

Abstract
Nitrile hydratase metalloenzymes are unique and important biocatalysts that are used industrially to produce high value amides from their corresponding nitriles. After more than three decades since their discovery, the mechanism of this class of enzymes is becoming clear with evidence from multiple recent studies that the cysteine-derived sulfenato ligand of the active site metal serves as the nucleophile that initially attacks the nitrile. Herein we describe the first direct evidence from solution phase catalysis that the source of the product carboxamido oxygen is the protein. Using 18O-labeled water under single turnover conditions and native high resolution protein mass spectrometry, we show that the incorporation of labeled oxygen into both product and protein is turnover-dependent and that only a single oxygen is exchanged into the protein even under multiple turnover conditions, lending significant support to proposals that the post-translationally modified sulfenato group serves as the nucleophile to initiate hydration of nitriles.
Keywords: enzyme, enzyme catalysis, enzyme mechanism, metalloenzyme, post-translational modification (PTM), nitrile hydratase, protein chemical modification
Introduction
Nitrile hydratases (NHases)2 catalyze the formation of amides from nitriles in aqueous, pH-neutral environments without any carboxylic acid byproducts (1, 2). These bacterial enzymes utilize trivalent iron or cobalt metal cations in this non-redox reaction, and uniquely, these metal ions are ligated directly to the protein backbone through amidate ligands and three cysteine sulfur ligands, of which two are post-translationally modified to the sulfinic and sulfenic acid (Fig. 1A) (3). The mechanism of hydration is slowly emerging through multiple studies and appears to involve the sulfenic acid oxygen as the nucleophile. This sulfenate oxygen is proposed to initially attack the metal-bound nitrile carbon atom, creating a cyclic intermediate that is then resolved through attack at the sulfenic sulfur either directly by water or from the neighboring axial thiolato sulfur (Fig. 1B) (4–6). Alternative mechanisms have also found support from time-resolved crystallographic and kinetic studies that involve an activated water molecule attacking the metal-bound nitrile (Fig. 1C) (7, 8).
FIGURE 1.

Active site structure and mechanism of NHase. A, the x-ray crystal structure of the cobalt-type NHase homolog from Pseudonocardia thermophila JCM 3095 (Protein Data Bank (PDB): 1IRE) shows the active site cobalt complex including two modified cysteine sulfur ligands oxidized to the sulfinic and sulfenic acids. The sequence shown is CTLCSC. B, potential mechanism of nitrile hydratase in which the sulfenate oxygen acts as the initial nucleophile to attack the nitrile carbon. C, potential mechanism of nitrile hydratase wherein a solvent water molecule serves as the initial nucleophile. (In B and C, bonds from enzyme to modified cysteine and amidate ligands are omitted for clarity.)
Early studies on sulfenato ligands in cobalt complexes demonstrated the nucleophilicity of the sulfenato oxygen (9, 10), and recently, this same characteristic was observed in NHases wherein the sulfenato oxygen reacted with electrophilic boronic acids (11). Quite tellingly, the activities of NHase, the related thiocyanate hydrolase, and NHase mimics were found to depend on the presence of these sulfenato ligands where oxidation to the sulfinate abrogated activity (12–14).
To find direct evidence of either a protein-based or a solvent-based nucleophile, we became interested in the observations made by Heinrich et al. (14) with an iron NHase mimic bearing two sulfenato ligands that is capable of hydrating nitriles with up to 50 turnovers. They discovered that labeled oxygen from solvent water was exchanged into one of these sulfenato groups only if the nitrile hydration reaction occurred. This provides a way to test the mechanistic paradigms shown in Fig. 1, B and C, with actual NHase enzymes. In 18O-labeled water, the mechanism shown in Fig. 1B predicts that a single turnover should result in a product with no isotopic enrichment as the [16O]oxygen originally from the sulfenato ligand is used and not the bulk solvent. This mechanism also predicts that after a single turnover in 18O-labeled water, the enzyme should then possess the labeled oxygen derived from solvent in the sulfenato group. By contrast, the mechanism in Fig. 1C predicts that solvent-derived oxygen would be incorporated on the first and all subsequent turnovers and that the protein would never become enriched with solvent-derived oxygen. Single turnover experiments to distinguish between these possibilities are challenging because of the relatively high catalytic efficiency of this class of enzyme with turnover rates for aromatic nitrile substrates ranging from 90 to 159 s−1 and turnover rates for aliphatic nitrile substrates approaching 1,000 s−1 and even 4,500 s−1 for a thermophilic NHase (15–18).
Toyocamycin nitrile hydratase (TNH) is a cobalt-type nitrile hydratase, encoded by the toyJKL genes of Streptomyces rimosus, which catalyzes the hydration of toyocamycin to sangivamycin (19). Interestingly, although homologous to other NHase proteins, TNH is a three-subunit hexameric protein complex (20) where the α-subunit is homologous to the α-subunit of other nitrile hydratases, and the β- and γ-subunits (ToyK and ToyL, respectively) are homologous to the N- and the C-terminal halves of what is the single β-subunit in other known NHases (19).
The active site of NHases is at the interface of the subunits with β-subunit residues within hydrogen-bonding distance of the modified cysteine ligands of the active site metal complex (4, 15). Despite the importance of the β-subunit for catalytic efficiency (21), the recombinantly produced α-subunit of TNH (ToyJ) is catalytically active in the absence of the β- and γ-subunits (16). Moreover, as purified, the α-subunit alone contains one equivalent of cobalt(III) and three additional oxygen atoms, as expected for a fully post-translationally modified NHase α-subunit. The main difference between ToyJ and the full complement of ToyJKL is that ToyJ alone has lower catalytic efficiency (kcat/Km) and specificity for toyocamycin, suggesting that the β-subunits generally, or the β- and γ-subunits specifically in TNHase, are responsible for the substrate specificity and increased catalytic efficiency of these enzymes (16).
ToyJ is a convenient model system with which to explore the mechanism of NHases as the slow turnover number (kcat = 0.44 ± 0.04 s−1) and high Km (15 ± 2 mm) allow for easily accessed single turnover conditions that would be challenging to achieve with the subunit-replete enzyme, ToyJKL (kcat = 159 ± 2 s−1 and Km = 2.8 × 10−2 ± 1 × 10−3 mm) (16). Foremost among the advantages of ToyJ is that it provides a means of testing the reaction without the need for large, and consequently costly, quantities of labeled water that would be required for stopped flow experiments. In this study, we have carried out 18O incorporation experiments using H218O and ToyJ, which unambiguously show that the source of the product amide oxygen is a non-exchangeable ToyJ-bound oxygen that, when taken in the context of other studies of NHases (4–6), is likely the nucleophilic oxygen of the cysteine-sulfenate.
Experimental Procedures
ToyJ was purified as described previously (16) except that 50 mm sodium chloride was included in each purification buffer. The concentration of active ToyJ, as determined by cobalt metal content, was quantified by inductively coupled plasma mass spectrometry performed by the Arizona Laboratory for Emerging Contaminants at the University of Arizona. The concentration of active sites in each assay was determined solely by the cobalt concentration.
Monitoring Turnover by ToyJ
Turnover of ToyJ was monitored as follows. Reactions were initiated by mixing 0.1 mm toyocamycin (Berry and Associates, Dexter, MI) in 50 mm potassium phosphate, pH 6.8, with 30 μm ToyJ in the same buffer, which had been lyophilized and resuspended in water. An aliquot of the reaction mixture (20 μl) was withdrawn at various times (from ∼10 s to 2 h) and mixed with 4 μl of 30% (v/v) trichloroacetic acid. The precipitated protein was removed by centrifugation. The residual substrate toyocamycin and resultant product sangivamycin were quantified using reverse phase HPLC analysis as described previously (22) except that 50 mm ammonium acetate was used in the mobile phase.
Incorporation of 18O into Sangivamycin and ToyJ
Theturnover-dependent isotopic contents of both ToyJ and the product amide, sangivamycin, were determined as follows. Substrate (at 2 μm, 5 μm, 10 μm, 20 μm, 50 μm, 0.2 mm, 0. 5 mm, 2 mm, 10 mm, and 20 mm) and enzyme (60 μm) solutions were prepared separately, each in half the final volume, flash-frozen with liquid nitrogen, and lyophilized to remove natural abundance water. All protein aliquots were flash-frozen just once after purification to maintain the highest possible activity. For each assay, the lyophilized protein and substrate were resuspended in equal volumes of 97% enriched [18O]water (Cambridge Isotopes, Tewksbury, MA) and were incubated separately for 1 h to ensure complete exchange. The reactions were initiated by pipetting the protein solution into the substrate solution, and the mixtures were incubated for 2 h, after which 10 μl were removed and flash-frozen for characterization of ToyJ by mass spectrometry. The remaining samples were applied to a 10-kDa centrifugal filter with modified polyethersulfone support to remove protein for analysis of the product.
The eluent containing the sangivamycin product was analyzed by HPLC using the gradient described above. The HPLC was coupled to a Thermo Fisher LTQ Orbitrap XL instrument for in-line mass analysis. The MS was set to focus on m/z 200 to 400 in the positive mode at 100,000 resolution.
To characterize 18O incorporation into the protein, samples of ToyJ taken from the assays were first buffer exchanged into 0.1 m ammonium acetate via Amicon ultra-0.5 centrifugal filter devices with 10-kDa cutoff (EMD Millipore, Billerica, MA). The final concentration of the protein was estimated to be ∼5 μm. Samples were infused directly into an Exactive Plus EMR mass spectrometer (Thermo Fisher Scientific). Glass capillaries (approximate outer diameter of 1.5–1.8 mm and wall thickness of 0.2 mm) for nano-electrospray ionization were pulled on a P-97 micropipette puller (Sutter Instruments, Hercules, CA) and filled with sample solution. A 0.9–1.1-kV ionization voltage was applied to a platinum wire inserted into the back of the capillary. Typical instrument settings for native mass spectrometry experiments were: m/z range 400–8000, in-source CID 20 V, HCD 20 V, resolution 140,000, microscan 10, fixed AGC, and injection time 200 ms, capillary temperature 275 °C, S-lens RF 101, source DC offset 25 V, injection flat pole DC 12 V, inter flat pole lens 10 V, bent flat pole DC 8 V, transfer multipole DC tune offset −20 V, C-trap entrance lens tune offset 2 V, trapping gas pressure 5. Protein mass spectra in the m/z range 1500–3200 were charge-deconvolved and deisotoped by Thermo Xtract software. The Xtract settings were: generate MH+ Masses Mode, resolution 100,000 at 200, S/N Threshold 10, fit factor 44%, remainder 25%, AveragineLowSulfur model, max charge 30.
The relative abundances of unlabeled and singly 18O-labeled ToyJ populations were estimated as follows. Theoretical isotopic envelopes were simulated by combining theoretical spectra of 1–99% singly 18O-labeled ToyJ with unlabeled ToyJ. The isotope distributions of 100% unlabeled ToyJ and the 100% singly 18O-labeled ToyJ were obtained from the Bruker Isotope Pattern software from the theoretical protein formula. The experimental profile data were processed to centroid data, noise-subtracted, and normalized. The processed experimental data were compared with the family of theoretical data, and the best fits were estimated by calculating the sum of squared deviation of the intensity for each of the peaks. The entire process is illustrated in Fig. 2 and coded in a Matlab script (see supplemental material).
FIGURE 2.

Analysis of ToyJ mass spectrometry data for incorporation of 18O. The steps used to analyze the mass spectra of ToyJ are depicted. In step 1, the experimental profile data were processed to centroid data, noise-subtracted, and normalized. In step 2, a library of theoretical envelopes corresponding to 1–99% singly 18O-labeled ToyJ with unlabeled ToyJ in the +8 charge state was constructed using Bruker Isotope Pattern software. In step 2, the processed experimental data were finally compared with the family of theoretical data, and the best fits were estimated by calculating the sum of squared deviation of the intensity for each of the peaks.
The turnover-dependent exchange of labeled water from bulk solvent to enzyme and eventually to substrate was modeled with KinTek Explorer software (23) using the following scheme.
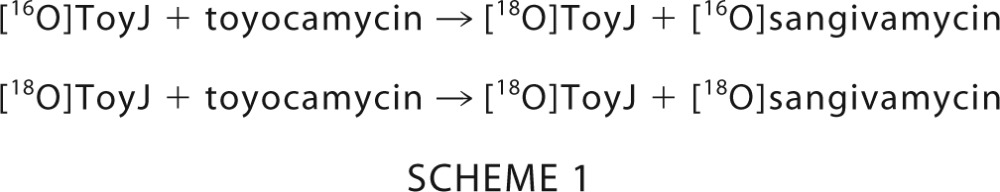 |
The concentrations of [16O]ToyJ, [18O]ToyJ, [16O]sangivamycin, and [18O]sangivamycin were computed as a function of constant [ToyJ] and 0.001–10 mm toyocamycin. In this model, we neglected the contribution to labeling of [16O]ToyJ by exchange of bulk solvent ± sangivamycin because the controls described above showed this to be negligible.
Results and Discussion
The key to distinguishing between mechanisms of nitrile hydratase proposed in the literature (4–8), those invoking an activated water molecule or sulfenate acting as the nucleophile, is to determine the source of the amide oxygen in the product. To this end, we tested whether it is feasible to carry out the reaction under single turnover conditions with ToyJ in a timescale allowing for hand mixing after being lyophilized and reconstituted in small volumes of 97% [18O]water. In these experiments, 30 μm ToyJ was incubated with 0.1 mm of the substrate, toyocamycin, and formation of the product, sangivamycin, was monitored by LC-MS. As expected given the sub-saturating concentration of substrate (Km = 15 ± 2 mm) (16), there is little turnover (4%) occurring in the reaction after 10 s, and quantitative conversion of toyocamycin to sangivamycin is achieved in ∼120 min (Fig. 3). Additionally, we tested the effect of lyophilization on the activity of the protein and determined that ToyJ retains ∼82% of the starting activity after lyophilization.
FIGURE 3.
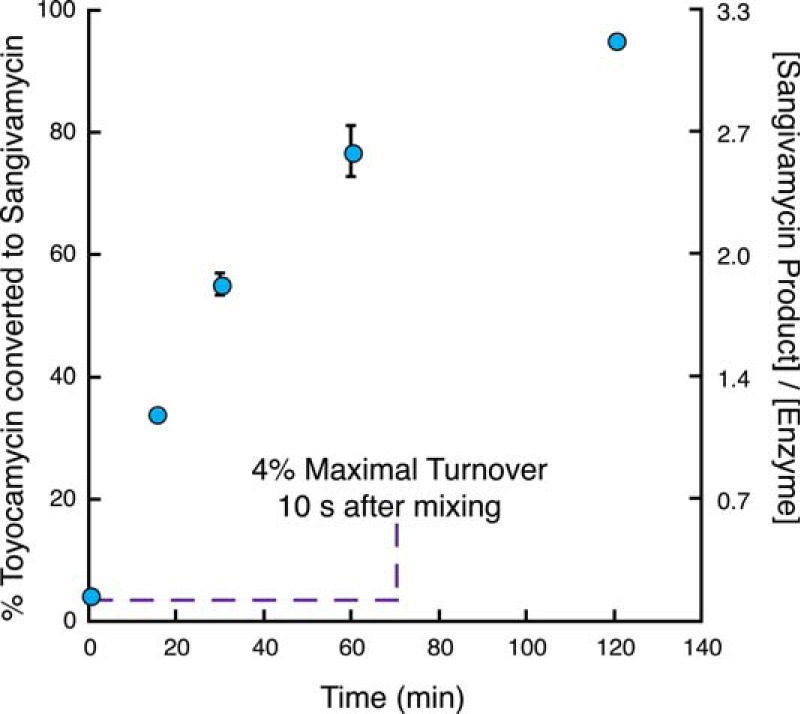
Time dependence of conversion of toyocamycin to sangivamycin. Quantitative conversion of 0.1 mm toyocamycin to sangivamycin is achieved in the presence of 30 μm ToyJ. Error bars indicate means ± S.E.
The source of the amide oxygen of sangivamycin was explored by carrying out the incubations under single and multiple turnover conditions by using a 24-fold excess of ToyJ active sites to toyocamycin to achieve single turnover, and up to a 400-fold excess of toyocamycin relative to ToyJ for multiple turnovers. The products were analyzed by LC-MS. The mass spectrum of natural abundance sangivamycin control sample (Fig. 4A) exhibits a base peak at m/z of 310.1146 m/z, exactly the theoretical m/z value for sangivamycin with the molecular formula [C12H15N5O5 + H]+. The peak corresponding to the presence of a single 18O [18O-M+H]+ appears at m/z 312.1189, exactly matching the theoretical m/z value for [C12H15N5O418O1 + H]+. This peak is 0.99 ± 0.02% as abundant as the base peak, which is consistent with the natural abundance of 18O in a molecule containing five oxygens (5 × 0.2% = 1%). The high resolution Orbitrap mass analyzer can resolve the 18O-containing peak from other relatively abundant isotope combinations giving a +2 peak, such as the species containing two 13C [C1013C2H15N5O5 + H]+ at m/z 312.1213 (expected abundance: 0.8%; observed abundance: 0.77 ± 0.01%), a single 13C and a single 15N [C1113C1H15N415N1O5 + H]+ at m/z 312.1150 (expected abundance: 0.2%; observed abundance: 0.20 ± 0.01%), and other lower abundance isotope combinations (highlighted in Fig. 4A) that are expected to be at or above a 0.02% abundance relative to the base peak at m/z 310.1146.
FIGURE 4.

Exchange of 18O from bulk solvent into sangivamycin is turnover-dependent. The mass spectra of sangivamycin in the +1 charge state are shown. The natural abundance peak is at m/z 310.1146, and that of the [18O]sangivamycin peak is at m/z 312.1189. The traces shown are for unlabeled sangivamycin (A), sangivamycin produced from 10 mm toyocamycin in 97% 18O-labeled water (B), sangivamycin produced from 1 μm toyocamycin in 97% 18O-labeled water (C), and control incubation of unlabeled sangivamycin with ToyJ in 97% 18O-labeled water (D). The high resolution spectra were obtained using an LTQ Orbitrap XL mass spectrometer.
When ToyJ is incubated with 10 mm toyocamycin in [18O]water, the base peak of the product sangivamycin shifts to m/z 312.1189 (Fig. 4B), indicating that turnover in [18O]water leads to incorporation of bulk solvent into the product. In contrast to this multiple turnover experiment, when a sub-stoichiometric concentration of substrate is used, such as 1 μm toyocamycin (Fig. 4C), the base peak for [M+H]+ of sangivamycin appears at m/z 310.1146, as it does with the natural abundance product. The singly 18O-labeled peak is slightly enriched when compared with that observed in the natural abundance sangivamycin control, 3.5 ± 0.8% when compared with 0.99 ± 0.02%. This may arise because a small percentage of enzymes may undergo multiple turnovers, and this effect can be amplified in the course of mixing. Nevertheless, the fact that nearly all the product contains 16O, when the reaction is carried out in 97% 18O-labeled water, shows that the newly installed oxygen in the product does not derive directly from bulk solvent.
Control reactions were carried out where the product amide sangivamycin was treated under assay conditions with 30 μm ToyJ in 97% enriched [18O]water. No exchange of 18O was observed, as the ratio of the 18O-containing peak relative to the base peak of unlabeled sangivamycin was indistinguishable from that of sangivamycin produced and incubated in natural abundance water (compare Fig. 4A and Fig. 4D). Indeed, the relative intensities of the various natural abundance isotopes at M+2 are identical to those observed with the unlabeled sangivamycin control. Therefore, incorporation of labeled water at neutral pH does not occur independent of catalysis on the time scale of the reactions.
A water molecule tightly bound in the active site is unlikely to be the source of this product carboxamido oxygen. The protein in these assays underwent lyophilization to remove natural abundance water and was also incubated for at least an hour after being resuspended in the labeled [18O]water before being added to substrate. Studies using 17O-labeled water and electron paramagnetic spectroscopy have shown that water molecules in the active site, specifically the active site metal-bound aquo or hydroxo ligand, are easily exchangeable (24, 25). Additionally, the active sites of NHases exist at the interface of the α- and β-subunits (26, 27), and because ToyJ in this experiment is monomeric (16), the active site is unlikely to harbor tightly non-exchangeable water molecules as it is probably exposed to the solvent. We note that no crystal structure of this specific NHase, TNH or ToyJKL, has been solved, but a recent study using multiple mass spectrometry techniques has shown that the structure of TNH is highly similar to other known structures of nitrile hydratases (28).
This series of experiments is most consistent with a mechanism whereby ToyJ is obligatorily labeled on a single turnover from bulk [18O]water and transfers the resulting 18O to product on subsequent turnovers. The solvent having been ruled out as the source of the product amide oxygen, we interrogated the protein using high-resolution mass spectrometry to determine whether there is indeed a turnover-dependent exchange of oxygen from solvent to enzyme after one turnover as would be expected if a single oxygen is transferred to product from enzyme.
ToyJ incubated in 97% H218O in the absence of substrate shows a mass envelope at the +8 charge state within 13 ppm of the theoretical natural abundance envelope for the fully post-translationally mature enzyme containing the cobalt complex with the sulfinic and sulfenic acid residues, which shows that there is no exchange of oxygen into the enzyme from labeled water in the time scale of the assays (Fig. 5A). However, ToyJ incubated in this manner but with the addition of 400-fold excess of the nitrile substrate, toyocamycin, shows an envelope shifted 0.25 m/z higher at the +8 charge state, which corresponds to a single 16O in the protein being exchanged for 18O (8 × 0.25 = 2 Da). This mass increase is within 13 ppm error of the theoretical values based on the isotopic distribution. The exchange of one oxygen upon multiple turnovers is shown clearly in the deconvolved spectra in Fig. 5B where ToyJ having undergone multiple turnovers is assigned a mass of 23,519.30 Da, which is 2.01 Da higher than ToyJ incubated in the absence of substrate at 23,517.29 Da. In accordance with the labeling of the product amide sangivamycin, ToyJ is clearly becoming labeled by bulk solvent only upon turnover.
FIGURE 5.

Exchange of 18O from bulk solvent into ToyJ is turnover-dependent. A, the deconvoluted mass spectra shown are of the +8 charge state of ToyJ. The envelope shifts 0.25 m/z units after multiple turnovers corresponding to the incorporation of just one labeled [18O]oxygen from solvent (8 × 0.25 = 2 Da). Black lines are the theoretical envelopes corresponding to ToyJ and singly 18O-labeled ToyJ. B, the deconvolved and deisotoped spectra of ToyJ from the 0 and 10 mm toyocamycin samples are shown. The ToyJ-sodium adducts are denoted with an asterisk. The pattern of sodium adducts is influenced by the varying spray conditions used. High resolution spectra were measured using an Exactive Plus EMR mass spectrometer.
Intriguingly, although ToyJ does not undergo exchange of oxygen with solvent water in the time scale of the assays, when ToyJ is incubated with the natural abundance product amide, sangivamycin, in labeled water, a slight increase in the average mass of this ToyJ is observed on the order of 10% of that which occurs with substrate nitrile. This suggests that the amide is able to bind in a catalytically similar fashion to poise the nucleophilic oxygen of ToyJ for exchange (Fig. 6).
FIGURE 6.

Possible mechanism of sangivamycin-induced exchange of labeled oxygen from solvent into ToyJ. Bonds from enzyme to modified cysteine and amidate ligands are omitted for clarity.
To further probe the source of the amide oxygen, we analyzed both sangivamycin and ToyJ under conditions where ToyJ was mixed with various concentrations of substrate nitrile ranging from sub-stoichiometric (1 μm) to excess (10 mm) relative to ToyJ. The mass spectra of ToyJ from these experiments are shown in Fig. 7, which also details the full m/z range used to acquire these data (Fig. 7A) as well as the zoomed-in range used for deconvolution (Fig. 7B) and a further zoomed-in region showing the isotopically resolved envelope of ToyJ in the +8 charge state (Fig. 7C). Remarkably, bulk 18O appears in the product and enzyme but only when approximately one turnover has been completed by the enzyme.
FIGURE 7.

ToyJ is labeled by 18O in a turnover-dependent manner. The full range mass spectrum containing ToyJ in the +8 and +9 charge states and the smaller range used for deconvolution to produce Fig. 8 are shown in A and B, respectively. C, the zoomed-in region of the isotopically resolved ToyJ in the +8 charge state after various numbers of turnovers with toyocamycin in 18O-labeled water. High resolution spectra were measured using an Exactive Plus EMR mass spectrometer.
The proportion of unlabeled to singly 18O-labeled ToyJ was extracted from analysis of the data detailed under “Experimental Procedures” and summarized in Fig. 2. The incorporation of 18O into the product was monitored by LC-MS as described above. The resulting data from both product amide and enzyme are overlaid in Fig. 8. Remarkably, the incorporation of bulk solvent into the protein (shown as purple squares) parallels the data on the appearance of 18O in sangivamycin (shown as red circles). These incorporation profiles were analyzed quantitatively using KinTek Explorer software (23) and a model that is depicted in Fig. 8 invoking labeling of ToyJ on each turnover to produce [18O]ToyJ. The data from the model (solid lines in Fig. 8) show that the incorporation of 18O from bulk solvent into sangivamycin mirrors the experimental observations for incorporation of 18O into both sangivamycin and ToyJ. We note that in the course of some of the experiments, after 2 h of incubation at room temperature, proteolytic fragments of ToyJ still containing the active site region corresponding to amino acids 32–214 are detectable in the mass spectra. Close examination of the mass envelopes for these proteins also showed the same turnover-linked increase in 18O content.
FIGURE 8.
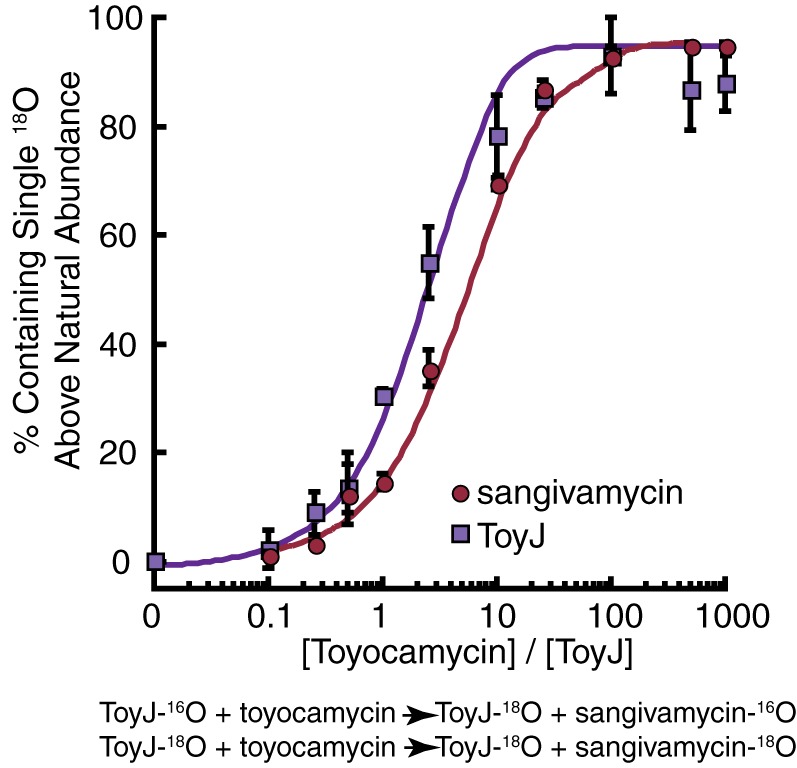
Turnover-dependent exchange of 18O from bulk solvent into ToyJ and sangivamycin. Turnover-dependent exchange of a single oxygen into enzyme, ToyJ, and the product amide, sangivamycin, was measured in the presence of various ratios of toyocamycin to ToyJ. Red circles indicate the percentage of the sangivamycin product that has incorporated an [18O]oxygen, and purple squares indicate the percentage of ToyJ that has incorporated a single[18O]oxygen after increasing numbers of turnovers. The solid curves are generated from theoretical values obtained from KinTek Explorer software according to the depicted kinetic model. Error bars indicate means ± S.E.
As we were preparing this manuscript, an elegant study using time-resolved crystallography and FTIR on the Rhodococcus iron-containing NHase was reported that showed the sulfenato oxygen is likely this protein-based nucleophile (6). Earlier time-resolved studies using an isonitrile substrate analog suggested the sulfenic oxygen may be acting as a general base to activate a water molecule to attack the isonitrile carbon (7). The 18O incorporation data in the present study (Fig. 8) rule out a solvent-derived nucleophile mechanism (Fig. 1C) and thus strongly support the mechanism involving the enzyme-based oxygen nucleophile in the form of a sulfenate nucleophile (Fig. 1B), which is supported by recent computational studies suggesting the sulfenato nucleophile mechanism (4, 5). This work provides the first direct evidence from the solution phase that a protein-based nucleophilic oxygen is the source of the carboxamido oxygen in product amides of NHases, representing the first evidence acquired from both product and enzyme directly.
Author Contributions
M. T. N., Y. S., V. H. W., and V. B. conceived of the experiments and wrote this paper. M. N. and Y. S. performed the experiments.
Supplementary Material
This work was supported by NIGMS of the National Institutes of Health Grants R01 GM72623 (to V. B.) and R01 GM113658 (to V. H. W.), and a Career Award in Biomedical Sciences from the Burroughs Wellcome Fund (to V. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as a Paper of the Week.

This article contains a supplemental Matlab script.
- NHase
- nitrile hydratase
- TNH
- toyocamycin nitrile hydratase.
References
- 1.Nagasawa T., Nanba H., Ryuno K., Takeuchi K., and Yamada H. (1987) Nitrile hydratase of Pseudomonas chlororaphis B23: purification and characterization. Eur. J. Biochem. 162, 691–698 [DOI] [PubMed] [Google Scholar]
- 2.Kobayashi M., and Shimizu S. (1998) Metalloenzyme nitrile hydratase: structure, regulation, and application to biotechnology. Nat. Biotechnol. 16, 733–736 [DOI] [PubMed] [Google Scholar]
- 3.Kovacs J. A. (2004) Synthetic analogues of cysteinate-ligated non-heme iron and non-corrin cobalt enzymes. Chem Rev. 104, 825–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hopmann K. H. (2014) Full reaction mechanism of nitrile hydratase: a cyclic intermediate and an unexpected disulfide switch. Inorg. Chem. 53, 2760–2762 [DOI] [PubMed] [Google Scholar]
- 5.Light K. M., Yamanaka Y., Odaka M., and Solomon E. I. (2015) Spectroscopic and computational studies of nitrile hydratase: insights into geometric and electronic structure and the mechanism of amide synthesis. Chem. Sci. 6, 6280–6294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamanaka Y., Kato Y., Hashimoto K., Iida K., Nagasawa K., Nakayama H., Dohmae N., Noguchi K., Noguchi T., Yohda M., and Odaka M. (2015) Time-resolved crystallography of the reaction intermediate of nitrile hydratase: revealing a role for the cysteine sulfenic acid ligand as a catalytic nucleophile. Angew. Chem. Int. Ed. Engl. 54, 10763–10767 [DOI] [PubMed] [Google Scholar]
- 7.Hashimoto K., Suzuki H., Taniguchi K., Noguchi T., Yohda M., and Odaka M. (2008) Catalytic mechanism of nitrile hydratase proposed by time-resolved x-ray crystallography using a novel substrate, tert-butylisonitrile. J. Biol. Chem. 283, 36617–36623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitra S., and Holz R. C. (2007) Unraveling the catalytic mechanism of nitrile hydratases. J. Biol. Chem. 282, 7397–7404 [DOI] [PubMed] [Google Scholar]
- 9.Adzamli I. K., Libson K., Lydon J. D., Elder R. C., and Deutsch E. (1979) Synthesis, characterization, and reactivity of coordinated sulfenic acids. Inorg. Chem. 18, 303–311 [Google Scholar]
- 10.Lydon J. D., and Deutsch E. (1981) Chemistry and reactivity of sulfur-bonded sulfenato-cobalt(III) complexes. Inorg. Chem. 21, 3180–3185 [Google Scholar]
- 11.Martinez S., Wu R., Sanishvili R., Liu D., and Holz R. (2014) The active site sulfenic acid ligand in nitrile hydratases can function as a nucleophile. J. Am. Chem. Soc. 136, 1186–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsujimura M., Odaka M., Nakayama H., Dohmae N., Koshino H., Asami T., Hoshino M., Takio K., Yoshida S., Maeda M., and Endo I. (2003) A novel inhibitor for Fe-type nitrile hydratase: 2-cyano-2-propyl hydroperoxide. J. Am. Chem. Soc. 125, 11532–11538 [DOI] [PubMed] [Google Scholar]
- 13.Arakawa T., Kawano Y., Katayama Y., Nakayama H., Dohmae N., Yohda M., and Odaka M. (2009) Structural basis for catalytic activation of thiocyanate hydrolase involving metal-ligated cysteine modification. J. Am. Chem. Soc. 131, 14838–14843 [DOI] [PubMed] [Google Scholar]
- 14.Heinrich L., Mary-Verla A., Li Y., Vaissermann J., and Chottard J. (2001) Cobalt(III) complexes with carboxamido-N and sulfenato-S of sulfinato-S ligands suggest that coordinated sulfenate-S is essential for the catalytic activity of nitrile hydratases. Eur. J. Inorg. Chem. 2203–2206 [Google Scholar]
- 15.Miyanaga A., Fushinobu S., Ito K., Shoun H., and Wakagi T. (2004) Mutational and structural analysis of cobalt-containing nitrile hydratase on substrate and metal binding. Eur. J. Biochem. 271, 429–438 [DOI] [PubMed] [Google Scholar]
- 16.Nelp M. T., Astashkin A. V., Breci L. A., McCarty R. M., and Bandarian V. (2014) The α subunit of nitrile hydratase is sufficient for catalytic activity and post-translational modification. Biochemistry 53, 3990–3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takarada H., Kawano Y., Hashimoto K., Nakayama H., Ueda S., Yohda M., Kamiya N., Dohmae N., Maeda M., and Odaka M. (2006) Mutational study on αGln90 of Fe-type nitrile hydratase from Rhodococcus sp. N771. Biosci. Biotechnol. Biochem. 70, 881–889 [DOI] [PubMed] [Google Scholar]
- 18.Pereira R. A., Graham D., Rainey F. A., and Cowan D. A. (1998) A novel thermostable nitrile hydratase. Extremophiles 2, 347–357 [DOI] [PubMed] [Google Scholar]
- 19.McCarty R. M., and Bandarian V. (2008) Deciphering deazapurine biosynthesis: pathway for pyrrolopyrimidine nucleosides toyocamycin and sangivamycin. Chem. Biol., 15, 790–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blackwell A. E., Dodds E. D., Bandarian V., and Wysocki V. H. (2011) Surface-induced dissociation reveals the quaternary substructure of a heterogeneous non-covalent protein complex. Anal. Chem. 83, 2862–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piersma S. R., Nojiri M., Tsujimura M., Noguchi T., Odaka M., Yohda M., Inoue Y., and Endo I. (2000) Arginine 56 mutation in the β subunit of nitrile hydratase: importance of the hydrogen bonding to the non-heme iron center. J. Inorg. Biochem. 80, 283–288 [DOI] [PubMed] [Google Scholar]
- 22.Pomerantz S. C., and McCloskey J. A. (1990) Analysis of RNA hydrolyzates by liquid chromatography. Methods Enzymol. 193, 796–824 [DOI] [PubMed] [Google Scholar]
- 23.Johnson K. A., Simpson Z. B., and Blom T. (2009) Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29 [DOI] [PubMed] [Google Scholar]
- 24.Sugiura Y., Kuwahara J., Nagasawa T., and Yamada H. (1987) Nitrile hydratase: the first non-heme iron enzyme with a typical low-spin Fe(III)-active center. J. Am. Chem. Soc. 109, 5848–5850 [Google Scholar]
- 25.Jin H., Turner I. M., Nelson M. J., Gurbiel R. J., Doan P. E., and Hoffman B. M. (1993) Coordination sphere of the ferric ion in nitrile hydratase. J. Am. Chem. Soc. 115, 5290–5291 [Google Scholar]
- 26.Nagashima S., Nakasako M., Dohmae N., Tsujimura M., Takio K., Odaka M., Yohda M., Kamiya N., and Endo I. (1998) Novel non-heme iron center of nitrile hydratase with a claw setting of oxygen atoms. Nat. Struct. Biol. 5, 347–351 [DOI] [PubMed] [Google Scholar]
- 27.Miyanaga A., Fushinobu S., Ito K., and Wakagi T. (2001) Crystal structure of cobalt-containing nitrile hydratase. Biochem. Biophys. Res. Comm. 288, 1169–1174 [DOI] [PubMed] [Google Scholar]
- 28.Song Y., Nelp M. T., Bandarian V., and Wysocki V. H. (2015) Refining the structural model of heterohexameric protein complex: surface induced dissociation and ion mobility provide key connectivity and topology information. ACS Cent. Sci. 1, 477–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.