

Abstract
For historical reasons, most proteomics workflows focus on MS/MS identification but consider quantification as the end point of a comparative study. The stochastic data-dependent MS/MS acquisition (DDA) gives low reproducibility of peptide identifications from one run to another, which inevitably results in problems with missing values when quantifying the same peptide across a series of label-free experiments. However, the signal from the molecular ion is almost always present among the MS1 spectra. Contrary to what is frequently claimed, missing values do not have to be an intrinsic problem of DDA approaches that perform quantification at the MS1 level. The challenge is to perform sound peptide identity propagation across multiple high-resolution LC-MS/MS experiments, from runs with MS/MS-based identifications to runs where such information is absent. Here, we present a new analytical workflow DeMix-Q (https://github.com/userbz/DeMix-Q), which performs such propagation that recovers missing values reliably by using a novel scoring scheme for quality control. Compared with traditional workflows for DDA as well as previous DIA studies, DeMix-Q achieves deeper proteome coverage, fewer missing values, and lower quantification variance on a benchmark dataset. This quantification-centered workflow also enables flexible and robust proteome characterization based on covariation of peptide abundances.
Label-free quantification (LFQ) is one of the most efficient approaches for quantifying proteome differences between multiple states of a biological system. LFQ aims to reproducibly identify and quantify peptides through multiple liquid-chromatography-coupled tandem mass spectrometry (LC-MS/MS) experiments. In the popular data-dependent acquisition (DDA) approach named Top-N DDA, the appearance of a peptide-like signal in a “survey” mass spectrum triggers a tandem mass spectrometry (MS/MS) event, targeting the (N) most-abundant precursor ions. Previous studies have shown that, due to the limited speed of a mass spectrometer, the majority of peptide ions detected in MS1 are not targeted in MS/MS, especially when a nonfractionated complex sample is analyzed (1, 2). This low sampling efficiency (<50%), combined with the stochastic nature of precursor selection and a limited efficiency of MS/MS identification (<70%) (3), frequently causes the absence of MS/MS identification for an individual peptide in some LC-MS/MS experiments (“runs”) within a larger dataset, even when replicate measurements are made (4). This deficiency is known as the missing value problem in LFQ. The problem significantly limits the size of the DDA-acquired proteomics dataset across which reliable quantification can be made for each protein (5, 6).
One of the causes of the missing value problem is the traditional focus on the process of identifying a peptide as opposed to its quantification. For historical reasons, peptide sequence identification has been considered the focal point and the most important step in the whole proteomics procedure, while quantification came as almost an afterthought (7, 8). This dominant proteomics paradigm can be characterized as the identification-centered approach, also known as a spectrum-centric approach (9). Only gradually the missing value problem has been identified as one of the biggest drawbacks of the DDA approach (4, 5). To address the reproducibility issue in MS/MS identification, several alternative data acquisition strategies had been suggested, including targeted (10) and semi-targeted (11, 12) approaches. However, none of the improved DDA strategies has solved the missing value problem anywhere close to the data-independent acquisition (DIA) (13, 14). The latter approach, however, typically provides somewhat lower depth and breadth of the proteome coverage than the DDA methods.
In our opinion, the DDA-associated missing value problem is caused by the sequential execution of two independent processes: peptide identification by MS/MS and its quantification by MS1. At first glance, performing MS1-based quantification simultaneously with MS/MS identification should provide an obvious solution to the missing value problem. Since MS1 spectra contain many more peptide ions than are selected for MS/MS in DDA (or identified in DIA), the peptide's mass information is practically always present when an identification is available (15). This information comes in several domains: accurate position on the m/z scale of the monoisotopic and other isotopic mass peaks and the position and the abundance profile on the retention time scale of the extracted ion trace for the above peaks, as well as the charge-state distribution of the analyte ions. Using this information, one can, at least in principle, identify the peptide ion even when MS/MS data are of low quality (16) or entirely missing (17).
In practice, during the last decade, features including monoisotopic m/z and retention time have been used for peptide identification, a strategy referred to as accurate mass and time tags (18–20). The accurate mass and time tag performs the feat of “peptide identity propagation” (PIP) from the LC-MS/MS runs with valid MS/MS information to those runs where such information is lacking. Today, one or another variant of the accurate mass and time tag-based PIP is employed in many MS1-based LFQ algorithms for analyzing DDA data (6, 21). MS feature matching (22, 23) and targeted extraction of ion chromatograms (XIC) (24, 25) represent examples of such variants. Although these algorithms are not free from certain generic drawbacks (26), they allow large-scale comparison of DDA-analyzed complex proteomes (23, 27, 28).
However, conventional PIP approaches only alleviate, but do not fully solve, the missing value problem. Given large enough sample cohorts and a set of analyzed peptides, we will encounter missing values. One might consider DIA as an alternative, but the process of signal extraction from DIA data is not fundamentally different from PIP. Therefore, while DIA reduces the occurrence of missing values, it is not completely absent in such data (29). It is, however, much more pronounced with DDA, regardless of which PIP procedure is used. On the other hand, DDA tends to identify more peptides and give deeper proteome coverage from the same sample than DIA, which is easy to understand, given the burden of peptide identification in DIA from severely convoluted data (2). When the size of comparative proteomics datasets becomes larger, the impact of the missing values becomes progressively worse. Resorting to imputations (i.e. qualified guesses) (30) cannot be considered satisfactory unless no other approaches are available.
Is all information stored in survey (MS1) mass spectra fully recovered by conventional PIP algorithms? It appears that some information domains have not yet been fully used, especially the peptide abundances. This fact reflects today's dominance of the identification-centered approach to proteomics. It has become a natural part of every modern proteomics study to report the false discovery rate (FDR) of its lists of identifications, but the discussion of coefficient of variation (CV)1 or even the FDR in peptide quantification—essential features of any quantification workflow—is still conspicuously missing in many current studies. An unfortunate consequence of such a miss is that sometimes MS/MS-inferred peptides with vastly deviating abundances in run-to-run or sample-to-sample comparisons are attributed to the same protein. These deviating peptides with questionable identities can drastically worsen the variances of protein abundances in the whole dataset and thus reduce the statistical power of the experiment (31).
In contrast, in a quantification-centered approach, peptide abundance is the central factor to be investigated (9, 15, 32–34). When peptide abundance is considered together with other parameters, such as RT difference and mass error, for the overall assessment of peptide reliability, deviating abundance behavior may result in exclusion of a given peptide from consideration (35). An expected abundance behavior should, on the other hand, strengthen the certainty of peptide identity. In other words, using only “well-behaved” peptides should enhance the quality of protein quantification by improving certainty in peptide identification and reducing the abundance variance, i.e. CV. The challenge is in inclusion in the quantitative assessment of the “goodness of behavior” of peptide abundances into the overall PIP scoring scheme. In this study, we will meet this challenge by introducing a new quantification-centered label-free workflow, DeMix-Q. It represents a LFQ-extension of the previously developed DeMix identification workflow designed for maximizing proteome coverage by identifying co-fragmented peptides (2). But in principle, DeMix-Q does not require DeMix for peptide identification and is compatible with any other peptide identification methods. Besides reducing quantification variations, DeMix-Q aims to significantly alleviate, if not eliminate, the missing value problem in comparative studies of many complex proteomes, while preserving the DDA advantage of higher proteome coverage. This is achieved by introducing a hybrid PIP method with a scoring function for quality control, which takes into account deviations from RT and m/z, as well as peptide abundance. For testing the new workflow, we selected the iPRG-2015 dataset (36) as an easily accessible, well-characterized and high-quality reference.
EXPERIMENTAL PROCEDURES
Preprocessing and MS/MS Identification
Raw LC-MS/MS data and the protein database were downloaded from the FTP server (ftp://iprg_study:ABRF329@ftp.peptideatlas.org/) of iPRG-2015 study (36). In the study, three replicates of four samples with the same amount (200 ng) of yeast digest were spiked with different concentrations of six exogenous marker proteins (Supplemental Table 1). The mixtures were digested by trypsin and analyzed by LC-MS/MS using an Orbitrap Q-Exactive mass spectrometer selecting the MS/MS precursors in the Top-10 DDA mode. Our DeMix workflow deconvoluted chimeric MS/MS spectra from the detected cofragmentation events for maximizing peptide identifications (2). The MS-GF+ (37) search engine (v10089) was used for matching the MS/MS spectra against the yeast database (6628 UniProt protein sequences), allowing up to two missed tryptic cleavage sites. Carbamidomethylation of Cys was set as a fixed modification, while acetylation of protein N terminus, oxidation of Met, and deamidation of Asn/Gln were set as variable modifications. A double-pass searching strategy was implemented. From the first-pass searching (10 ppm precursor tolerance), confident MS/MS identifications (<1% FDR) were used as software lock-masses for mass scale recalibration and mass error estimation. IDPicker (v3.1.643) (38) was used to merge the second-pass identifications (.mzid files) at maximum 1% peptide-spectral match FDR and with minimum two distinct peptides for protein inference. COMPASS (v. 1.0.4.5) (39) was used to assign peptide sequences to protein groups using the principle of maximum parsimony.
Retention Time and Mass Scale Recalibration
Reliable peptide-spectral matches were converted to OpenMS-compatible format and employed for aligning multiple LC-MS/MS experiments using MapAlignerIdentification from the OpenMS proteomics pipeline (TOPP) (40, 41). One individual run that gave the largest number of peptide identifications was chosen as a reference. The RT scales in all other runs were transformed to the scale of the reference run by aligning common peptide identifications. Next, using InternalCalibration with 5 ppm mass tolerance, the mass scale in each experiment was recalibrated to theoretical peptide masses. As a result, a new set of mzML files containing only MS1 (full-range) spectra was generated, in which the scales of RT and m/z for all runs were very similar. This processed dataset was then used as the basis for all following procedures (Fig. 1).
Fig. 1.

Overview of DeMix-Q data processing workflow. Processes are colored in blue for TOPP, yellow for the search engine, and green for the postprocessing programs developed in-house. Internal processes are highly flexible and can be replaced by alternative tools or simply be skipped. In the latter case, DeMix-Q may become a traditional feature-, XIC-, or MS/MS-based quantification workflow. Note that MS1-based quantification procedures, including feature detection and between-run propagation, are independent of peptide identification and can even be done in the absence of MS/MS.
Feature Detection and Matching Across Runs (Feature-Based PIP)
Here, an LC-MS feature can be defined as a peptide-like XIC pattern assembled from a cluster of raw MS peaks (41, 42). Each feature has information of its RT and m/z coordinates, as well as its integrated ion-current and charge state. Recalibrated MS1 spectra were loaded into FeatureFinderCentroided in TOPP for assembling chromatographic feature maps (m/z tolerance 0.01 Da, min spectra 5, feature min score 0.6). All features listed in the feature maps were de facto quantified independently and reported with integrated ion-current. Features were tentatively associated to peptide sequences using available MS/MS identifications. Afterward, the FeatureLinkerUnlabeledQT pipeline in TOPP was used for grouping features by similarity with a user-defined quality threshold. Here, we used RT difference <180 s and mass difference < 5 ppm. This process served as feature-based identity propagation. As a result, features that matched across multiple experiments were linked into a single consensus map (a.k.a. reference map or master map) (40). In this consensus map, each consensus feature contained at least one subelement (the best-matched feature from a single run), with reference information on RT (centroid of the feature chromatographic shape), m/z, and integrated ion-current (called “intensity” in OpenMS). A fully quantified consensus feature contains the maximum possible number of subelements that equals to the total number of LC-MS runs; otherwise, the consensus feature contains missing values in one or more runs.
Recovering Missing Values by Targeted XIC (Ion-Based PIP) with Quality Control
Ion-based PIP considers only the existence of molecular ions in a given (RT ± ΔRT, m/z ± Δm/z) window, regardless of the chromatographic peak shape, its abundance, and the isotopic pattern. Therefore, ion-based PIP is more sensitive than the feature-based method. However, this advantage comes at the price of lower reliability, and thus future-based approaches are normally preferred. Here, ion-based PIP was used only for recovering remaining missing values after feature-based PIP (i.e. the consensus feature map). For each consensus feature, local maxima (apexes) of the monoisotopic peaks (M) and the 13C isotopic peaks (M + 1) were extracted in each individual run using EICExtractor of TOPP, with a matching window of RT ± 1 min and m/z ± 5 ppm around the consensus reference location (Fig. 2A). Since both RT and m/z were recalibrated and aligned, such a narrow matching window did not result in substantial loss of useful data (false negatives). The geometric average of ion intensities of the two peaks () was used as an estimate of the feature abundance in the corresponding run. This calculation served as a quality checkpoint, which required both isotopic peaks to be traced with nonzero intensities.
Fig. 2.

Scoring the feature-quantity assignments. (A) XIC and deviation factors. For each reference feature in every individual run, the precursor ion is traced in MS1 spectra. Within a matching window of RT ±1 min and m/z ±5 ppm, the ion with maximum intensity is picked. By comparing the apex of its monoisotopic peak with the consensus reference RT and m/z, the deviation factors ΔT1 and ΔM1 are obtained. Comparison to the apex of the monoisotopic peak with the apex of the 13C isotopic peak gives the deviation factors ΔT2 and ΔM2. The geometric average of ion intensities I1 and I2 represents the feature quantity, which is used to calculate the CV between the replicate runs. Five deviation factors are combined by the scoring function in Equation (1). (B) Score distribution and target-decoy comparison for FDR estimation. Consensus features linked across all runs by OpenMS are considered to be reliable “target” features. “Decoy” features are generated by arbitrarily shifting the target features' RT and m/z values. Any XIC extracted for a “decoy” feature is assumed to be false. In each individual run, the number of decoy hits does not exceed 5% of target hits, corresponding to FDR of <5%. This threshold is applied to feature-quantity assignments in the process of missing value recovery.
Central to the DeMix-Q approach, based on extracted m/z, RT, and ion intensities of the two isotopic MS1 peaks (M and M+1), a scoring scheme was established and applied to every feature in every LC-MS run, combining five deviation factors (Fig. 2A):
- ΔT1, the RT difference between the consensus feature and the apex of the monoisotopic peak (M);
- ΔT2, the RT difference between the two isotopic apexes (M and M+1);
- ΔM1, the deviation of the monoisotopic peak (M) from its theoretical mass;
- ΔM2, the difference of mass deviations between the two isotopic peaks (M and M+1);
- CV of extracted abundances in replicate runs.
Deviations (ΔT1 and ΔM1) between the consensus feature and the extracted monoisotopic peak reflect between-run variances. Relative deviations (ΔT2 and ΔM2) between the two isotopic peaks indicate within-run inconsistencies, with an assumption that the extracted 13C isotopic peak (M+1) should have the same deviations as the monoisotopic peak (M). The last factor (CV) penalizes the features that were not reliably quantified in the replicate runs. Since these five factors have different units and intervals of changes, they were all normalized by their own standard deviations and thus converted to unitless quantities that can be simply combined. One overall score function combining the five deviation factors was formulated as a negative logarithm of pooled variances, with larger variation resulting in a lower score:
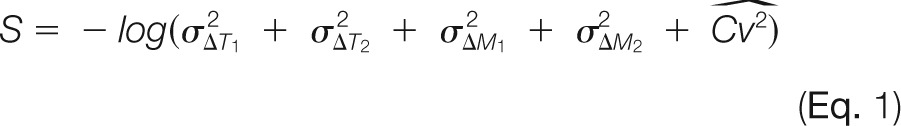 |
Here, the five deviation factors were assumed having equal weights for the reason of simplicity. However, one could imagine expansions of this work that assign weights that are optimal by some criteria using more rigorous statistical methods or machine-learning techniques, e.g. Percolator (43).
A target-decoy method was then applied to estimate the false discovery rate (FDR) for a given scoring cutoff. Assuming that complete features grouped by the FeatureLinkerUnlabeledQT are most statistically reliable, a reference feature set (“target”) was generated by selecting all fully quantified features with the maximum number of subelements. A false feature set (“decoy”) was generated from the target set by shifting each target feature outside its original extraction window through alteration of the retention time (+5 min) and m/z (+50 ppm), with small random noise being added. The score distribution of the decoy features formed a null-score distribution (Fig. 2B). FDR was then estimated as the fraction of all assignments that passed a given score threshold: The number of decoy matches was divided by the total number of target matches. In this study, a 5% FDR threshold was applied for XIC-feature assignments. The features that failed to pass the threshold were considered missing (zero-intensity).
Special treatment was then given to “gray zone” features, which were incompletely quantified by feature-based PIP and yet most likely present in ion-based PIP. For such features (subelements), missing abundances were estimated from a k-nearest neighbors (KNN) regression (k = 5), averaging abundances from other quantified features having the most similar XIC patterns in the same run. After filling missing values, the consensus map was further filtered by removing features that failed to be quantified in at least one replicate in each sample. Lastly, the remaining missing values were imputed as having the lowest detectable feature abundance in order to avoid extreme ratios (or divisions by zero) in sample-to-sample comparison. This approach explicitly assumes that every protein is present in every sample. Although this assumption is demonstrably untrue in case of spike-in or knock-down, it provides useful approximation in a great majority of proteomics studies.
Intensity Recalibration
Interrun (batch effects) and intrarun systematic biases (e.g. due to sample loading, column temperature, ESI current stability, etc.) can greatly affect analytical accuracy in label-free experiments (21, 44, 45). In order to correct fold-changes induced by systematic biases, a rescaling of feature abundances was performed, using a time-dependent median-shift approach. Chromatographic features from the consensus map were sorted by the retention order, then a sliding window (step size = 50 features) containing 500 adjacent consensus features was used to compare the local median abundance of all features with those of the subset of features from each individual run (one-versus-all). This yielded a set of local median shifts for each run, based on which, a nonlinear median-shift was estimated as a function of retention time by another k-nearest neighbors regression (k = 15). The abundance of every feature in every single run was normalized by correcting the predicted median shift (Fig. 3 and Supplementary Fig.).
Fig. 3.

Intensity recalibration by correcting median shift. In diagonal subfigures, nonlinear fluctuations of ion intensity in individual runs are reflected in the RT-dependent median-shift curves. The lower-left part shows systematic errors in between-run comparisons of feature abundances, before correction. The upper-right part shows the effect of median-shift correction, where the between-run abundance differences (fold-changes) become approximately zero-centered along the whole RT range. Each subfigure has normalized RT (0–8000 s) as x axis and feature abundance ratio (–2.0 to 2.0 in log2 scale) as y axis. Correction of three replicate runs of one sample is demonstrated; the comparisons between all 12 runs are shown in Supplementary Fig.
Detecting and Quantifying Differential Proteins
Since most protein groups have multiple peptides quantified in all runs, quantitative proteomic data bear similarity with gene expression microarray data, with peptide abundances being equivalent to probe intensities. Like microarray probes, peptide abundances quantified by LFQ are supposed to reflect the concentrations of their respective proteins and have linear responses to abundance changes. In theory, if one protein has an abundance difference between the samples, all its constituent peptides should show the same level of abundance difference, giving rise to strong covariation between the abundances of all peptides. Thus, a strong covariation of a peptide abundance with that of other same-protein peptides means “well-behaving” of a given peptide and higher certainty in both its identity and quantification.
In the literature, we found no LFQ algorithm that would measure and utilize peptide covariations. However, giving the basic similarity between proteomics and transcriptomics, tools developed for microarray analysis should also work for quantitative proteomics. In this study, an in-house implementation of factor analysis (Zhang et al., manuscript in preparation) was adapted for detecting differential proteins (i.e. proteins with abundances varying from sample to sample, as opposed to background proteins with unchanged abundances).
All identified features were grouped by peptide sequence, with summed abundances from all charge states. By applying the factor analysis to maximize covariation signals of peptides, a signal-to-noise ratio was obtained for a group of peptide “probes.” This parameter was used to decide whether a peptide group (as a protein) is “informative”: Proteins with signal-to-noise ratio < 1 were considered “noninformative” and thus excluded from differential analysis. Protein expression values summarized from peptide abundances were labeled by sample identities, and their sample-to-sample abundance changes were tested using one-way analysis of variance: Proteins with p values lower than 0.05 were reported as differential.
Comparison to Traditional Methods
MS/MS spectral counts (SpC) were exported from IDPicker after integrating reliable identifications. MaxQuant analysis was performed with default instrumental parameters and database setting. The option of match-between-runs was enabled for feature-based identity propagation, with 20 min alignment window and 2 min matching tolerance. Peptide abundances were retrieved from the resultant peptide.txt file (intensity column), which was filtered by 1% FDR based on posterior error probability (PEP) values (Supplemental Table 2). Skyline MS1-filtering (XIC) results provided in the iPRG-2015 study materials were processed as follows: Nonunique peptides were excluded from the analysis, and the abundances of the first two isotopic peaks (M and M+1) were utilized. The OpenMS results were exported right after generating the consensus map (feature-based PIP), without filling missing values by targeted XIC (ion-based PIP). For each method, peptide quantification variation (CV) was calculated using peptides that were quantified in all runs.
RESULTS
A Combined Identity Propagation Method Substantially Alleviates the Missing Value Problem
We compared missing values in peptide abundances quantified in the iPRG-2015 dataset by four common analytical methods as well as DeMix-Q. The four common methods can be grouped into three categories: MS/MS-based spectral counting (SpC) without identity propagation; MS1-level LFQ with feature-based PIP (OpenMS and MaxQuant), and with ion-based PIP (Skyline).
As shown in Fig. 4, SpC that does not employ identity propagation was greatly affected by the DDA randomness, yielding more than 40% missing values in peptide abundances. Only about one-fourth (26%) of all peptides could be found in all 12 LC-MS runs. The steeply declining trend of common quantifications with the dataset size makes SpC less suitable for deep proteome profiling in large-scale experiments. In contrast, OpenMS and MaxQuant that apply feature-based PIP reduced the fraction of missing values to smaller but still nonnegligible figures: 15% and 13%, respectively. The number of commonly quantified peptides increased to over 60%, i.e. more than double compared with SpC. Skyline that applies ion-based PIP without matching chromatographic features showed a very high sensitivity, providing negligible 1.5% missing values and over 90% peptides quantified in all runs. DeMix-Q that combines two complementary PIP approaches to achieve highly sensitive signal extraction together with reliable feature matching gave 2.8% missing values and quantified over 86% of peptides across all 12 runs (Fig. 4B).
Fig. 4.
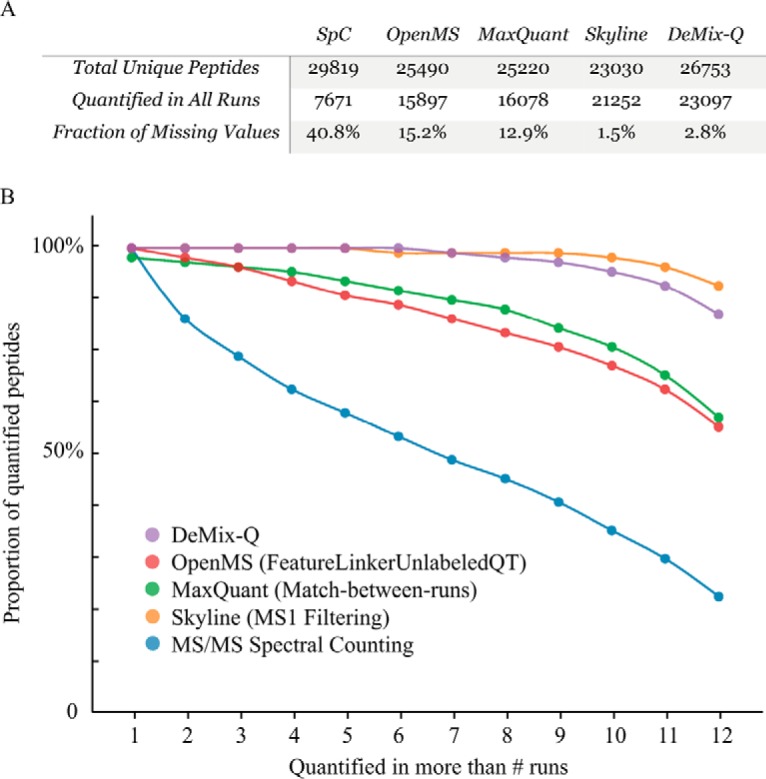
Comparison of five quantification approaches in terms of missing values. (A) Number of unique peptides and fraction of missing values in respective approach. (B) The fraction of peptides quantified in all runs drops as a function of sample size but with different rates for different quantification approaches.
Formally, Skyline (MS1 filtering) outperformed all other methods in terms of missing values. However, as mentioned before, ion-based PIP does not have a quality threshold, thus providing results with uncertain FDR. In contrast, DeMix-Q applies quality thresholds in feature-quantity assignments, which reflects in a somewhat higher fraction of missing values. This drawback should be outweighed by DeMix-Q superiority in quantification: Uncontrolled matching in Skyline may result in false assignment, thus introducing a large abundance of variations. This prediction was tested on the distributions of quantification variances discussed below.
A Quantification-Centered Workflow Provides High Coverage with Low Variance
DeMix-Q quantified in total 26,753 unique peptides representing 2912 proteins, with at least two distinct constituent peptides per protein (Supplemental Table 1). Notably, we found that MS/MS-based identifications explained only one-third (44,024/129,641) of the total chromatographic features that were quantified and matched across experiments (Fig. 5). This highlights the great potential of exploiting the hidden majority of quantified features. One way of using these features is for correcting the systematic bias in measured ion intensities, which is likely to be the largest source of peptide abundance variation. In this study, we applied a KNN regression of RT-dependent median shift to centralize the ion intensity variations around zero-fold change (Fig. 3 and Supplementary Fig.). As one can see from the regression curves, system biases are mostly nonlinear functions of RT. After normalization, all pairwise comparisons of feature abundances between runs showed zero-centered distributions. By correcting the systematic bias, we obtained significantly lower quantification variance compared with other quantification methods. From DeMix-Q, the median CV calculated for 23,097 fully quantified peptides in all 12 runs was 11.6%, distinctively lower than in other methods (Fig. 6). When using the average values from three replicate experiments for each sample, the median CV of peptide abundances among the four samples (background proteins) was only 6.0%.
Fig. 5.
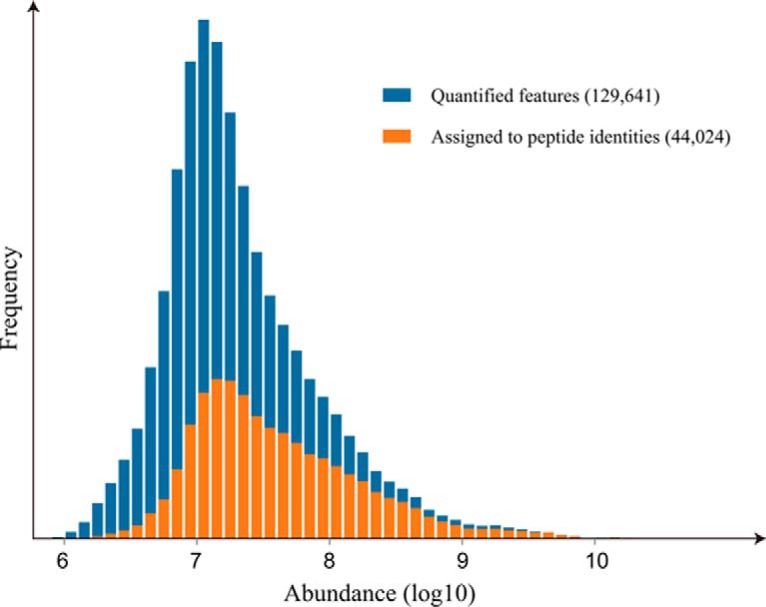
Abundance distribution of chromatographic features. Only around one-third of features quantified in DeMix-Q were assigned to MS/MS-based peptide identifications, which is in accordance with the fact that most features were not targeted by DDA and identified by MS/MS but were recorded by full-range MS. Features with higher abundances have higher identification rate, which also reflects the intensity-dependent bias of MS/MS data acquisition.
Fig. 6.
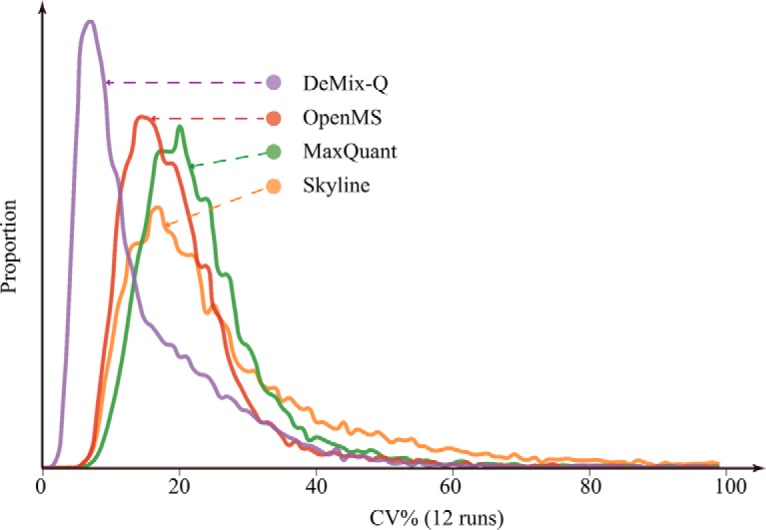
Comparison of CV distributions. DeMix-Q provided distinctively lower median CV than other methods, while quantifying the largest number of peptides across all runs. This is achieved primarily due to the introduction of scoring and FDR filtering that prevented large variances from false extractions, and the RT-dependent abundance correction that reduced systematic variability.
In contrast to that, peptides abundance quantified by Skyline showed higher CVs on average and a long tail of peptides with CV > 40% (Fig. 6). As mentioned above, direct XIC methods do not ensure the correctness of signal extraction. Wrongly extracted XIC would introduce large variances in peptide quantification and lead to a long tail in the CV distribution.
Accurate and Robust Protein Quantification
So far, we performed the peptide-level quantification by aggregating precursors' ion currents. However, the estimation of protein abundances is not as easy as aggregating peptide abundances. It is widely known—although not often discussed—that peptides originating from the same protein can give vastly different abundances in LC-MS, varying orders of magnitude. Moreover, it is less than certain that the LC-MS signal of all peptides scales linearly and has the same slope with protein abundance variation. As a result, it becomes problematic to reliably estimate relative protein abundance by simply averaging or aggregating abundances of the multitude of peptides attributed to that protein. This is because the abundance variances of a few intensive peptides may significantly affect the result, suppressing the signals from other peptides. Therefore, wrong identity assignment of intensive chromatographic features poses higher risks for quantification than that of less intensive ones. For this reason, an identification-centered approach has a stringent requirement of identity correctness (46), but a quantification-centered method should be able to cope with possible misidentification using quantitative information (35).
As an example, Fig. 7 shows the quantitative behavior of peptides (top-10 and bottom-10, respectively) from the spiked-in protein bovine serum albumin (sp P02769 ALBU_BOVIN). Although peptides were reproducibly quantified across the runs, some peptides (mostly with low abundance) did not reflect the known difference of protein concentrations (11: 0.6: 10: 500). In particular, the seventh highest-abundance peptide (LGEYGFQNALIVR) showed a strong deviation from both known protein concentrations as well as behavior of other peptides. As a rule, low-abundant peptides showed a smaller dynamic range of abundance differences compared with known values and more-abundant peptides. While the exact nature of such behavior remains unclear and should be thoroughly investigated, it was more likely due to instrumental effects rather than inaccurate data processing.
Fig. 7.

Responses from peptide abundances to actual protein concentration differences. The abundances of the 10 most-abundant peptides from a spiked-in bovine serum albumin (left) well correlated with the known protein concentrations, except for the outlier peptide (blue). However, the least-abundant 10 peptides (right) turned out to be less responsive to the actual protein concentration changes. The expected abundances (dark gray lines) were calculated: for sample 4 (that has the highest spike-in amount, 500 fmol)—as the average log2 peptide abundances (33.8 for top-10 and 28.5 for bottom-10 peptides); for samples 1, 2, and 3—as the corresponding log2 fold-changes compared with sample 4 (–5.5, –9.7, and –5.64, respectively).
Considering this example. It is clear that any robust protein quantification algorithm should be able to cope with a fraction of incorrect identifications, as well as with differences in peptide signal responses to the protein abundance changes. One approach would be to design such an algorithm based on specifics (not thoroughly known yet) of the peptide responses in LC-MS. Another, perhaps more pragmatic, approach would be to borrow an existing tried-and-proved algorithm from a related research area. Since transcriptomics is at least a decade older than proteomics, the problem of inconsistences of probe signals was addressed in microarray analysis workflows some time ago (47). With more than nine peptides quantified per protein group on average and with the benefit of solving the missing value problem for a great majority of peptides, our data mimicked a “protein microarray,” with peptide abundances posing as probe signal intensities.
Applying the factor analysis and analysis of variance for selecting proteins with varying abundances between the samples, we discovered all six spiked-in proteins with high certainty. In contrast, the same procedure applied to quantification results from three other PIP-based algorithms missed one or more proteins (Fig. 8A and Supplemental Table 1), while also (for Open-MS) yielding more false positives. After protein summarization, the sample-to-sample protein ratios showed a linear correlation with expected values up to over ninefold abundance difference (Fig. 8B).
Fig. 8.

Differential protein detection and quantification. (A) Signal-to-noise ratios of calling differential proteins by factor analysis. DeMix-Q showed high sensitivity and specificity for identifying the six spiked marker proteins (signal-to-noise ratio > 1 and one-way analysis of variance p value < 0.05). (*) Using nonredundant MS/MS dataset, all six proteins were identified by DeMix-Q as differential. (B) Estimation of protein abundance ratios. Abundances of the six proteins were pairwise compared between the four samples (color coded). The quantification accuracy of DeMix-Q is reflected in the good agreement between the estimated and expected fold-changes ranging from 0.14 to 9.7.
One could hypothesize that sensitive and reliable identity propagation may eliminate the need for redundancy in MS/MS identifications. We tested this hypothesis by simulating a sparse dataset, keeping MS/MS from only one of the 12 runs and eliminating all redundant identifications. Despite certain reduction in the total number of quantified proteins due to DeMix-Q quality control, all six spiked proteins were still correctly recalled (Fig. 8A). This result more than any previously discussed one demonstrates the possibility of a paradigm change from identification-centered to quantification-centered proteomics.
DISCUSSION
We demonstrated in this study a new quantification-centered workflow for label-free proteomics that practically solves the DDA-induced missing value problem that haunted shotgun proteomics for years. By taking advantage of the high quality of modern LC-MS/MS, we integrated two types of PIP methods into the workflow. This enabled unbiased and reproducible quantification across many runs even when only a single set of MS/MS identification was available. This workflow is particularly suitable for DDA, where quantified features compose a much larger population than MS/MS-identified species.
The demonstration of the “redundancy of redundant MS/MS peptide identifications” highlighted the potential of PIP in reliable quantification. This result may herald a new strategy in label-free shotgun proteomics, emphasizing the acquisition of higher-quality MS1 spectra and deeper proteome coverage rather than larger number of redundant MS/MS spectra. One way of exploring this new strategy is by segmenting the mass range for precursor selection (48); another one is by applying the exclusion list aggregated from previous runs to subsequent runs.
In some respects DeMix-Q is similar to a sequential window acquisition of all theoretical fragment-ion spectra-MS processing workflow but propagates MS1 data instead of MS/MS information. Similar ideas have been presented at the American Society for Mass Spectrometry 2015 conference by Shen et al. (ThP449) and by Reiter et al. (Wednesday oral section B am 08:30), which indicates that the time for a change is ripe. Shen et al. also claimed solving the missing value problem by separating the procedure of MS1-based quantification from MS/MS identification, while Reiter et al. demonstrated a robust MS1-only approach for whole-proteome analysis in which the peptide identities are purely inferred from a predicted accurate mass and time tag library. Regardless of the similarities and differences between these approaches and DeMix-Q, they all are based on the three fundamental quality factors essential for reliable analysis: stable retention time, high resolution, and high mass accuracy.
Contrary to common belief, proteome analysis applying DIA (e.g. sequential window acquisition of all theoretical fragment-ion spectra) is not free from the missing value problem. A recent study (29) quantified 80% of the 18,600 yeast peptides belonging to 2333 proteins (including one-hit wonders) in four sequential window acquisition of all theoretical fragment-ion spectra-MS replicate runs. Compared with 86% of 26,753 peptides we quantified here in 12 runs, the DIA study did not demonstrate any advantage. In our case, DDA required less experimental time and produced both deeper proteomics analysis as well as fewer missing values. Theoretically speaking, this is not too surprising given the lower burden on reliable peptide identification and chromatographic feature extraction with DDA. Compared with the typical 2 Th isolation window in DDA, a highly multiplexed 20 Th window used in DIA penalizes identification of low-abundant peptides that give fewer fragment peaks (2, 9). The intrinsic advantage of DDA has, however, not yet been fully realized in practice, mainly because of the missing value problem. Releasing the full power of MS1 in a quantification-centered data processing workflow should lead to deeper and more accurate label-free proteomics. In principle, DeMix-Q should not be limited to DDA data and could also be adapted to solve the missing value problem in DIA analysis, e.g. by propagating identities of peptide fragments instead of precursors. However, due to the increased spectral complexity and the loss of precursor selectivity, identity propagation in DIA might require a more sophisticated scoring scheme for controlling false discoveries.
Finally, we strongly believe that time has come to put quantification in the center of comparative proteomics workflow. Unlike the traditional identification-centered proteomics that is very strict in terms of peptide identification but has a somewhat lax attitude to peptide quantification, new protein quantification algorithms should be able to tolerate incorrect peptide identifications because further abundance-based filtering will eliminate wrong assignments. This may allow for the revision of certain dogmas of identification-centered proteomics, such as 1% FDR for peptide-spectral matches. A deeper understanding of the error propagation in mass spectrometry experiments might allow for more flexible treatment of error rates (49), which could have a significant positive effect on the depth of the quantitative proteome analysis. More advanced quantification algorithms that will take into account the variance and covariance of peptide and protein abundance in multiple experiments urgently need to be developed. Given the data similarity between shotgun proteomics and gene expression microarrays, we can learn much from the more mature area of transcriptomics by microarrays. But of course, sooner or later, superior proteomics-specific algorithms will be developed.
Supplementary Material
Footnotes
Author contributions: B.Z. designed the research; B.Z. performed the research; B.Z. and L.K. contributed new reagents or analytic tools; B.Z. analyzed data; and B.Z. and R.A.Z. wrote the paper.
* This research was funded by Knut and Alice Wallenberg Foundation (Grant KAW 2010.0022 to RZ).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- CV
- coefficient of variation
- DDA
- data-dependent acquisition
- DIA
- data-independent acquisition
- EIC/XIC
- extracted ion chromatography
- FDR
- false discovery rate
- LC-MS/MS
- liquid chromatography coupled to tandem mass spectrometry
- LFQ
- label-free quantification
- MS1
- primary or survey mass spectrometry
- PIP
- peptide identity propagation
- TOPP
- The OpenMS Proteomics Pipeline.
REFERENCES
- 1.Michalski A., Cox J., and Mann M. (2011) More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J. Proteome Res. 10, 1785–1793 [DOI] [PubMed] [Google Scholar]
- 2.Zhang B., Pirmoradian M., Chernobrovkin A., and Zubarev R. A. (2014) DeMix workflow for efficient identification of cofragmented peptides in high resolution data-dependent tandem mass spectrometry. Mol. Cell. Proteomics 13, 3211–3223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pirmoradian M., Budamgunta H., Chingin K., Zhang B., Astorga-Wells J., and Zubarev R. A. (2013) Rapid and deep human proteome analysis by single-dimension shotgun proteomics. Mol. Cell. Proteomics 12, 3330–3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tabb D. L., Vega-Montoto L., Rudnick P. A., Variyath A. M., Ham A. J., Bunk D. M., Kilpatrick L. E., Billheimer D. D., Blackman R. K., Cardasis H. L., Carr S. A., Clauser K. R., Jaffe J. D., Kowalski K. A., Neubert T. A., Regnier F. E., Schilling B., Tegeler T. J., Wang M., Wang P., Whiteaker J. R., Zimmerman L. J., Fisher S. J., Gibson B. W., Kinsinger C. R., Mesri M., Rodriguez H., Stein S. E., Tempst P., Paulovich A. G., Liebler D. C., and Spiegelman C. (2010) Repeatability and reproducibility in proteomic identifications by liquid chromatography-tandem mass spectrometry. J. Proteome Res. 9, 761–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Domon B., and Aebersold R. (2010) Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721 [DOI] [PubMed] [Google Scholar]
- 6.Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., and Mann M. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marcotte E. M. (2007) How do shotgun proteomics algorithms identify proteins? Nat. Biotechnol. 25, 755–757 [DOI] [PubMed] [Google Scholar]
- 8.Nesvizhskii A. I., Vitek O., and Aebersold R. (2007) Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat. Methods 4, 787–797 [DOI] [PubMed] [Google Scholar]
- 9.Ting Y. S., Egertson J. D., Payne S. H., Kim S., MacLean B., Käll L., Aebersold R., Smith R. D., Noble W. S., and MacCoss M. J. (2015) Peptide-centric proteome analysis: An alternative strategy for the analysis of tandem mass spectrometry data. Mol. Cell. Proteomics 14, 2301–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peterson A. C., Russell J. D., Bailey D. J., Westphall M. S., and Coon J. J. (2012) Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteomics 11, 1475–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graumann J., Scheltema R. A., Zhang Y., Cox J., and Mann M. (2012) A framework for intelligent data acquisition and real-time database searching for shotgun proteomics. Mol. Cell. Proteomics 11, M111.013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bailey D. J., McDevitt M. T., Westphall M. S., Pagliarini D. J., and Coon J. J. (2014) Intelligent data acquisition blends targeted and discovery methods. J. Proteome Res. 13, 2152–2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gillet L. C., Navarro P., Tate S., Röst H., Selevsek N., Reiter L., Bonner R., and Aebersold R. (2012) Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11, O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egertson J. D., Kuehn A., Merrihew G. E., Bateman N. W., MacLean B. X., Ting Y. S., Canterbury J. D., Marsh D. M., Kellmann M., Zabrouskov V., Wu C. C., and MacCoss M. J. (2013) Multiplexed MS/MS for improved data-independent acquisition. Nat. Methods 10, 744–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.America A. H., and Cordewener J. H. (2008) Comparative LC-MS: A landscape of peaks and valleys. Proteomics 8, 731–749 [DOI] [PubMed] [Google Scholar]
- 16.The M., and Käll L. (2015) MaRaCluster: A fragment rarity metric for clustering fragment spectra in shotgun proteomics. J. Proteome Res. [DOI] [PubMed] [Google Scholar]
- 17.Mueller L. N., Brusniak M. Y., Mani D. R., and Aebersold R. (2008) An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J. Proteome Res. 7, 51–61 [DOI] [PubMed] [Google Scholar]
- 18.Smith R. D., Anderson G. A., Lipton M. S., Masselon C., Pasa-Tolic L., Shen Y., and Udseth H. R. (2002) Review: The use of accurate mass tags for high-throughput microbial proteomics. OMICS 6, 61–90 [DOI] [PubMed] [Google Scholar]
- 19.Pasa-Tolic L., Masselon C., Barry R. C., Shen Y., and Smith R. D. (2004) Proteomic analyses using an accurate mass and time tag strategy. BioTechniques 37, 621–624, 626–633, 636 passim [DOI] [PubMed] [Google Scholar]
- 20.Moruz L., Hoopmann M. R., Rosenlund M., Granholm V., Moritz R. L., and Käll L. (2013) Mass fingerprinting of complex mixtures: Protein inference from high-resolution peptide masses and predicted retention times. J. Proteome Res. 12, 5730–5741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyutvinskiy Y., Yang H., Rutishauser D., and Zubarev R. A. (2013) In silico instrumental response correction improves precision of label-free proteomics and accuracy of proteomics-based predictive models. Mol. Cell. Proteomics 12, 2324–2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaffe J. D., Mani D. R., Leptos K. C., Church G. M., Gillette M. A., and Carr S. A. (2006) PEPPeR, a platform for experimental proteomic pattern recognition. Mol. Cell. Proteomics 5, 1927–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geiger T., Wehner A., Schaab C., Cox J., and Mann M. (2012) Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol. Cell. Proteomics 11, M111.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schilling B., Rardin M. J., MacLean B. X., Zawadzka A. M., Frewen B. E., Cusack M. P., Sorensen D. J., Bereman M. S., Jing E., Wu C. C., Verdin E., Kahn C. R., Maccoss M. J., and Gibson B. W. (2012) Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: Application to protein acetylation and phosphorylation. Mol. Cell. Proteomics 11, 202–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bateman N. W., Goulding S. P., Shulman N. J., Gadok A. K., Szumlinski K. K., MacCoss M. J., and Wu C. C. (2014) Maximizing peptide identification events in proteomic workflows using data-dependent acquisition (DDA). Mol. Cell. Proteomics 13, 329–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith R., Ventura D., and Prince J. T. (2015) LC-MS alignment in theory and practice: A comprehensive algorithmic review. Brief Bioinform. 16, 104–117 [DOI] [PubMed] [Google Scholar]
- 27.Khan Z., Bloom J. S., Garcia B. A., Singh M., and Kruglyak L. (2009) Protein quantification across hundreds of experimental conditions. Proc. Natl. Acad. Sci. U.S.A. 106, 15544–15548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neilson K. A., Ali N. A., Muralidharan S., Mirzaei M., Mariani M., Assadourian G., Lee A., van Sluyter S. C., and Haynes P. A. (2011) Less label, more free: Approaches in label-free quantitative mass spectrometry. Proteomics 11, 535–553 [DOI] [PubMed] [Google Scholar]
- 29.Selevsek N., Chang C. Y., Gillet L. C., Navarro P., Bernhardt O. M., Reiter L., Cheng L. Y., Vitek O., and Aebersold R. (2015) Reproducible and consistent quantification of the Saccharomyces cerevisiae proteome by SWATH-mass spectrometry. Mol. Cell. Proteomics 14, 739–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Webb-Robertson B. J., Wiberg H. K., Matzke M. M., Brown J. N., Wang J., McDermott J. E., Smith R. D., Rodland K. D., Metz T. O., Pounds J. G., and Waters K. M. (2015) Review, evaluation, and discussion of the challenges of missing value imputation for mass spectrometry-based label-free global proteomics. J. Proteome Res. 14, 1993–2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clough T., Thaminy S., Ragg S., Aebersold R., and Vitek O. (2012) Statistical protein quantification and significance analysis in label-free LC-MS experiments with complex designs. BMC Bioinformatics 13, S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiener M. C., Sachs J. R., Deyanova E. G., and Yates N. A. (2004) Differential mass spectrometry: A label-free LC-MS method for finding significant differences in complex peptide and protein mixtures. Anal. Chem. 76, 6085–6096 [DOI] [PubMed] [Google Scholar]
- 33.Rinner O., Mueller L. N., Hubálek M., Muller M., Gstaiger M., and Aebersold R. (2007) An integrated mass spectrometric and computational framework for the analysis of protein interaction networks. Nat. Biotechnol. 25, 345–352 [DOI] [PubMed] [Google Scholar]
- 34.Finney G. L., Blackler A. R., Hoopmann M. R., Canterbury J. D., Wu C. C., and MacCoss M. J. (2008) Label-free comparative analysis of proteomics mixtures using chromatographic alignment of high-resolution muLC-MS data. Anal. Chem. 80, 961–971 [DOI] [PubMed] [Google Scholar]
- 35.Forshed J., Johansson H. J., Pernemalm M., Branca R. M., Sandberg A., and Lehtiö J. (2011) Enhanced information output from shotgun proteomics data by protein quantification and peptide quality control (PQPQ). Mol. Cell. Proteomics 10, M111.010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.ABRF (2015) iPRG 2015 Study http://www.abrf.org/research-group/proteome-informatics-research-group-iprg.
- 37.Kim S., and Pevzner P. A. (2014) MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma Z. Q., Dasari S., Chambers M. C., Litton M. D., Sobecki S. M., Zimmerman L. J., Halvey P. J., Schilling B., Drake P. M., Gibson B. W., and Tabb D. L. (2009) IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering. J. Proteome Res. 8, 3872–3881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wenger C. D., Phanstiel D. H., Lee M. V., Bailey D. J., and Coon J. J. (2011) COMPASS: A suite of pre- and post-search proteomics software tools for OMSSA. Proteomics 11, 1064–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weisser H., Nahnsen S., Grossmann J., Nilse L., Quandt A., Brauer H., Sturm M., Kenar E., Kohlbacher O., Aebersold R., and Malmström L. (2013) An automated pipeline for high-throughput label-free quantitative proteomics. J. Proteome Res. 12, 1628–1644 [DOI] [PubMed] [Google Scholar]
- 41.Kohlbacher O., Reinert K., Gröpl C., Lange E., Pfeifer N., Schulz-Trieglaff O., and Sturm M. (2007) TOPP–the OpenMS proteomics pipeline. Bioinformatics 23, e191–197 [DOI] [PubMed] [Google Scholar]
- 42.Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 43.Käll L., Canterbury J. D., Weston J., Noble W. S., and MacCoss M. J. (2007) Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 [DOI] [PubMed] [Google Scholar]
- 44.Rudnick P. A., Wang X., Yan X., Sedransk N., and Stein S. E. (2014) Improved normalization of systematic biases affecting ion current measurements in label-free proteomics data. Mol. Cell. Proteomics 13, 1341–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Riper S. K., de Jong E. P., Higgins L., Carlis J. V., and Griffin T. J. (2014) Improved intensity-based label-free quantification via proximity-based intensity normalization (PIN). J. Proteome Res. 13, 1281–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Silva J. C., Gorenstein M. V., Li G. Z., Vissers J. P., and Geromanos S. J. (2006) Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol. Cell. Proteomics 5, 144–156 [DOI] [PubMed] [Google Scholar]
- 47.Aittokallio T., Kurki M., Nevalainen O., Nikula T., West A., and Lahesmaa R. (2003) Computational strategies for analyzing data in gene expression microarray experiments. J. Bioinform. Comput. Biol. 1, 541–586 [DOI] [PubMed] [Google Scholar]
- 48.Vincent C. E., Potts G. K., Ulbrich A., Westphall M. S., Atwood J. A. 3rd, Coon J. J., and Weatherly D. B. (2013) Segmentation of precursor mass range using “tiling” approach increases peptide identifications for MS1-based label-free quantification. Anal. Chem. 85, 2825–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Serang O., and Käll L. (2015) Solution to statistical challenges in proteomics is more statistics, not less. J. Proteome Res. 14, 4099–4103 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.