (G) Brusatol viability is plotted against cell doubling time. R2 and p value calculated using linear regression. (H) Brusatol viability is plotted against eIF6 mRNA levels. R2 and p value calculated using linear regression.
DISCUSSION
Nrf2 plays a dual role tumorigenesis, both preventing and enhancing progression of tumors. Mice lacking Nrf2 show increased numbers of tumors induced by chemical carcinogens (e.g. benzo[a]pyrene-induced gastric cancer (27) or 7,12-dimethylbenz(a)anthracene/12-O-tetradecanoylphorbol-13-acetate induced skin tumors (28)) when crossed to genetically engineered mouse tumor models of colon cancer (29). These data have spurred development of Nrf2 activators as chemopreventative agents. In fact, several well-established antioxidants, cytoprotective, and putative chemopreventive agents have been shown to act via inhibition of KEAP1 and consequential activation of Nrf2 (reviewed in (30)). Most of these activate Nrf2 through the covalent modification of key cysteine residues in KEAP1 that lie between the Nrf2 binding sites and the KEAP1 dimerization domain. Modification of these cysteine residues impedes the ability of KEAP1 to ubiquitinate Nrf2 (31). However, such reactive compounds are likely to modify many cysteine-containing proteins in the cell in a manner that could mask potential therapeutic benefits of activating Nrf2. In contrast, more recent efforts to activate Nrf2 have focused on identification of small molecules that specifically interfere with the KEAP1/Nrf2 binding interface. High-throughput screening has identified several such compounds (32, 33), which might allow more selective Nrf2 activation.
In contrast to its role in anticancer protective mechanisms, it has recently become clear that this pathway is also constitutively active in several human tumors owing to mutations in KEAP1 or Nrf2 itself (34). This finding has spurred interest in developing inhibitors of this pathway as therapeutic drugs. In contrast to the abundance of Nrf2 activators, there are fewer examples of Nrf2 inhibitors. Ascorbic acid (35), luteolin (36), and all trans retinoic acid (37) have all been shown to inhibit Nrf2 activity in ARE-luciferase reporter assays and alter the expression of endogenous Nrf2 target genes. Brusatol seemed initially attractive as a Nrf2 inhibitor, as its effectiveness at depleting Nrf2 protein levels at concentrations <100 nm, its effectiveness in both WT and mutant KEAP1-expressing cells, and its sensitization of cancer cells to chemotherapy in vitro and in vivo (11, 12, 38). While brusatol was previously demonstrated to inhibit total protein synthesis in rabbit reticulocyte lysates (39), the concentrations used were high (25–100 μm), leaving open the possibility that additional mechanisms were engaged at the lower concentrations used to inhibit Nrf2 protein levels. Indeed, Olayanju et al. (12) came to the conclusion that brusatol did not inhibit global translation, as short-lived proteins such as cyclin A, p53, and survivin were not depleted by brusatol treatment, although a positive control for inhibition of protein synthesis such as cycloheximide was not used in these experiments. While our Western blotting confirmed relatively minor effects on these example proteins, our MS analysis showed that the vast majority of the proteins decreased in abundance following brusatol treatment. Of the few proteins whose abundance increased following brusatol treatment, many were components of the ribosome, suggesting that brusatol may directly engage the translational machinery. A pulse analysis using a methionine analogue to monitor newly synthesized proteins confirmed that nascent protein synthesis is essentially abolished following brusatol treatment. A previous study suggested that brusatol inhibited formation of the first peptide bond between puromycin and [3H]methionyl-transfer RNA bound to the initiation complex (40). Why this mechanism would cause increased levels of ribosomal components is not clear. The most straightforward explanation would be that brusatol binds to the 80S ribosome and stabilizes the complex thereby decreasing the turnover of proteins associated with it. An alternative hypothesis is that intact ribosomes are precipitated by sample centrifugation and not visualized by Western blotting or MS analysis. In this model, disruption of intact ribosomes by brusatol allows their visualization. In support of this, ribosomal proteins appear to increase in abundance as the cells are heated and the effects of brusatol treatment are lost at the higher temperatures, suggesting that heating cells could also disrupt these complexes similar to brusatol treatment. Further experiments would be required to delineate the precise mechanisms. Interestingly, eIF6 levels predict response to brusatol (Fig. 7F), suggesting that this protein might be involved in the action of brusatol. Although it remains a formal possibility, our melting curve data failed to provide data to support the conclusion that eIF6 is the direct protein target of brusatol (Supplemental Fig. 1).
The cellular thermal shift assay has emerged as a powerful method to demonstrate target engagement in cells (13, 41), as well as to determine cellular selectivity of well-characterized compounds (21). An extension of this method is its use to find unknown targets of compounds with potent cellular activity. Brusatol was an appealing candidate for testing this approach given the therapeutic relevance of the Nrf2 pathway in lung cancer and its effectiveness at decreasing Nrf2 protein levels. Our Western blot analyses demonstrated that the few proteins showing subtle shifts in melting temperatures upon brusatol treatment could not be validated by orthogonal approaches. False negatives are inevitable from screening approaches. In mass-spectrometry-based CETSA assays, the two most plausible explanations behind the inability to identify a single protein target for brusatol would be incomplete proteome coverage or the possibility that brusatol–protein engagement does not alter thermal stability. An alternative reason is that the target of this compound is not a single protein but, rather, a member of a cellular macromolecular complex. Nonetheless, the richness of the MS data collected, particularly at the 38 °C starting temperature point, provided clues to a likely mechanism of action for brusatol. Thermal proteomic profiling by mass spectrometry is therefore positioned to become a mainstay for target identification and characterization in drug development.
Supplementary Material
Acknowledgments
We thank Jeff Settleman and Shiva Malek for thoughtful and critical discussions. We thank Joachim Rudolph for help in brusatol extraction. We thank gCell for supply of cell lines and the small molecule cell profiling group gCSI for analyzing cellular viability in response to brusatol and silvestrol. We also thank the bioinformatics group, especially Chris Klijn and Florian Gnad, for annotating KEAP1 mutations in NSCLC cell lines.
Footnotes
Author contributions: S. Vartanian prepared samples for MS analysis, performed Western blotting and Taqman analysis of target proteins/genes, and ran the methionine pulse labeling experiments. T. Ma ran the MS analyses. K. Yu analyzed the MS data and developed algorithms for visualization and interpretation. J. Lee created and validated the Nrf2 inducible knockdown cell lines. P. Haverty performed the predictive modeling of brusatol sensitivity. All authors were involved in the conception and design of this study, and co-wrote the manuscript.
 This article contains supplemental material Supplemental Tables 1–4 and Supplemental Figs. 1 and 2.
This article contains supplemental material Supplemental Tables 1–4 and Supplemental Figs. 1 and 2.
Conflict of interest: S. Vartanian, T. Ma, J. Lee, P. Haverty, D. Kirkpatrick, K. Yu, and D. Stokoe are paid employees of Genentech.
1 The abbreviations used are:
- KEAP1
- kelch-like ECH-associated protein 1
- DCM
- dichloromethane
- NMR
- nuclear magnetic resonance
- RT
- room temperature
- ACN
- acetonitrile
- ID
- inner diameter
- SPS
- synchronous precursor selection
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- VSVG
- vescicular stomatitis virus glycoprotein
- CMV
- cytomegalovirus
- RABL5
- Rab-like protein 5
- HPCL1
- Hippocalcin-like protein 1
- PYRG2
- pyrimidine reductaseG2
- ARE
- anti-oxidant response element
- CETSA
- cellular thermal shift assay
- NADPH
- nicotinamide adenine dinucleotide phosphate
- NSCLC
- non-small cell lung cancer
- TMT
- tandem mass tags
- UTR
- untranslated region.
REFERENCES
- 1.Rachakonda G., Xiong Y., Sekhar K. R., Stamer S. L., Liebler D. C., and Freeman M. L. (2008) Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem. Res. Toxicol. 21, 705–710 [DOI] [PubMed] [Google Scholar]
- 2.Tong K. I., Kobayashi A., Katsuoka F., and Yamamoto M. (2006) Two-site substrate recognition model for the Keap1-Nrf2 system: A hinge and latch mechanism. Biol. Chem. 387, 1311–1320 [DOI] [PubMed] [Google Scholar]
- 3.Nishizawa M., Kataoka K., Goto N., Fujiwara K. T., and Kawai S. (1989) v-maf, a viral oncogene that encodes a “leucine zipper” motif. Proc. Natl. Acad. Sci. U.S.A. 86, 7711–7715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malhotra D., Portales-Casamar E., Singh A., Srivastava S., Arenillas D., Happel C., Shyr C., Wakabayashi N., Kensler T. W., Wasserman W. W., and Biswal S. (2010) Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 38, 5718–5734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chorley B. N., Campbell M. R., Wang X., Karaca M., Sambandan D., Bangura F., Xue P., Pi J., Kleeberger S. R., and Bell D. A. (2012) Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 40, 7416–7429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirotsu Y., Katsuoka F., Funayama R., Nagashima T., Nishida Y., Nakayama K., Engel J. D., and Yamamoto M. (2012) Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 40, 10228–10239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research, N. (2014) Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research, N. (2012) Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hast B. E., Cloer E. W., Goldfarb D., Li H., Siesser P. F., Yan F., Walter V., Zheng N., Hayes D. N., and Major M. B. (2014) Cancer-derived mutations in KEAP1 impair NRF2 degradation but not ubiquitination. Cancer Res. 74, 808–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh A., Boldin-Adamsky S., Thimmulappa R. K., Rath S. K., Ashush H., Coulter J., Blackford A., Goodman S. N., Bunz F., Watson W. H., Gabrielson E., Feinstein E., and Biswal S. (2008) RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 68, 7975–7984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ren D., Villeneuve N. F., Jiang T., Wu T., Lau A., Toppin H. A., and Zhang D. D. (2011) Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. U.S.A. 108, 1433–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olayanju A., Copple I. M., Bryan H. K., Edge G. T., Sison R. L., Wong M. W., Lai Z. Q., Lin Z. X., Dunn K., Sanderson C. M., Alghanem A. F., Cross M. J., Ellis E. C., Ingelman-Sundberg M., Malik H. Z., Kitteringham N. R., Goldring C. E., and Kevin Park B. (2014) Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radical Biol. Med. 78, 202–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez Molina D., Jafari R., Ignatushchenko M., Seki T., Larsson E. A., Dan C., Sreekumar L., Cao Y., and Nordlund P. (2013) Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87 [DOI] [PubMed] [Google Scholar]
- 14.Savitski M. M., Reinhard F. B., Franken H., Werner T., Savitski M. F., Eberhard D., Martinez Molina D., Jafari R., Dovega R. B., Klaeger S., Kuster B., Nordlund P., Bantscheff M., and Drewes G. (2014) Proteomics. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346, 1255784. [DOI] [PubMed] [Google Scholar]
- 15.Yang Z., Xie H., and Wang J. (1996) Chemical studies of the active antitumor components from the fruits of Brucea javanica. Natural Product Research and Development 1996, 35–39 [Google Scholar]
- 16.Sim K. Y., Sims J. J., and Geissman T. A. (1968) Constituents of Brucea sumatrana J. Organic Chem. 33, 429–431 [Google Scholar]
- 17.Beausoleil S. A., Villén J., Gerber S. A., Rush J., and Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 18.Meerbrey K. L., Hu G., Kessler J. D., Roarty K., Li M. Z., Fang J. E., Herschkowitz J. I., Burrows A. E., Ciccia A., Sun T., Schmitt E. M., Bernardi R. J., Fu X., Bland C. S., Cooper T. A., Schiff R., Rosen J. M., Westbrook T. F., and Elledge S. J. (2011) The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 108, 3665–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friedman J., Hastie T., and Tibshirani R. (2010) Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 [PMC free article] [PubMed] [Google Scholar]
- 20.Kuhn M., Wing J., Weston S., Williams A., Keefer K., Engelhardt A., Cooper T., Mayer Z., Kenkel B., Benesty M., Lescarbeau R., Ziem A., Scrucca L., Tang Y., and Candan C. (2015) caret: Classification and regression training. R package version 6.0–58 [Google Scholar]
- 21.Jafari R., Almqvist H., Axelsson H., Ignatushchenko M., Lundbäck T., Nordlund P., and Martinez Molina D. (2014) The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 9, 2100–2122 [DOI] [PubMed] [Google Scholar]
- 22.Bloom J., Amador V., Bartolini F., DeMartino G., and Pagano M. (2003) Proteasome-mediated degradation of p21 via N-terminal ubiquitinylation. Cell 115, 71–82 [DOI] [PubMed] [Google Scholar]
- 23.Choudhary G. S., Tat T. T., Misra S., Hill B. T., Smith M. R., Almasan A., and Mazumder S. (2015) Cyclin E/Cdk2-dependent phosphorylation of Mcl-1 determines its stability and cellular sensitivity to BH3 mimetics. Oncotarget 6, 16912–16925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larance M., Ahmad Y., Kirkwood K. J., Ly T., and Lamond A. I. (2013) Global subcellular characterization of protein degradation using quantitative proteomics. Mol. Cell. Proteomics 12, 638–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song W., Wang Y., Yu Z., Vera C. I., Qu J., and Lin Q. (2010) A metabolic alkene reporter for spatiotemporally controlled imaging of newly synthesized proteins in Mammalian cells. ACS Chem. Biol. 5, 875–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindqvist L., and Pelletier J. (2009) Inhibitors of translation initiation as cancer therapeutics. Future Medicinal Chem. 1, 1709–1722 [DOI] [PubMed] [Google Scholar]
- 27.Ramos-Gomez M., Kwak M. K., Dolan P. M., Itoh K., Yamamoto M., Talalay P., and Kensler T. W. (2001) Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 98, 3410–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu C., Huang M. T., Shen G., Yuan X., Lin W., Khor T. O., Conney A. H., and Kong A. N. (2006) Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res. 66, 8293–8296 [DOI] [PubMed] [Google Scholar]
- 29.Cheung K. L., Lee J. H., Khor T. O., Wu T. Y., Li G. X., Chan J., Yang C. S., and Kong A. N. (2014) Nrf2 knockout enhances intestinal tumorigenesis in Apc(min/+) mice due to attenuation of anti-oxidative stress pathway while potentiates inflammation. Mol. Carcinog. 53, 77–84 [DOI] [PubMed] [Google Scholar]
- 30.Magesh S., Chen Y., and Hu L. (2012) Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med. Res. Rev. 32, 687–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang D. D., and Hannink M. (2003) Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 23, 8137–8151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang Z. Y., Lu M. C., Xu L. L., Yang T. T., Xi M. Y., Xu X. L., Guo X. K., Zhang X. J., You Q. D., and Sun H. P. (2014) Discovery of potent Keap1-Nrf2 protein–protein interaction inhibitor based on molecular binding determinants analysis. J. Med. Chem. 57, 2736–2745 [DOI] [PubMed] [Google Scholar]
- 33.Hu L., Magesh S., Chen L., Wang L., Lewis T. A., Chen Y., Khodier C., Inoyama D., Beamer L. J., Emge T. J., Shen J., Kerrigan J. E., Kong A. N., Dandapani S., Palmer M., Schreiber S. L., and Munoz B. (2013) Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg. Med. Chem. Lett. 23, 3039–3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leinonen H. M., Kansanen E., Pölönen P., Heinaniemi M., and Levonen A. L. (2014) Role of the keap1-nrf2 pathway in cancer. Adv. Cancer Res. 122, 281–320 [DOI] [PubMed] [Google Scholar]
- 35.Tarumoto T., Nagai T., Ohmine K., Miyoshi T., Nakamura M., Kondo T., Mitsugi K., Nakano S., Muroi K., Komatsu N., and Ozawa K. (2004) Ascorbic acid restores sensitivity to imatinib via suppression of Nrf2-dependent gene expression in the imatinib-resistant cell line. Exp. Hematol. 32, 375–381 [DOI] [PubMed] [Google Scholar]
- 36.Tang X., Wang H., Fan L., Wu X., Xin A., Ren H., and Wang X. J. (2011) Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs. Free Radical Biol. Med. 50, 1599–1609 [DOI] [PubMed] [Google Scholar]
- 37.Wang X. J., Hayes J. D., Henderson C. J., and Wolf C. R. (2007) Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc. Natl. Acad. Sci. U.S.A. 104, 19589–19594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tao S., Wang S., Moghaddam S. J., Ooi A., Chapman E., Wong P. K., and Zhang D. D. (2014) Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 74, 7430–7441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Willingham W. Jr., Stafford E. A., Reynolds S. H., Chaney S. G., Lee K. H., Okano M., and Hall I. H. (1981) Mechanism of eukaryotic protein synthesis inhibition by brusatol. Biochim. Biophys. Acta 654, 169–174 [DOI] [PubMed] [Google Scholar]
- 40.Liou Y. F., Hall I. H., Okano M., Lee K. H., and Chaney S. G. (1982) Antitumor agents XLVIII: Structure-activity relationships of quassinoids as in vitro protein synthesis inhibitors of P-388 lymphocytic leukemia tumor cell metabolism. J. Pharma. Sci. 71, 430–435 [DOI] [PubMed] [Google Scholar]
- 41.Tan B. X., Brown C. J., Ferrer F. J., Yuen T. Y., Quah S. T., Chan B. H., Jansson A. E., Teo H. L., Nordlund P., and Lane D. P. (2015) Assessing the efficacy of Mdm2/Mdm4-inhibiting stapled peptides using cellular thermal shift assays. Sci. Reports 5, 12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Griffith J. P., Kim J. L., Kim E. E., Sintchak M. D., Thomson J. A., Fitzgibbon M. J., Fleming M. A., Caron P. R., Hsiao K., and Navia M. A. (1995) X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell 82, 507–522 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.