

Abstract
Methodology for sequence analysis of ∼150 kDa monoclonal antibodies (mAb), including location of post-translational modifications and disulfide bonds, is described. Limited digestion of fully denatured (reduced and alkylated) antibody was accomplished in seconds by flowing a sample in 8 m urea at a controlled flow rate through a micro column reactor containing immobilized aspergillopepsin I. The resulting product mixture containing 3–9 kDa peptides was then fractionated by capillary column liquid chromatography and analyzed on-line by both electron-transfer dissociation and collisionally activated dissociation mass spectrometry (MS). This approach enabled identification of peptides that cover the complete sequence of a murine mAb. With customized tandem MS and ProSightPC Biomarker search, we verified 95% amino acid residues of this mAb and identified numerous post-translational modifications (oxidized methionine, pyroglutamylation, deamidation of Asn, and several forms of N-linked glycosylation). For disulfide bond location, native mAb is subjected to the same procedure but with longer digestion times controlled by sample flow rate through the micro column reactor. Release of disulfide containing peptides from accessible regions of the folded antibody occurs with short digestion times. Release of those in the interior of the molecule requires longer digestion times. The identity of two peptides connected by a disulfide bond is determined using a combination of electron-transfer dissociation and ion–ion proton transfer chemistry to read the two N-terminal and two C-terminal sequences of the connected peptides.
Monoclonal antibodies (mAbs)1 and related biological molecules constitute one of the most rapidly growing classes of human therapeutics. These large proteins (Fig. 1) have molecular weights near 150 kDa and are composed of two identical ∼50 kDa heavy chains (HC) and two identical ∼25 kDa light chains (LC) (1). They also contain at least 16 disulfide bonds that maintain three-dimensional structure and biological activity (2). Although sharing similar secondary protein structures, different mAbs differ greatly in the sequence of variable regions, especially in the complementarity determining regions (CDRs) which are responsible for the diversity and specificity of antibody-antigen binding. Changes to the mAb structure introduced during the manufacturing process or storage may influence the therapeutic efficacy, bio-availability and -clearance, and immunogenic properties and thus alter drug safety (3–5). Comprehensive characterization of mAbs primary structure, post-translational modifications (PTMs), and disulfide linkages is critical to the evaluation of drug efficacy and safety, as well as understanding the structure/function relationships (4, 6). Presented in this work is a novel protein analytical platform that consists of innovative methods for mass spectrometry (MS) characterization of mAbs. The methodology reported here will have a dramatic impact on the whole field of antibody characterization.
Fig. 1.

Diagram of a murine monoclonal antibody structure.
Typical MS characterization of proteins uses a “Bottom-Up” approach. This method involves tryptic digestion of the protein(s) into small peptides (mostly below 2500 Da) followed by high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) analyses of the resulting peptides (7). Although sensitive for MS analysis, small tryptic peptides often have issues such as weak retention in liquid chromatography, difficulties in assigning peptides to specific gene products, and loss of combinatorial PTM information (8). Recent years have seen developments in direct MS analysis of intact proteins (often called “Top-Down” MS). Despite increasing success in characterization of small to medium-sized proteins, MS analysis of intact proteins larger than 50 kDa, including mAbs, is still unsatisfactory because of inefficient gas-phase protein fragmentation and complex fragment ions that restrict efficient data interpretation (9, 10).
A “compromise” between Bottom-Up and Top-Down approaches is the “Middle-Down” (or “Middle-Up”) method. Middle-Down analysis typically involves proteolysis using proteases (e.g. Lys-C) or chemicals that hydrolyze proteins at a single type of amino acid residue. This approach aims to generate 3–15 kDa peptides which are compatible with high resolution MS/MS analysis on a chromatographic time scale. The Middle-Down approach inherits some of the advantages of Top-Down analysis, yet has less demanding instrumental requirements compared with intact protein MS in achieving sufficient signal-to-noise ratio (S/N) of fragment ions for sequence mapping (11–15).
However, limitations of currently available tools for Middle-Down protein analysis are also obvious. First, none of the twenty amino acids is evenly distributed along a polypeptide. Protein digestion at single-type amino acid residues can still produce very small (<1000 Da) or ultra large (>15 kDa) peptides, which deviates from the original intention of the Middle-Down approach (16). Second, the enzymatic digestion efficiency is often low for proteins with highly folded structure or low solubility. Although high concentrations of chaotropic agents such as 8 m urea are often used for protein denaturation, this harsh condition quickly deactivates many commonly used proteases. Third, traditional data-dependent ETD or electron-capture dissociation MS/MS analyses adopt a single reaction parameter for gas-phase dissociation and select only several abundant ions regardless of their charge states. As these methods were previously optimized for tryptic peptide ions that typically carry +2 or +3 charges, they are incompatible with the analysis of large, highly charged peptides that require optimized ETD to achieve high sequence coverage and PTM mapping (12).
Herein we report a “time-controlled” proteolysis method for tailored Middle-Down MS analysis of mAb. To hydrolyze the 150 kDa mAb into large peptides for HPLC-MS analysis, we fabricated a capillary enzyme reactor column that contains a specified length of immobilized protease (supplemental Fig. S1 and S2A). Precise control of the sample flow rate leads to defined digestion time of the substrate protein in the reactor. A short digestion time results in a small number of “cuts” along the protein chain and consequently the formation of large peptides (supplemental Fig. S2B). The Bruening group previously demonstrated a similar concept using a nylon membrane electrostatically adsorbed with pepsin or trypsin. Pushing a protein solution (protein dissolved in 5% formic acid solution) through the membrane-based enzyme reactor in less than 1 s breaks the protein into large peptides that facilitate sequence mapping of horse apomyoglobin (17 kDa) and bovine serum albumin (66 kDa) by infusion electrospray ionization MS/MS (17). The advantages of their enzyme disc include simple preparation procedures, as well as the low back pressure in the thin disc that allows for rapid sample flow rate. In our present work, we designed a more robust enzyme reactor that digests alkylated or native mAb into 3–12 kDa peptides in a buffer containing 8 m urea (a condition incompatible with most widely used proteases), and characterized their amino acid sequences, PTMs, as well as the disulfide linkages using HPLC-MS/MS.
We chose a rarely used protease, aspergillopepsin I, for the enzyme reactor. Aspergillopepsin I, also known as Aspergillus saitoi acid proteinase, generally catalyzes the hydrolysis of substrate proteins at P1 and P1′ of hydrophobic residues, but also accepts Lys at P1 (18). There are several innovative aspects of employing this enzyme: (1) Aspergillopepsin I is active in 8 m urea at pH 3–4 for at least 1 h. This extreme chaotropic condition may disrupt the higher-order structure of proteins to a great extent and allows for easy access of the protease to most regions of the substrate protein once the disulfide bonds are reduced. (2) Compared with proteases with dual- or single-type amino acid specificity, aspergillopepsin I provides more cleavage sites along an unfolded substrate protein. Allowing limited time for the substrate protein to interact with immobilized aspergillopepsin I should generate large peptides with a relatively narrow size distribution because of similar numbers of missed cleavages on these peptides. (3) The enzyme reactor automatically “quenches” proteolysis as the sample flows out of the column. This is in great contrast to in-tube digestion using solubilized proteases that are active in acidic conditions. In the latter case, digestion is difficult to quench or control because of the sustained enzymatic activity in an acidic condition. (4) Compared with electrostatic or hydrophobic interactions for enzyme immobilization, covalent conjugation of the protease onto porous beads should prevent the replacement of enzymes by upcoming substrate proteins. (5) The enzyme beads can be stored at 4 °C for at least half a year once water is removed, allowing the production of hundreds of disposable enzyme reactors from one batch of beads. In addition, we introduced a new cysteine (Cys) alkylation reagent, N-(2-aminoethyl)maleimide (NAEM) for protein MS analysis. This reagent improves ETD (19) of peptides containing Cys residues by adding a basic, readily protonated side chain to thiol groups.
The above features of our new strategy led to the generation of large, highly charged peptides that cover the entire murine mAb. Analyzing ETD and collisionally activated dissociation (CAD) fragments from the most abundant large peptides by ProSightPC revealed near complete sequence coverage of the mAb and multiple PTMs. Furthermore, we digested the native mAb into large fragments of disulfide-bonded peptides using time-controlled digestion. The ETD/ion-ion proton transfer (IIPT) technique (20) allowed facile identification of the N- and C-terminal sequences of two disulfide-bonded peptides and localization of the disulfide bond(s) within/connecting different mAb domains.
EXPERIMENTAL PROCEDURES
Materials
Unless otherwise stated, materials in this work were obtained from Sigma-Aldrich (St. Louis, MO).
Enzyme Reactor Fabrication
Aspergillopepsin I (protease from Aspergillus saitoi, Type XIII) was immobilized on 20 μm aldehyde-functionalized particles (POROS® AL 20 μm Self Pack® Media, Life Technologies, Grand Island, NY) by reductive amination under “salting out” conditions. See the supplemental information section 1.1 for details. Particles immobilized with enzyme were suspended in water and packed into a 360 μm o.d. × 150 μm i.d. fused silica capillary (Polymicro Technologies, Phoenix, AZ) to form a 2–35 cm long enzyme reactor. Supplemental Table S1 lists specific column lengths for different samples.
Sample Preparation and Protein Digestion
Supplemental information section 1.3 describes the detail procedures for protein digestion. Briefly, apomyoglobin from equine skeletal muscle was dissolved in the digestion buffer (pH 3.9 containing 8 m urea) at 0.2 μg/μl and pressure-loaded through the enzyme reactor at different flow rates to achieve different digestion times.
To digest the murine IgG1 mAb (Waters, Milford, MA) with the enzyme reactor for peptide mapping, the disulfide bonds of mAb were reduced with tris(2-carboxyethyl)phosphine (TCEP) and alkylated with NAEM at pH 6.8 in 8 m urea. The NAEM-alkylated mAb was acidified and diluted to 0.2 μg/μl in a buffer containing 8 m urea (final pH 3.9), followed by on-column digestion with different digestion times. The same mAb was also reduced and alkylated with iodoacetamide (IAM) as a control sample for on-column digestion with aspergillopepsin I and in-tube digestion with Lys-C (Roche Diagnostics, Indianapolis, IN). To digest the native mAb for disulfide localization, the intact mAb was dissolved in the digestion buffer for time-controlled on-column digestion with aspergillopepsin I. An aliquot of each native mAb digest was reduced with TCEP to separate the disulfide-linked peptides.
Chromatography and Mass Spectrometry
An Agilent Technologies (Palo Alto, CA) 1100 Series binary HPLC system was interfaced with a Thermo Scientific™ Orbitrap Velos Pro™ Hybrid Ion Trap-Orbitrap Mass Spectrometer (San Jose, CA) for online separation of protein digests. One pmol of the protein digest was pressure-loaded onto a 360 μm o.d.×150 μm i.d. fused silica capillary precolumn packed with 11 cm long POROSHELL 300SB-C18 (5 μm diameter, Agilent). After desalting, the precolumn was connected to an analytical column (360 μm o.d. × 50 μm i.d. capillary packed with 16 cm of the same material) that was integrated with a laser-pulled nanoelectrospray emitter tip (21). Peptides were eluted at a flow rate of 100 nL/min using the following gradient: 0–25% B for 5 min, 25–60% B for 105 min, 60–100% B for 4 min (A = 0.3% formic acid in water; B = 0.3% formic acid, 72% acetonitrile (Mallinckrodt, Inc., Paris, KY), 18% isopropanol and 9.7% water).
Mass spectrometric analyses of apomyoglobin and mAb sequences consisted of an HPLC-MS experiment with full MS Orbitrap scans for sample evaluation (Experiment I), followed by a multi-segment HPLC-MS/MS experiment with ETD MS/MS Orbitrap scans (Experiment II) targeted on the most abundant large peptides (3–9 kDa) selected from Experiment I. For each selected large peptide, the ion with the highest charge state (but with sufficient intensity) was selected as the precursor ion for ETD MS/MS analysis. ETD reaction time was set based on the following formula, tETD = 50 ms × (3/charge state)2. To obtain maximum mAb sequence coverage, an HPLC-CAD MS/MS experiment was performed as Experiment III in a similar way as in Experiment II to generate complimentary peptide sequence information. To characterize the location of mAb disulfide bonds, ETD/IIPT and ion multi-fill techniques (22) were performed on the on-column generated disulfide-containing mAb fragments to identify the N- and C-terminal sequences of the two disulfide-bonded peptides. See supplemental information section 1.4 for detailed MS methods.
In addition to the on-column time-controlled digestions, a Lys-C digest of the murine mAb was generated in a tube and then analyzed using data-dependent HPLC MS/MS. See supplemental information section 3 for details.
Data Analysis
Targeted ETD MS/MS spectra for each of the pre-selected peptides were merged (if there was more than one scan per peptide) and extracted from the raw file of the HPLC-MS/MS Experiment II using Xcalibur™ 2.1 (Thermo Scientific). The same procedure was performed for CAD spectra obtained from Experiment III of the mAb analysis. Each extracted ETD and CAD spectrum was then searched against the single protein database, horse myoglobin (accession number P68082, see supplemental Fig. S6) or murine mAb IgG1 (sequence provided by Waters, see supplemental Fig. S7), using ProSightPC 3.0 in BioMarker Search mode. Initial search parameters included: 2.1 Da precursor tolerance, 2.1 Da fragment tolerance, monoisotopic precursor and fragment ion type, Δm mode on, disulfide off, and a user-defined fixed modification of Cys with NAEM (monoisotopic +140.05857 Da, only for mAb analysis). Identified peptides with the lowest E-Values (E-Value threshold 1.0) and <0.01 Da precursor mass error were directly accepted. Identified peptides with +1.0 Da mass difference from the theoretical MW were considered for possible deamidation on Asn, and the deamidation sites were further identified by rematching the overall MS/MS fragment ions after manually adding +0.9840 Da on potential Asn residue(s) suggested by Δm function. Unidentified peptides were further searched by increasing the precursor tolerance to 20 Da to allow for Met oxidation or N-terminal pyroGlu from Gln (pyroglutamic acid formation of N-terminal Gln), or to 2000 Da to allow for glycosylation. Newly identified peptides were further verified by rematching the fragment ions in the program after manually adding either +15.9949 Da (oxidation) on potential Met residue(s) suggested by Δm function or +1444.5338 Da (fucosylated biantennary (-2 galactose) oligosaccharide, i.e. G0F) on potential Asn residue(s) suggested by Δm function. The identified PTM sites of all modified peptides were manually verified. To calculate sequence coverage, the c-, z-, b- and y-type fragment ions assigned by ProSightPC 3.0 with mass error within ±15 ppm were accepted. The mass error for the remaining fragment ions was recalculated by allowing a ±1.00 or ±2.00 unit Da mass shift (because of incorrect monoisotopic peak selection by the software, or electron transfer without dissociation (23)), and the ions with their new mass error within ±15 ppm were accepted after manual verification.
As a comparison, MS data from the mAb Lys-C digest obtained from data-dependent HPLC MS/MS were searched against the murine mAb reference sequence using Open Mass Spectrometry Search Algorithm (OMSSA, version 2.1.8) (24). See supplemental information section 3 for details.
RESULTS
Time-Controlled Proteolysis with Aspergillopepsin I
To generate 3–12 kDa fragments from protein samples, we constructed a micro-column enzyme reactor using fused silica (150 μm i.d.) packed with the protease, aspergillopepsin I, covalently linked to 20 μm particles. This protease is active under acidic conditions and exhibits broad specificity with preference for hydrolysis of amide bonds at the N terminus of hydrophobic residues such as Val, Ile, and Leu, and at the C terminus of Lys (18). It also has the unusual property of being active in 8 m urea for at least 1 h. Under these conditions most protein substrates, once their disulfide bonds (if any) are reduced, will be completely denatured and easily digested. Operation of the enzyme reactor involves flowing a solution of the protein sample in pH 3.9 buffer containing 8 m urea under constant back pressure through the packed fused silica column (supplemental Fig. S2A). This produces a constant flow rate and allows the digestion (residence) time in the enzyme reactor to be calculated as a function of the flow rate and the length of the packed enzyme reactor (see supplemental information section 1.2 for details).
On-column Digestion and Sequence Analysis of Horse Apomyoglobin
Shown in supplemental Fig. S5 are total ion current (TIC) chromatograms from micro-capillary HPLC-MS spectra recorded on peptides produced from the standard, 17 kDa protein, horse apomyoglobin, with three different digestion times (2.8, 0.77, and 0.3 s) in the micro column reactor. Small peptides dominate the spectra acquired at the longest reaction time (2.8 s), intact apomyoglobin remains undigested at the shortest reaction time (0.3 s), and peptides in the 2–8 kDa mass range are both abundant and well spread out across the chromatogram for the intermediate digestion time (0.77 s). Shown in Fig. 2A is the base peak chromatogram for the peptides generated with 0.77 s digestion. Major large peptides are labeled with apomyoglobin amino acid sequence numbers deduced from ETD spectra recorded on each parent ion population. Fig. 2B displays the ETD spectrum recorded on the +7 ion at m/z 619.89 that corresponds to the last 40 residues (114–153) in the protein (all multiply charged ions have been converted to +1 ions by Xcalibur Xtract). Observed fragment ions of type c and z· are labeled below the spectrum. Total sequence coverage for apomyoglobin observed with two peptide fragments 1–69 and 70–153 was 86%, and with 5 peptides, 1–31, 32–69, 70–113, 114–153, and 105–153 was 97% (supplemental Fig. S6).
Fig. 2.

A, Base peak chromatogram of apomyoglobin digest generated by 0.77 s time-controlled digestion, with the 3–9 kDa base peak peptides labeled using apomyoglobin sequence number. Inset is the MS1 spectrum at the elution time corresponding to peptide 114–153, and ions corresponding to peptide 114–153 are labeled with different charge states. B, The spectrum of apomyoglobin peptide 114–153 after converting the original ETD MS/MS spectrum to +1 ions by Xcalibur Xtract (some fragment ions are lost after Xtract convertion). Under the spectrum is the sequence coverage by c and z· ions assigned by ProSite PC 3.0 using the original MS/MS data (manually verified).
On-column Digestion and Sequence Analysis of a Murine mAb
For on-column digestion of reduced and alkylated mAb in 8 m urea, several reaction times were evaluated (Fig. 3) as described above for apomyoglobin. From this data, a digestion time of 5.7 s was selected because HPLC-MS (Experiment I) of the resulting product mixture showed that more than three dozen peptides with masses in the 3–10 kDa range eluted within the chromatographic time window from 30–105 min. Intact protein was not detected.
Fig. 3.
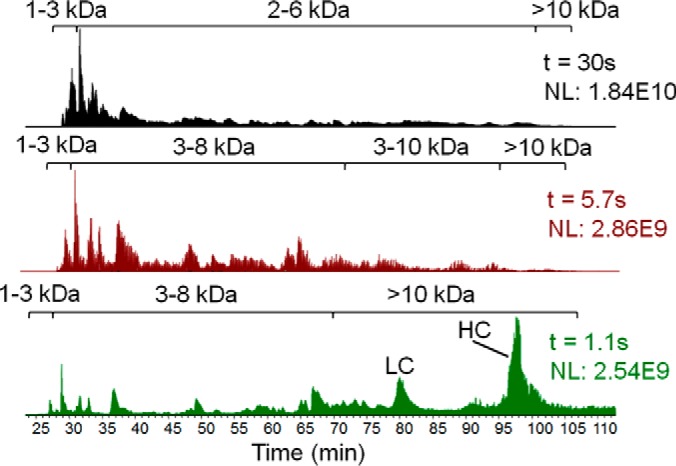
TIC chromatograms of HPLC-Orbitrap MS for mAb digests produced from size-controlled digestion with three different digestion times. The digestion time (t) and normalized ion count (NL) are indicated in each TIC chromatogram. Peptides sizes and undigested mAb LC and HC are labeled within each chromatogram.
To acquire sequence information for peptides in the 3–10 kDa range, we divided the TIC chromatogram into 8 time segments (supplemental Fig. S3) and selected ions representing the most abundant large peptides at specific m/z values in each segment for targeted analysis by ETD during a second HPLC-MS/MS experiment (Experiment II). Generation of a list of the ions to be targeted involved averaging of the mass spectra (full MS) acquired in a particular segment of Experiment I, deconvolution of the clusters of multiply charged ions into single peaks corresponding to the monoisoptic molecular weights of each component, and selection of a m/z window that would only contain highly charged ions from one of the peptides to be targeted. An example of ETD sequencing of HC 37–77 is presented in supplemental Fig. S4.
Using the above approach, 39 large peptides were selected based on their abundance from Experiment I. These peptides were targeted for ETD during 8 time segments of a second HPLC MS/MS experiment (Experiments II), as well as for CAD the same way in a third HPLC MS/MS experiment (Experiment III). These peptides and their PTMs (if any) were successfully identified by ProSightPC software using Biomarker mode search. Among these peptides, four peptides (LC1–52, 53–110, 111–148 and 149–219) cover the entire LC, and 9 peptides (HC1–36, 37–83, 84–148, 149–210, 211–260, 261–276, 277–319, 320–371 and 372–441) cover the entire HC. MS/MS spectra acquired on 6 LC peptides and 14 HC peptides provided 98 and 94% sequence coverage of these two mAb components, respectively (supplemental Fig. S7). In addition, spectra recorded on 8 of the 39 peptides detected one or more PTMs. Shown in Fig. 4A is an average of the peptide signals (displayed as monoisotopic molecular masses) that were detected in mass spectra recorded during Segment I-3 (supplemental Fig. S3) of the TIC chromatogram. Peptides targeted for sequence analysis by ETD MS/MS and identified by ProSightPC and by manual interpretation are labeled with the appropriate amino acid sequence numbers. Note that there are three groups of signals in the spectrum that contain peaks that differ in mass by 162 Da. This mass difference corresponds to that of hexose and suggests that all nine of the signals are likely to come from glycopeptides. Shown in Fig. 4B is the ETD mass spectrum recorded on m/z 840.1 (+8 charge state) ions from the peptide of MW 6708 Da. For display purposes, all of the multiply charged ions in the ETD spectrum have been converted to singly-charged ions by the Xcalibur Xtract program. ProSightPC and manual interpretation both assigned the peptide sequence as residues 277–319 of the HC with an extra 1444 Da attached to Asn292. This is the expected mass of the N-linked, displayed in Fig. 4A. Addition of one and two galactose residues to this structure would generate the expected G1F and G2F structures and explain the signals observed at molecular masses at 6870 Da and 7032 Da.
Fig. 4.

A, Averaged full MS (ions converted to monoisotopic MW by Xcalibur Xtract) of Segment I-3 (see supplementalFig. S3) of mAb digest. Peptides labeled with sequence identities are those picked for targeted MS/MS. The three groups of peptides colored green, red and blue correspond to three large peptides with potential N-glycans. B, ETD MS/MS of the peptide of 6708 Da MW in Fig. 4A. All of the ions in the original MS/MS spectrum were converted to +1 charge state by Xcalibur Xtract with c and z· fragments labeled. Below the spectrum is shown the sequence coverage and N-glycosylation assigned by ProSitePC 3.0 (with manual verification) based on the original MS/MS spectrum. More ions were labeled in the peptide sequence than in the spectrum because some fragment ions were lost in Xcalibur Xtract deconvolution.
In addition to N-glycosylation, we also found significant (∼40%) deamidation of an Asn residue to Asp and isoAsp on the HC. Because of this modification, peptide HC 80–148 eluted as three adjacent peaks as shown in supplemental Fig. S8A, with their monoisotopic masses differing by 1 Da (NH→O) (supplemental Fig. S8B) (25). ProSightPC and manual interpretation of CAD spectra of the three peaks confirmed the deamidation site to be Asn138 on HC (supplemental Fig. S8C). We noticed that the deamidation level of Asp138 in the HC of this commercial murine mAb is consistent in different digests from the same mAb sample. However, the level increased greatly in digests from later mAb sample batches. The deamidation of HC Asp138 should not be affected to this extent by sample preparation and MS analysis, as they were performed in acidic conditions except for the alkylation procedure carried out at pH 7 for only 10 min. It is known that acidic condition can minimize mAb deamidation (26). Other PTMs defined by targeted analysis of the 3–9 kDa peptides in the digest include oxidation of multiple Met residues in LC and HC to the corresponding sulfoxide, as well as conversion of Gln to pyroglutamate at the N terminus of HC. Table I lists all the identified PTMs. Supplemental Table S2 lists the relative quantification of each identified PTM.
Table I. Summary of identified mAb PTMs.
| PTMs and Disulfide Bonds | Site |
|---|---|
| Met oxidation | LC Met4 |
| HC Met304, Met49 a, Met140 a, Met353 a, Met363 a, Met393 a | |
| Pyruglutamate formation | HC N-term Gln |
| Deamidation | HC Asn138 |
| N-linked glycosylation (G0F, G1F, G2F) | HC Asn292 |
| Disulfide linkages | LC Cys23-Cys93 b, LC Cys139-Cys199 b, HC Cys22-Cys95 b, HC Cys145-Cys200 b, HC Cys256-Cys316 b, HC Cys362-Cys420 b, LC Cys219-HC Cys220 b, HC Cys222-HC' Cys222, HC Cys225-HC' Cys225, HC Cys227-HC' Cys227 |
a Low level Met oxidation verified by targeted MS/MS analysis on low level peptides with MW +16 Da higher than identified unmodified peptides.
b This disulfide bond appears twice in an IgG molecule due to two identical copies of LC and HC.
Charge Enhancement on Cys Improves MAb Sequence Coverage by ETD
In this work, we introduced a novel reagent, NAEM, for Cys alkylation prior to protein digestion (supplemental Fig. S9). NAEM alkylates Cys residues completely in only 10 min. Compared with Cys alkylation with the widely used iodoacetamide (IAM), reacting sulfhydryl groups with NAEM increases the charge state of the protein. Placing a positively charged amino group on Cys side chains facilitates fragmentation and sequence analysis of nearby residues in the LC and HC by ETD MS. For example, alkylation of LC1–52 (5767 Da) Cys with NAEM adds one more charge to this peptide compared with the IAM-alkylated form (Fig. 5A and 5B). Alkylation of Cys with NAEM yielded a twofold improvement in the sequence coverage of LC1–52 using ETD (Fig. 5C). Many of the newly appearing c and z· fragments come from CDR1 close to the Cys residue. In another example, replacing IAM with NAEM on LC111–148 enhanced charge state by +1, and increased the sequence coverage from 10.5% to 89.5%. Similarly, alkylating the 5 Cys of HC211–260 with NAEM instead of IAM enhanced the charge state distribution by +3 and improved peptide sequence coverage upon ETD from 53 to 73%. Supplemental Table S3 lists the comparison of NAEM- and IAM-alkylated Cys-containing peptides in sequence coverage upon ETD.
Fig. 5.

Comparison of charge state distributions of peptide LC 1–52 alkylated with NAEM (A) and IAM (B). The influence of increased charge on this peptide for ETD sequence mapping is displayed in C, with the underline showing CDR1.
Comparison with In-tube Lys-C Digestion, Data-dependent MS/MS and Traditional Enzyme-based Database Search
To compare our new Middle-Down methodology with conventional peptide mapping method, we performed in-tube Lys-C digestion for the murine mAb followed by data-dependent HPLC MS/MS analyses. We then searched the ETD and CAD data against the in silico digested reference mAb sequence by OMSSA to identify mAb peptides and their PTMs. Details can be found in supplemental information section 3.
We chose Lys-C for comparison because, as opposed to aspergillopepsin I, Lys-C digests protein at a single type of amino acid residue (Lys) and is widely used for producing peptides larger than tryptic peptides. Displayed in supplemental Fig. S10 is the TIC chromatogram of the Lys-C peptides. The first 1/8 portion (40–50 min) of the chromatogram is dominated by peptides smaller than 3 kDa, which are too small to provide Middle-Down benefits. On the other hand, the last 1/3 portion of the chromatogram is occupied by peptides over 11 kDa with poor separation resolution. Compared with 3–9 kDa peptides, these ultra-large peptides are less effective in producing sequence information in data-dependent MS/MS, as can be seen from the frequent false positive identifications of peptides (or their PTMs) over 8 kDa upon OMSSA search (supplemental information_2.xlsx).
Although the OMSSA search identified Lys-C peptides that cover the entire mAb, the sequence coverage for many Lys-C peptides obtained from data-dependent MS/MS is low and PTMs can be difficult to localize. For example, the Lys-C peptide HC254–315 (8.7 kDa) eluted as a base peak in HPLC-MS (supplemental Fig. S10). However, ETD and CAD scans from the data-dependent MS/MS contain fragment ions that allow verification of only 56% of the peptide sequence and do not fully cover the site (Asn292) modified with glycan G0F (supplemental Fig. S11). In contrast, our new Middle-Down approach covered 90% sequence of a 6.7 kDa peptide (HC277–319) using only ETD, with confident assignment of glycan on Asn292 (Fig. 4). In another example, ETD and CAD scans from the data-dependent MS/MS yielded 41% sequence coverage of Lys-C peptide LC1–50, including 31% of CDR1 (LC24–39) (supplemental Fig. S12). In contrast, our new Middle-Down approach mapped 90% sequence of peptide LC1–52, including 94% of CDR1.
Location of Disulfide Bonds
Sequence analysis of the reduced and alkylated mAb revealed 5 and 12 Cys residues in the LC and HC, respectively. Because the intact antibody has two HCs and two LCs, the total number of Cys residues in the molecule is 34 and the likely number of disulfide bonds is 17. To specify the location of disulfide linkages, we evaluated multiple reaction times for on-column digestion of the intact mAb. We searched mass spectra (after deconvolution to the monoisotpic molecular weights) recorded on the digestion products for large peptides that were not detected in the data set generated from the post-TCEP reduction sample of the same digest (see supplemental Fig. S13 for an example). We then targeted these unique peptides (nonreduced form) for sequence identification. A combination of ETD and gas phase IIPT reactions on these target ions allowed us to read amino acid sequences from the two N termini and two C termini of the disulfide-bonded peptides (22).
As might be expected, reaction times required to digest the tightly folded, and disulfide bond constrained, intact mAb in 8 m urea were substantially longer than that required for the fully reduced, alkylated, denatured molecule. Digestion times were also different for the different domains of the antibody structure. Most accessible were the N-terminal variable domain of LC (VL) and C-terminal constant domain of HC (CH3), both of which released disulfide-bond containing peptides after 12 s on the reactor column. Those in the other domains, i.e. VH, CL plus CH1, and CH2 plus the hinge region required 93, 260 and 740 s, respectively, on the enzyme reactor column before being released (supplemental Fig. S14). All of the 17 disulfide bonds of the murine IgG were successfully characterized.
Shown in supplemental Fig. S15A is the ETD mass spectrum recorded on the +12 charge state ion ((M+12H)+12 = 985.5) of the disulfide-connected peptides (MW 11,814) released from the intact mAb after 12 s on the enzyme reactor. To acquire this spectrum, +12 ions were allowed to accept an electron from fluoranthene radical anions and then dissociate at the various amide bonds into a collection of fragment ions of type c and z· from both of the attached peptides. These fragment ions can carry up to 11 protons, and many of them (such as the c+10 and z·+10 ions) are fragments from one peptide but with the Cys linked to the entire chain of the second peptide (see supplemental Fig. S15C and S15D). To simplify this mixture, highly charged fragments were allowed to react with a second gas phase anion, SF6−· that functions as a base and removes protons and thus charge from the c and z· ions. The result is a mass spectrum that contains ions carrying only +1 or +2 charges which are distributed across the 4000 m/z range of the Orbitrap mass spectrometer (Fig. 6). To enhance the S/N of fragment ions in this spectrum, we used C-trap multiple fill technology to store products of 15 ETD/IIPT reactions before the ions were scanned in the Orbitrap (22). Observed ions were more than sufficient to identify the two disulfide peptides as LC 1–52 and LC 53–108. Because each of the two peptides contains only one Cys residue, the disulfide bond was assigned as LC Cys23-Cys93. Note that capture of an electron into the disulfide bond produces fragments that correspond to protonated forms of the two peptides that were previously disulfide-linked. Doubly charged ions at m/z 2880 and 3022 in Fig. 6 are the result of this phenomenon and confirm the identity of the connected peptides. The same protocol was employed to identify the disulfide bond connections presented in Table I and supplemental Fig. S14. The ETD/IIPT mass spectra used to make these assignments are displayed in supplemental Fig. S16.
Fig. 6.

Identification of disulfide-containing peptide LC1–52 and LC53–108 by ETD (3 ms)/IIPT (90 ms) with 15 multi-fills in C-trap prior to Orbitrap MS/MS analysis. The inset shows the sequence and disulfide linkage of LC1–52 and LC53–108, and the detected fragment ions. Fragment ions labeled in blue represent those coming from LC 1–52, and red from LC 53–108.
DISCUSSION
Structural characterization of mAbs by MS is challenging because of the high complexity of IgG mAbs in both the primary structure and higher order structures. Time-controlled on-column digestion with aspergillopepsin I of the reduced, alkylated mAb in 8 m urea narrows the size of major peptides to 3–9 kDa. Peptides in this MW range have higher sequence coverage compared with small tryptic peptides, and are spread across a wide range in the chromatogram generated from porous shell-type C18 silica particles. On the other hand, these large peptides are more compatible with online high resolution MS/MS analysis than intact proteins. Although aspergillopepsin I favors protein hydrolysis at hydrophobic and Lys residues, the probabilities of hydrolysis at these sites are not completely equal. Otherwise the time-controlled digestion would generate a much higher number of 3–9 kDa mAb peptides with highly overlapped sequences and equivalent quantities. Based on the most abundant large peptides produced from apomyoglobin and mAb in this work, the most frequent hydrolysis occurs at the N terminus of Val, Leu, and Ile, and the C terminus of Lys. The following calculations can also reflect the partially controlled protease specificity. The average MW of the 39 most abundant large peptides from murine mAb targeted for MS/MS analysis is 5485 Da. With this MW as a “standard” Middle-Down peptide length, covering the whole sequence of LC and HC (combined MW 76.5 kDa) would require at least 14 peptides of this average MW with no overlapping sequences. The 14 hypothesized peptides should contain 11 missed cleavages on average if considering only Val/Leu/Ile/Lys as potential hydrolysis sites. If the chance of hydrolysis is equal for each of the four amino acid residues, the actual number of resulting peptides with this size would be ∼160. This number can greatly increase if additional types of amino acid residues (e.g. Gly, Trp, Phe) are considered as potential hydrolysis sites. However, in our work the actual number of major large peptides selected for HPLC-MS/MS analysis is only 39. These peptides cover the entire mAb primary structure and led to 95% sequence coverage by MS/MS, suggesting controllable sample complexity for mAb sequencing. Moreover, these peptides provide some overlapping sequences which are beneficial for protein sequencing.
Taking advantage of the highly reproducible retention time on the POROSHELL C18 column (within 0.3 min for each peptide in the HPLC MS experiments I, II and III), we analyzed the 39 preselected large mAb peptides using a customized multi-segment MS/MS method. Selecting the highest charge state of each peptide for ETD with tailored ETD reaction time maximizes the fragmentation efficiency and avoids wasting MS/MS scans on the same peptide with other charge states.
We improved ETD of many Cys-containing peptides by derivatizing Cys residues with an amino group prior to digestion. As over a dozen disulfide bonds are evenly distributed within different domains of an IgG molecule, this strategy improves the overall sequence coverage of the mAb. It is worth noting that some studies correlating IgG secondary structure to the primary structure information found that CDR-L1 and -L3 always begin immediately after a Cys residue, and CDR-H1 and -H3 always begin only a few amino acid residues after a Cys (27). Considering the close proximity of Cys to CDRs, placing a positively charged amino group on Cys residue(s) may facilitate mapping nearby CDR sequences by ETD.
In our new methodology, we verified peptide sequence from the MS/MS data using a ProSightPC Biomarker search (no enzyme search). This search mode first identifies a candidate peptide (any portion of a protein in the database) matching an observed precursor mass, and then compares the theoretical fragment ion masses of the candidate peptide to the observed fragment ion masses. By including Δm function to the N terminus and C terminus of candidate peptides in Biomarker search, we observed the pattern of fragments with and without Δm (e.g. +0.9840 Da for deamidation), and successfully identified one or more PTMs on multiple large peptides. The Δm function avoids incorporating multiple variable modifications into search algorithm. The latter is widely adopted in traditional database search programs based on in silico digestion of candidate protein(s) (e.g. SEQUEST, Mascot, and OMSSA). This traditional search mode has been proved to be successful for identifying small tryptic peptides. However, the computational complexity greatly increases with the growth of peptide length when adding multiple types of PTMs in the variable modification list, and may result in frequent false positive identifications. This can be seen from the OMSSA search result of Lys-C peptides in our comparison study, which will be discussed below.
In contrast to our time-controlled on-column digestion which produces mainly 3–9 kDa peptides from alkylated mAb, in-tube Lys-C digestion generated peptides with a wider size distribution (0.6–15 kDa). Data-dependent MS/MS of these Lys-C peptides followed by OMSSA search is successful in identifying peptides below 5 kDa. However, false positive identification appeared more often for larger peptidesespecially those with MW above 10 kDa (see supplemental information section 3, and supplemental information_2.xlsx). The sequence coverage for some identified large peptides is also low after manual examination of their merged MS/MS spectra. These observations can be explained by: (1) instrumental recognition of co-eluted highly charged peptides from a single MS1 scan is sometimes unsuccessful, and therefore does not trigger MS/MS scans for the unrecognized peptides, (2) fixed ETD reaction time (50 ms in the Lys-C digest comparison study) is incompatible with the charge state of some peptides, and (3) identification of peptide sequence or PTMs is based on program search using a single MS/MS scan, which often produces an insufficient number of fragment ions from a large peptide.
Although our new Middle-Down methodology optimizes peptide size and the quality of MS/MS scans, a drawback of our method is the requirement of pre-selection of several tens of highly abundant large peptides (assuming they can cover the entire mAb) for targeted HPLC MS/MS analysis. This strategy, although successful for single protein characterization, is somewhat arbitrary in selecting representative peptides and can be time consuming for analyzing protein mixtures (e.g. two or more mAbs). To improve MS analysis efficiency, a future method can include instrumental control code that allows for advanced data-dependent MS/MS features, such as (1) automatic selection of only the precursor ion with the highest charge state (if above an intensity threshold) for each peptide, and (2) automatic setting of ETD reaction time tETD according to the reciprocal relationship of tETD and peptide charge state to optimize ETD kinetics.
In the characterization of mAb disulfide linkages, gradually extending the time for on-column digestion of native mAb yielded large disulfide-bonded peptides first from the most flexible, terminal domains, then from the more rigid, interior domains. This stepwise protocol separates different disulfide bonds into multiple samples according to the disulfide locations. This strategy simplifies the search for disulfide bonds by “amplifying” the small disulfide bond information to a large piece of disulfide-bonded peptides. The latter typically elutes as a strong peak in TIC chromatogram and can be easily discerned by comparing the nonreduced and reduced digest samples. Because the previous experiments confirmed the mAb sequence, and each peptide in the disulfide-bonded peptides normally have only one Cys (except for the hinge region), localization of the disulfide bond is equivalent to the identification of the two disulfide-bonded peptides. ETD/IIPT fits this need perfectly. This technique separates the N- and C-terminal +1 and +2 fragment ions (appearing in the low m/z region) from the large fragment ions (m/z beyond 4000 upon IIPT). The former fragment ions are readily determined thereby enabling identifying the two disulfide-bonded peptides.
It can be expected that, with the continuous progress in MS (including scan rate, gas-phase fragmentation, instrumental control, etc.) as well as in peptide separation, this unique enzyme reactor will work as a flexible protein “cutter” and fit its role in future analytical platforms. Our methodology is likely to be very useful for the characterization of proteins with highly folded native structure or low solubility, such as membrane proteins.
Supplementary Material
Acknowledgments
We thank Prof. Neil Kelleher (Northwestern University) for providing ProSightPC 3.0 for data analysis.
Footnotes
Author contributions: L.Z., D.F.H., and W.-H.W. designed research; L.Z., S.A.U., and W.-H.W. performed research; A.M.E. and W.-H.W. contributed new reagents or analytic tools; L.Z., J.S., D.F.H., and W.-H.W. analyzed data; L.Z., M.M.R., D.F.H., and W.-H.W. wrote the paper; D.L.B. and W.-H.W. contributed to software application.
* This work was supported by National Institutes of Health grants AI 033993 (DFH) and GM 037537 (DFH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- mAb
- monoclonal antibody
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- HPLC
- high-performance liquid chromatography
- m/z
- mass-to-charge ratio
- ETD
- electron-transfer dissociation
- CAD
- collisionally activated dissociation
- IIPT
- ion-ion proton transfer
- TIC
- total ion current
- S/N
- signal-to-noise ratio
- CDR
- complementarity determining region
- HC
- heavy chain
- LC
- light chain
- PTM
- post-translational modification
- G0F
- fucosylated biantennary (-2 galactose) oligosaccharide
- G1F
- fucosylated biantennary (-1 galactose) oligosaccharide
- G2F
- fucosylated biantennary oligosaccharide
- NAEM
- N-(2-aminoethyl)maleimide
- IAM
- iodoacetamide
- TCEP
- tris(2-carboxyethyl) phosphine
- OMSSA
- Open Mass Spectrometry Search Algorithm.
REFERENCES
- 1.Beck A., Sanglier-Cianferani S., and Van Dorsselaer A. (2012) Biosimilar, biobetter, and next generation antibody characterization by mass spectrometry. Anal. Chem. 84, 4637–4646 [DOI] [PubMed] [Google Scholar]
- 2.Liu H., and May K. (2012) Disulfide bond structures of IgG molecules structural variations, chemical modifications and possible impacts to stability and biological function. Mabs 4, 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernandez L., Kalume D., Calvo L., Mallo M., Vallin A., and Roepstorff P. (2001) Characterization of a recombinant monoclonal antibody by mass spectrometry combined with liquid chromatography. J. Chromatogr. B 752, 247–261 [DOI] [PubMed] [Google Scholar]
- 4.Wright A., and Morrison S. L. (1997) Effect of glycosylation on antibody function: implications for genetic engineering. Trends Biotechnol. 15, 26–32 [DOI] [PubMed] [Google Scholar]
- 5.Roberts G. D., Johnson W. P., Burman S., Anumula K. R., and Carr S. A. (1995) An integrated strategy for structural characterization of the protein and carbohydrate components of monoclonal-antibodies: application to anti-respiratory syncytial virus mab. Anal. Chem. 67, 3613–3625 [DOI] [PubMed] [Google Scholar]
- 6.Zhang Z., Pan H., and Chen X. (2009) Mass spectrometry for structural characterization of therapeutic antibodies. Mass Spectrom. Rev. 28, 147–176 [DOI] [PubMed] [Google Scholar]
- 7.Bensimon A., Heck A. J. R., and Aebersold R. (2012) Mass spectrometry-based proteomics and network biology. Annu. Rev. Biochem. 81, 379–405 [DOI] [PubMed] [Google Scholar]
- 8.Lanucara F., and Eyers C. E. (2013) Top-down mass spectrometry for the analysis of combinatorial post-translational modifications. Mass Spectrom. Rev. 32, 27–42 [DOI] [PubMed] [Google Scholar]
- 9.Fornelli L., Damoc E., Thomas P. M., Kelleher N. L., Aizikov K., Denisov E., Makarov A., and Tsybin Y. O. (2012) Analysis of intact monoclonal antibody IgG1 by electron transfer dissociation orbitrap FTMS. Mol. Cell. Proteomics 11, 1758–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mao Y., Valeja S. G., Rouse J. C., Hendrickson C. L., and Marshall A. G. (2013) Top-down structural analysis of an intact monoclonal antibody by electron capture dissociation-fourier transform ion cyclotron resonance-mass spectrometry. Anal. Chem. 85, 4239–4246 [DOI] [PubMed] [Google Scholar]
- 11.Garcia B. A. (2010) What does the future hold for top down mass spectrometry? J. Am. Soc. Mass Spectrom. 21, 193–202 [DOI] [PubMed] [Google Scholar]
- 12.Kalli A., Sweredoski M. J., and Hess S. (2013) Data-dependent middle-down nano-liquid chromatography-electron capture dissociation-tandem mass spectrometry: an application for the analysis of unfractionated histones. Anal. Chem. 85, 3501–3507 [DOI] [PubMed] [Google Scholar]
- 13.Wu S., Kim J., Hancock W. S., and Karger B. (2005) Extended range proteomic analysis (ERPA): a new and sensitive LC-MS platform for high sequence coverage of complex proteins with extensive post-translational modifications comprehensive analysis of beta-casein and epidermal growth factor receptor (EGFR). J. Proteome Res. 4, 1155–1170 [DOI] [PubMed] [Google Scholar]
- 14.Wu C., Tran J. C., Zamdborg L., Durbin K. R., Li M., Ahlf D. R., Early B. P., Thomas P. M., Sweedler J. V., and Kelleher N. L. (2012) A protease for ‘middle-down’ proteomics. Nat. Methods 9, 822–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cannon J., Lohnes K., Wynne C., Wang Y., Edwards N., and Fenselau C. (2010) High-throughput middle-down analysis using an orbitrap. J. Proteome Res. 9, 3886–3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laskay U. A., Lobas A. A., Srzentic K., Gorshkov M. V., and Tsybin Y. O. (2013) Proteome digestion specificity analysis for rational design of extended bottom-up and middle-down proteomics experiments. J. Proteome Res. 12, 5558–5569 [DOI] [PubMed] [Google Scholar]
- 17.Tan Y., Wang W., Zheng Y., Dong J., Stefano G., Brandizzi F., Garavito R. M., Reid G. E., and Bruening M. L. (2012) Limited proteolysis via millisecond digestions in protease-modified membranes. Anal. Chem. 84, 8357–8363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rawlings N. D., and Salvesen J. S. (2013) Handbook of Proteolytic Enzymes, 3rd Ed., pp. 135–141, Elsevier, London, UK [Google Scholar]
- 19.Syka J. E. P., Coon J. J., Schroeder M. J., Shabanowitz J., and Hunt D. F. (2004) Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 101, 9528–9533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coon J. J., Ueberheide B., Syka J. E. P., Dryhurst D. D., Ausio J., Shabanowitz J., and Hunt D. F. (2005) Protein identification using sequential ion/ion reactions and tandem mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 102, 9463–9468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Udeshi N. D., Compton P. D., Shabanowitz J., Hunt D. F., and Rose K. L. (2008) Methods for analyzing peptides and proteins on a chromatographic timescale by electron-transfer dissociation mass spectrometry. Nat. Protoc. 3, 1709–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson L. C., English A. M., Wang W., Bai D. L., Shabanowitz J., and Hunt D. F. (2015) Protein derivatization and sequential ion/ion reactions to enhance sequence coverage produced by electron transfer dissociation mass spectrometry. Int. J. Mass Spectrom. 377, 617–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hunt D. F., Shabanowitz J., and Bai D. (2015) Peptide sequence analysis by electron transfer dissociation mass spectrometry: a web-based tutorial. J. Am. Soc. Mass Spectrom. 26, 1256–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X. Y., Shi W. Y., and Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 25.Yang H., and Zubarev R. A. (2010) Mass spectrometric analysis of asparagine deamidation and aspartate isomerization in polypeptides. Electrophoresis 31, 1764–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen G. (2013) Characterization of Protein Therapeutics using Mass Spectrometry, pp.187–189, Springer, New York, NY [Google Scholar]
- 27.Martin A. C. R. (2010) Chapter 3: Protein Sequence and Structure Analysis of Antibody Variable Domains from Antibody Engineering (Volume 2), 2nd Ed., pp.33–51, Springer, New York, NY [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.