

Abstract
Periplasmic substrate-binding proteins (SBPs) bind to the specific ligand with high affinity and mediate their transport into the cytoplasm via the cognate inner membrane ATP-binding cassette proteins. Because of low sequence identities, understanding the structural basis of substrate recognition by SBPs has remained very challenging. There are several structures available for the ligand-bound sugar SBPs, but very few unliganded structures are reported. No structural data are available for sugar SBPs from Pseudomonas sp. to date. This study reports the first high resolution crystal structures of periplasmic glucose-binding protein from Pseudomonas putida CSV86 (ppGBP) in unliganded form (2.5 Å) and complexed with glucose (1.25 Å) and galactose (1.8 Å). Asymmetric domain closure of ppGBP was observed upon substrate binding. The ppGBP was found to have an affinity of ∼ 0.3 μm for glucose. The structural analysis showed that the sugars are bound to the protein mainly by hydrogen bonds, and the loss of two strong hydrogen bonds between ppGBP and galactose compared with glucose may be responsible for lowering its affinity toward galactose. The higher stability of ppGBP-glucose complex was also indicated by an 8 °C increase in the melting temperature compared with unliganded form and ppGBP-galactose complex. ppGBP binds to monosaccharide, but the structural features revealed it to have an oligosaccharide-binding protein fold, indicating that during evolution the sugar binding pocket may have undergone structural modulation to accommodate monosaccharide only.
Keywords: ABC transporter, glucose transport, protein structure, Pseudomonas, structure-function, substrate specificity, sugar transport, periplasmic glucose-binding protein, x-ray crystallography, sugar binding pocket
Introduction
The periplasmic substrate-binding proteins (SBPs)5 are involved in the transport of a large variety of ligands (1–4) via the ATP-binding cassette (ABC) transporters, which are located in the inner membrane (2). The SBPs specifically bind to their cognate ligand with high affinity, enabling rapid response even in the presence of very low concentrations of the ligand (3). The SBP-ligand complex binds to the cognate inner membrane ABC transporter and transmit a transmembrane signal for ATP hydrolysis by the ATPase to energize the transport process (4, 5). This leads to opening of the channel formed by the transmembrane subunits of the ABC transporter (2, 6). These properties of substrate specificity and high binding affinity have led to the use of SBPs for the design of reagentless biosensors (7, 8), allosteric control elements (9) and enzymes (10).
Despite low sequence similarity among various SBPs, the overall three-dimensional fold is quite similar with significant differences in the binding pocket (6, 11). The overall three-dimensional structures of the SBPs are composed of two α/β-domains with each domain having a β-sheet core surrounded by α-helices and are connected by a hinge region (11). The class I SBPs have a β2β1β3β4β5-sheet topology in both domains. The class II SBPs have β2β1β3βnβ4-sheet topology where n is the first crossover from the N-terminal domain to the C-terminal domain or vice versa (12). The class III SBPs have a single α-helix in the hinge region between the two domains (13). Recently, with the increase in the number of SBP structures deposited in the Protein Data Bank, the SBPs were reclassified into six clusters (A–F) based on structural alignment (6) in which monosaccharide- and disaccharide-binding proteins are grouped in clusters B and D, respectively. The unliganded SBPs predominantly exist in the open conformation with their domains separated from each other. The ligand binding induces closure of these domains through asymmetric hinge-bending motions, termed the “Venus fly trap” mechanism (14). Although the substrate-binding proteins have been well studied, there are very few structures reported where both the apo and sugar-bound forms are available depicting the conformational changes upon substrate binding in the protein. Among monosaccharide-binding proteins, only the Escherichia coli glucose/galactose-binding protein (ecGGBP) structure has been determined in unliganded and glucose-complexed forms (15). Because the SBPs have low sequence identity (less than 20%), phylogeny analysis based on multiple sequence alignments is not very reliable (6). However, the structural fold similarities among the various SBPs may be considered as a better parameter to study the evolutionary relatedness of the proteins belonging to this superfamily (2). Therefore, structural studies on SBPs from new organisms are essential.
Pseudomonas putida CSV86, a Gram-negative soil isolate, preferentially utilizes aromatics and organic acids over glucose (16). The repression of the glucose transport system in the presence of aromatics and organic acids was found to be responsible for this novel property (16, 17). The draft genome sequence of this organism has been determined (18, 19). It is proposed that the components of glucose transport include an outer membrane protein, OprB (17); a periplasmic glucose-binding protein, GBP (20, 21); and a putative glucose ABC transporter (64).6 The detailed mechanism of sugar transport in Pseudomonas sp. is unknown. The glucose-binding protein of P. putida CSV86 (ppGBP) has low sequence identity with other SBPs for which structure is known. The structural and biochemical studies of ppGBP will provide detailed insights into the sugar binding mechanism and therefore aid in understanding sugar transport in Pseudomonas sp. Although there is no direct evidence of involvement of the glucose transport system in pathogenesis of Pseudomonas sp., in P. aeruginosa it has been reported that the transport of carbon sources is required for biosynthesis of virulence factors such as alginate (22). Therefore, information pertaining to the structure and function of the components of this transport system is important. Structures of the unliganded and ligand-bound forms of ppGBP will reveal the ligand-induced conformational changes.
In this study, we report the first high resolution crystal structures of ppGBP from Pseudomonas sp. in unliganded form and its complex with glucose and galactose. The structural analysis and site-specific mutation studies revealed the basis for substrate specificity of ppGBP. We have compared the mode of sugar binding in SBPs for the first time and observed striking differences. The structural comparison also indicated that ppGBP is a close relative of monosaccharide-binding protein from thermophilic bacteria.
Experimental Procedures
Protein Purification
Purification of recombinant ppGBP was performed as described earlier (21, 23). Briefly, a single colony of pET28a-ppGBP-transformed E. coli BL21(DE3) was grown overnight at 37 °C in LB medium (10 ml) supplemented with kanamycin (30 μg/ml). The culture (1%, v/v) was reinoculated into LB medium (1 liter) containing kanamycin (30 μg/ml), grown at 37 °C to an optical density of 1.0 at 600 nm, and induced by addition of isopropyl 1-thio-β-d-galactopyranoside (100 μm) for 4 h. Cells were harvested (8000 × g for 10 min) and resuspended in binding buffer (10 mm Tris-Cl, pH 7.5, 1 mm MgCl2). Cell-free lysate was prepared by sonication (three cycles/g of cells; 1-s pulse; 1-s interval; cycle duration, 30 s; output, 10 watts) followed by centrifugation (20,000 × g for 20 min). The supernatant was subjected to nickel-nitrilotriacetic acid affinity chromatography followed by Superdex S-200 gel filtration chromatography as described earlier (21, 23). Purity of the protein was assessed by SDS-PAGE (12%) (24). The protein concentration was determined by the Bradford method (25).
Crystallization of Unliganded and Complexed ppGBP
Purified ppGBP was concentrated up to 20 mg/ml for setting up crystallization. The ppGBP crystals complexed with glucose were obtained as described earlier (23). The purified protein was incubated on ice for 30 min with a 10 molar excess of galactose/glucose prior to crystallization for ppGBP-sugar complex formation. The unliganded ppGBP and glucose- and galactose-complexed ppGBP were crystallized in the condition of 0.1 m phosphate citrate buffer and 2 m ammonium sulfate at pH 4.6 by the hanging drop vapor diffusion method at 295 K. Upon optimization of this condition, the best crystals appeared in the same precipitant at pH 4.8. The glucose-complexed and galactose-complexed ppGBP crystals attained the maximum size (0.6 × 0.6 × 0.6 mm) in 2 days, whereas the unliganded ppGBP crystals attained maximum size (0.1 × 0.4 × 0.1 mm) in 2 weeks.
X-ray Diffraction, Data Collection, and Processing
The unliganded and galactose-complexed crystals were cryoprotected using the reservoir solution also containing glycerol (30%). The crystals were briefly transferred to the cryoprotectant solution and then to a liquid nitrogen stream at 100 K. The diffraction data sets from these crystals were collected by rotation method. X-ray diffraction data for unliganded ppGBP crystal were collected at the Protein Crystallography Facility, Indian Institute of Technology Bombay using CuKα radiation generated by a Rigaku Micromax 007HF generator equipped with R-Axis IV++ detector. Diffraction data from the galactose-bound ppGBP crystals were collected at the Advanced Centre for Treatment, Research and Education in Cancer (Navi Mumbai, India) using CuKα radiation generated by a Bruker MICROSTARgenerator equipped with a MAR345 image plate detector. The ppGBP-glucose complex crystals were cryoprotected using the reservoir solution also containing glycerol (30%). The crystals were briefly transferred to the cryoprotectant solution and then subsequently flash frozen in liquid nitrogen. A frozen crystal was then transferred to a liquid nitrogen stream at 100 K. Diffraction data were collected at BM14 of the European Synchrotron Radiation Facility (Grenoble, France) using a MarCCD detector. The high resolution data were collected with a 0.1° oscillation with a 1.5-s exposure time. The data for the ppGBP-glucose complex were truncated by omitting images collected near the end of the data collection because those images showed signs of radiation damage. The data sets from apo, glucose-, and galactose-complexed crystals were indexed and integrated using XDS (26). The intensities were converted to structure factors with the program modules F2MTZ and CAD of CCP4 (27). The data collection statistics of these crystals are presented in Table 1.
TABLE 1.
Data collection and refinement statistics
| ppGBP | ppGBP-glucose | ppGBP-galactose | |
|---|---|---|---|
| Data collectiona | |||
| Wavelength (Å) | 1.5418 | 0.9763 | 1.5418 |
| Resolution range (Å) | 20–2.5 (2.6–2.5) | 40–1.25 (1.35–1.25) | 20–1.8 (1.9–1.8) |
| Space group | P21212 | P21212 | P21212 |
| Unit cell constants | |||
| a, b, c (Å) | a = 84.4, b = 154.6, c = 60.6 | a = 102.94, b = 119.05, c = 66.60 | a = 102.6, b = 118.8, c = 66.6 |
| α, β, γ (°) | α = β = γ = 90 | α = β = γ = 90 | α = β = γ = 90 |
| Measured reflections | 204,052 (22,393) | 1,403,213 (250,017) | 770,047 (112,572) |
| Unique reflections | 28,191 (3,094) | 224,933 (45,701) | 76,051 (11,226) |
| Mean I/σ(I) | 9.25 (2.25) | 16.41 (2.71) | 19.97 (3.66) |
| Completeness (%) | 99.7 (100) | 99.7 (99.2) | 99.9 (99.9) |
| Rmerge (%) | 22.5 (90.2) | 5.2 (66.6) | 11.5 (77.7) |
| Rmeas (%)b | 24.2 (97.1) | 5.6 (73.6) | 12.1 (81.8) |
| CC1/2 (%)c | 98.8 (73.3) | 99.9 (83) | 99.9 (88.8) |
| Redundancy | 7.24 (7.24) | 6.24 (5.47) | 10.12 (10.02) |
| Refinement | |||
| Resolution (Å) | 20–2.5 | 40–1.25 | 20–1.8 |
| Number of reflections (working set/test set) | 26,741/1,408 | 213,608/11,245 | 72,247/3,803 |
| R-factor (%) | 18.1 | 12.2 | 17.7 |
| Rfree (%) | 25.3 | 15.2 | 22.9 |
| Number of atoms | |||
| Protein | 5,936 | 6,266 | 6,043 |
| Solvent | 247 | 1,042 | 1,155 |
| Glucose | 0 | 24 | 0 |
| Galactose | 0 | 0 | 24 |
| r.m.s.d. | |||
| Bond lengths (Å) | 0.012 | 0.012 | 0.019 |
| Bond angles (°) | 1.529 | 1.599 | 1.902 |
| Isotropic average B-factor (Å2) | |||
| Chain A | 24.69 | 24.59 | 20.77 |
| Chain B | 30.02 | 19.93 | 23.75 |
| Solvent | 33.04 | 41.88 | 29.37 |
| Glucose/galactose | 0 | 11.50 | 12.19 |
| Estimated coordinate error (Å) | |||
| Based on maximum likelihood | 0.23 | 0.03 | 0.11 |
| Based on Rfree | 0.31 | 0.04 | 0.13 |
| Protein geometry (MolProbity) | |||
| Ramachandran plot favored (%) | 98 | 98 | 98 |
| Ramachandran plot allowed (%) | 2 | 2 | 2 |
| Ramachandran plot outliers (%) | 0 | 0 | 0 |
| Protein Data Bank code | 5DVF | 5DVI | 5DVJ |
a Values in parentheses correspond to highest resolution shell.
b Rmeas = Σhkl√n/(n − 1)Σj = 1n|Ihkl,j − 〈Ihkl〉|/ΣhklΣjIhkl,j where Ihkl is the average of symmetry-related observations of a unique reflection.
c CC, Pearson correlation coefficient of two “half” datasets.
Structure Determination, Model Building, and Refinement
The structure of ppGBP-glucose complex was solved using the molecular replacement method. The crystal structure of Thermus thermophilus glucose-binding protein (ttGBP; Protein Data Bank code 2B3B), which has an amino acid sequence identity of 27% with ppGBP, was used as a search model. The ttGBP model structure was modified using CHAINSAW (28) by using ppGBP and ttGBP sequence alignments as a reference. Calculation of the Matthews coefficient (29) indicated the presence of two molecules in the asymmetric unit. The correct orientation of two ppGBP molecules in the asymmetric unit was found by Phaser (30) using the modified ttGBP structure. After the first cycle of refinement of the model in REFMAC5 (31), the sigma-A-weighted Fo − Fc electron density maps indicated the presence of glucose molecules in the substrate binding pocket of ppGBP. After modeling the sugar, iterative cycles of refinement with REFMAC5 and manual model building in the electron density map using Coot (32) were carried out. Glycerol, sulfate, phosphate, and water molecules were progressively added at peaks of electron density higher than 3σ in sigma-A-weighted Fo − Fc maps while monitoring the decrease of Rfree and improvement of the overall stereochemistry of the model. The local anisotropy was modeled with translation-libration-screw (TLS) parameters by dividing the protein molecule into three TLS groups (26–135, 136–392, and 393–421 for chain A; 26–140, 141–295, and 296–421 for chain B) (33). Restrained and TLS refinements were performed at the initial cycles of refinement. In the later stages, anisotropic refinements (without TLS) were carried out taking into account the individual anisotropic displacement parameters for all the atoms. Three N-terminal residues in both chains in the asymmetric unit could not be modeled as electron density was missing for these residues.
The phases for the ppGBP-galactose structure were obtained by performing rigid body refinement of the ppGBP-glucose structure without glucose using REFMAC5 as both complexed form crystals belonged to the same space group and had almost identical cell dimensions. After the first cycle of refinement, the sigma-A-weighted Fo − Fc electron density map indicated the presence of galactose in the sugar binding pocket. After modeling the galactose, iterative cycles of structure refinement by REFMAC5 and model building were carried out using Coot. Water molecules were added to the structure, and alternate conformations of residues were built using Coot. The local anisotropy was modeled with TLS parameters by dividing the protein into three TLS groups (26–84, 85–402, and 403–421 for chain A; 26–238, 239–266, and 267–421 for chain B). Three N-terminal residues in both chains in the asymmetric unit could not be modeled as electron density was missing for these residues.
The unliganded ppGBP diffraction data were collected using a home x-ray source. The high Rmeas of this data set was mainly due to the weak reflections at higher resolution shells. Despite the high Rmeas value, incorporation of data to 2.5 Å (with mean I/σ(I) = 2.25) improved the quality of the electron density map. Two molecules of ppGBP were present in the asymmetric unit of the unliganded crystal form. The structure solution for unliganded ppGBP structure was performed through the molecular replacement module of Phaser using the glucose-bound ppGBP structure as a search model. After obtaining the initial phase, the partial protein model was built by Buccaneer (34), which could uniquely allocate 778 (98.2%) residues to both chains in the asymmetric unit. We attempted refinement of the structure by incorporating NCS restraints; however, it failed to improve the R-factor values. Furthermore, this model was refined by REFMAC5, and manual model building was done by visual inspection of the electron density map in Coot. The local anisotropy was modeled with TLS parameters by dividing the protein molecule into three TLS groups (26–59, 60–141, and 142–421 for chain A; 26–74, 75–330, and 331–421 for chain B). Water molecules were added manually in Coot. Six residues (64–69) of chain A and seven residues (64–70) of chain B could not be modeled in the unliganded ppGBP structure because of poor quality of the electron density map. All figures were generated using PyMOL.
Site-directed Mutagenesis and [14C]Glucose Binding Assay
Site-directed mutagenesis was performed to generate alanine-substituted ppGBP mutants at residues Gln90, Trp250, Trp270, Asn301, and Asp303 using pET28a-ppGBP (21). Mutants were confirmed by DNA sequencing. The mutant proteins were overexpressed in E. coli BL21(DE3) and purified using nickel-nitrilotriacetic acid affinity chromatography to homogeneity. The glucose binding activity of wild-type and mutant proteins was measured using a [14C]glucose binding assay as described earlier (16, 21).
Circular Dichroism and Thermal Denaturation Studies
Far-UV CD spectra of recombinant ppGBP and its mutants in binding buffer were recorded between 198 and 260 nm in a 0.1-cm-path length cuvette using a spectropolarimeter (Jasco J-810, Germany) with the following parameters: 25 °C; response, 2 s; sensitivity, 100 millidegrees; scan speed, 100 nm/min. The data were processed by smoothing and subtraction of spectra obtained from binding buffer alone. Ellipticity values (millidegrees) were recorded as a function of wavelength.
Thermal denaturation profiles were determined by measuring CD signals at 222 nm (0.1-cm path length) as a function of temperature using 5 μm protein (10 mm Tris-Cl, pH 7.5, 1 mm MgCl2) in the presence or absence of 1 mm sugars (glucose or galactose). Protein sample with 1 mm sugars was incubated for 15 min prior to collecting spectral data. Each measurement includes a 3-s averaging time for data collection and a 180-s equilibration period at each temperature. Data were processed using a two-state model, which accounts for the native and denatured baseline slopes to determine the Tm values (35).
Determination of Substrate Binding Constant
The sugar binding constant of ppGBP was determined by surface plasmon resonance (SPR) spectroscopy method (Biacore S200, GE Healthcare) as described earlier (36). Recombinant wild-type ppGBP (100 μg/ml or 2.2 μm in 10 mm HEPES, pH 7.4, 150 mm NaCl, 0.05% Biacore Surfactant P20) was immobilized onto a carboxymethyl dextran (CM5) surface using a nonspecific N-ethyl-N-(3-dimethylaminopropyl)carbodiimide hydrochloride/N-hydroxysuccinimide amine coupling method as described previously for E. coli maltose-binding protein (ecMBP) (37). The CM5 chip was activated with N-ethyl-N-(3-dimethylaminopropyl)carbodiimide hydrochloride/N-hydroxysuccinimide for 7 min. The recombinant ppGBP in 10 mm sodium acetate, pH 4.0, was injected at a 10 μl/min flow rate for 720 s. A pulse of 1 m ethanolamine, pH 8.0, was injected to quench the reaction and to remove nonspecifically bound protein. A surface of 6580 response units was achieved. Various glucose and galactose concentrations (0.05, 0.1, 0.2, 0.4, 0.8, 1.6, 3.2, 6.25, 12.5, 25, 50, and 100 μm) were used for the binding studies. The relative SPR signal in the presence of 100 μm glucose was found to be +16.0 response units, which was greater than the SPR signal for buffer (+0.5 response unit) run over the immobilized protein surface. Saturation was achieved at 6.25 μm glucose. All sugar injections (glucose and galactose) were introduced at a 30 μl/min flow rate with a contact time and dissociation time of 90 and 240 s, respectively. Injections of different sugar concentrations were performed two times and alternated between the protein and blank surfaces. The binding studies were performed twice with two batches of purified protein. The data were corrected with the appropriate blank and analyzed by a steady state one-site binding saturation model.
Evolutionary and Structural Adaptation Studies
The ppGBP-glucose complex structure was submitted to the DALI server (38) without ligand coordinate. Several structures with similar folds were suggested by the server of which a few sugar-bound structures were chosen to build the structure-based phylogeny tree using VMD software (39). All structural superpositions (Cα atoms) were carried out between ppGBP-glucose complex and other sugar-bound cluster B and D SBPs using Coot.
Results and Discussion
Structural Fold of ppGBP
The crystal structures of unliganded ppGBP and its complex with glucose or galactose (Fig. 1, A and B) have been determined at 2.5-, 1.25-, and 1.8-Å resolutions, respectively. In this study, the reported ppGBP-glucose complex structure is the highest resolution crystal structure (1.25 Å) so far among the SBPs belonging to cluster D. In the crystal structure, 396 of 399 amino acids of the recombinant ppGBP are correctly built in the complexed forms, and 390 residues in chain A and 389 residues in chain B are unambiguously defined in the unliganded form. The quality of the ppGBP structures after fitting the final models to the electron density maps and refinement is reflected by parameters (Table 1) such as R-factor and departure from stereochemical standard parameters. Because of the close detector distance, the data from ppGBP-glucose complex crystal were collected with 0.1° rotation per image, and the crystal was exposed to the synchrotron beam for longer time. This caused radiation damage to the crystal that is reflected by partial disruption of disulfide bonds in the structure. Despite having high resolution, ppGBP-glucose complex has a lesser number of water molecules compared with ppGBP-galactose complex. The observed disappearance of water molecules in the protein structure might be due to radiation damage, which has also been reported in the literature previously (40). The interactions and orientations of glucose and galactose in the sugar binding pocket of ppGBP are unambiguously defined by the clear electron density map (Fig. 1B). Sequence analysis (Fig. 2) showed that ppGBP has low sequence identity with that of other structurally characterized monosaccharide-binding proteins. However, the residues at the sugar binding pocket are quite similar in ppGBP, ttGBP, and a glucose-binding protein from Ochrobactrum anthropi (oaGBP). The overall structural fold of ppGBP (Fig. 1, A and C) is composed of two α/β-domains. The N-terminal (28–141, 302–337, and 343–352) and C-terminal domains (142–301, 338–342, and 353–419) are composed of five β-strands and flanked by 10 α-helices each. The β-sheet core is formed in the order of β2β1β3βnβ4 where n is β-sheet 9, forming the first crossover from the C-terminal to the N-terminal domain (Fig. 1). The hinge is composed of three strands (141–144, 298–301, and 342–343), which form the link between the two domains. Arrangements of β-strands show that the overall structural fold of ppGBP is similar to that reported for cluster D SBPs (6, 12). The structural comparison (Table 2) shows that ppGBP has a high level of structural similarity to disaccharide- and oligosaccharide-binding proteins such as trehalose/maltose-binding protein from Thermococcus litoralis (tlMBP), the maltose/maltodextrin-binding protein GacH from Streptomyces glaucescens, ecMBP, trisaccharide-binding protein from S. pneumoniae, and maltose-binding protein from Alicyclobacillus acidocaldarius. Structural superposition (Cα atoms) of ppGBP-glucose complex with other monosaccharide-bound SBPs from E. coli, Salmonella enterica serovar Typhimurium, and Thermotoga maritima belonging to cluster B resulted in high root mean square deviation (r.m.s.d.) values of 4.02, 4.3, and 4.1 Å, respectively. Despite being a glucose-binding protein, ppGBP has low structural similarity with monosaccharide-binding proteins except ttGBP (Protein Data Bank code 2B3B; r.m.s.d. = 1.66 Å) and oaGBP (Protein Data Bank code 4R2B; r.m.s.d. = 1.10 Å) of cluster D SBPs. SBPs belonging to cluster D bind to a large variety of substrates such as carbohydrates (maltose, glucose, and galacturonide), spermidine, putrescine, thiamine, tetrahedral oxyanion (phosphate and sulfate), and ferric or ferrous iron. Cluster D SBPs are all slightly larger (molecular mass >40 kDa) than cluster B SBPs (∼35 kDa) and contain one small extra subdomain as described for the ecMBP (41). Although ppGBP has overall similar structural topology to cluster D SBPs, closer inspection reveals striking differences in the sugar binding pocket. In cluster D SBP structures (ecMBP and tlMBP), a wide sugar binding pocket allows accommodation of oligosaccharides (1). In ppGBP, the sugar binding pocket is narrowed by the presence of one helix (189–204) and two loops (64–60 and 376–381) (discussed under “Structural Adaptations in ppGBP to Facilitate Monosaccharide Binding”). This structural modulation of the binding pocket volume in ppGBP allows binding of only monosaccharides like glucose/galactose. Similar structural features have also been reported for ttGBP (1) and galactose-specific substrate-binding protein AcbH from Actinoplanes sp. (42). Because of low sequence identities (<20%) and structural similarities with ppGBP, cluster B monosaccharide-binding proteins were excluded from the construction of the structure-based phylogeny tree (Fig. 3). The phylogeny analysis and structural superposition with cluster B and D SBPs show that ppGBP is more similar to thermophilic SBPs and disaccharide-binding proteins than to cluster B monosaccharide-binding proteins.
FIGURE 1.

Crystal structure of ppGBP. A, overall structural fold of ppGBP. The β-sheets of the N- and C-terminal domains are shown in purple color to highlight their arrangements. Three strands at the hinge region are shown in blue. Glucose is shown as a ball and stick. B, the sigma-A-weighted Fo − Fc omit electron density maps of glucose (yellow) and galactose (cyan) contoured at 3.0σ level with the final refined models superimposed. C, the topology diagram showing the arrangement of secondary structural elements. The α-helices are shown as cylinders, and β-sheets are shown as arrows.
FIGURE 2.

Amino acid sequence alignment of ppGBP with oaGBP, ttGBP, AcbH, and ecGGBP. The alignment was done using ClustalW (62) and is presented along with secondary structural elements of ppGBP on the top using ESPript 3.0 (63) software. The residues of ppGBP, oaGBP, and ttGBP involved in glucose/galactose binding are enclosed within a red box. The conserved and similar residues are shown in boxes with red and yellow backgrounds, respectively.
TABLE 2.
Sequence and structural comparison of ppGBP with other sugar-binding SBPs
Structural superpositions were done between Cα atoms of ligand-bound SBPs and ppGBP-glucose complex in Coot.
| SBP (Ref.) | Source | Protein Data Bank code | r.m.s.d. (Å) | Sequence identity (%) |
|---|---|---|---|---|
| Glucose-binding (this study) | P. putida CSV86 | 5DVI | ||
| Glucose transporter | O. anthropi | 4R2B | 1.2 | 48 |
| Glucose/galactose-binding (1) | T. thermophilus | 2B3B | 1.7 | 28 |
| Maltose-binding (43) | T. litoralis | 1EU8 | 2.4 | 18 |
| Galactose-binding (42) | Actinoplanes sp. | 3OO6 | 2.6 | 22 |
| ABC transporter (44) | Caldanaerobius sp. | 4G68 | 2.6 | 22 |
| Maltose-binding (45) | E. coli | 1NL5 | 2.6 | 18 |
| Maltose-binding (46) | A. acidocaldarius | 1URG | 2.7 | 20 |
| Xylobiose-BxlE complex | Streptomyces thermovidaceae | 3VXC | 2.7 | 22 |
| Xylooligosaccharide SBP (47) | Bifidobacterium animalis | 4C1U | 2.8 | 21 |
| Sugar ABC transporter | Streptococcus pneumoniae | 2I58 | 3.0 | 18 |
| Maltose-binding (48) | Caldanaerobius polysaccharolyticus | 4R9G | 3.1 | 17 |
| Maltodextrin-binding (49) | Thermoactinomyces vulgaris | 2ZYM | 3.1 | 16 |
| Trisaccharide transporter (50) | S. pneumoniae | 2W7Y | 3.3 | 21 |
| Sugar transporter | Bacillus licheniformis | 4RK9 | 3.6 | 13 |
| Sugar ABC transporter (51) | S. glaucescens | 3K02 | 3.6 | 20 |
| Glucose/galactose-binding (52) | E. coli | 1GUB | 4.0 | 16 |
| Glucose/galactose-binding (53) | T. maritima | 2FN8 | 4.1 | 16 |
| Glucose/galactose-binding (54) | S. enterica Typhimurium | 1GCG | 4.3 | 18 |
FIGURE 3.

Structure-based phylogeny tree. The phylogeny tree depicts the evolutionary relatedness among cluster D SBPs.
Substrate Binding Pocket of ppGBP
Both ppGBP-glucose and ppGBP-galactose complexes were crystallized in the same condition with almost identical unit cell dimensions (Table 1). The well resolved electron density maps for the sugars (Fig. 1B) were visible in two molecules of ppGBP-sugar complex structures. The comparison of the Cα atoms of A chain with those of B chain of ppGBP-glucose complex produces an r.m.s.d. value of 0.5 Å. The comparison of the Cα atoms of A chain with those of B chain of ppGBP-galactose complex also produces an r.m.s.d. value of 0.5 Å. This indicates that A and B chains in both complexes are almost identical with small differences. However, when the Cα atoms of ppGBP-glucose complex A chain are compared with the Cα atoms of ppGBP-galactose complex A chain, the r.m.s.d. value is 0.3 Å. The same r.m.s.d. value (0.3 Å) is obtained when the Cα atoms of B chains of both complexes are compared. When the Cα atoms of AB and BA pair of chains of ppGBP-glucose and ppGBP-galactose complex were compared, each comparison produced an r.m.s.d. value of 0.5 Å. The average r.m.s.d. value of structural comparison between ppGBP-glucose and ppGBP-galactose complex is 0.4 Å. These structural superpositions do not indicate any major structural differences for the main chain atoms of both complexes.
The asymmetric unit of the ppGBP-glucose complex crystal contained two protein molecules, each complexed with glucose. In the sugar binding pocket, β-d-glucose is bound in a pyranose 4C1 full chair conformation (Figs. 1B and 4A). The hydroxyl groups of the glucose are placed equatorially with respect to the ring and form hydrogen bonds with the polar amino acid side chains (Table 3). The C6 hydroxyl group of glucose also forms polar contacts with the ppGBP residues. Twelve residues present in the substrate binding pocket, Trp35, Trp36, Glu41, Gln90, Lys92, Trp270, Trp250, Asn301, Asp303, Lys339, His379, and Gly68, are directly involved in glucose binding. Most of the residues form polar contacts with glucose by the side chains, and only one hydrogen bond is mediated through a main chain -NH group (Gly68). The side chain of Asn380 forms a hydrogen bond with the C1 hydroxyl group of glucose via a water molecule. Apart from these polar interactions, the indole side chain of Trp270 provides a non-polar stacking interaction with glucose. The importance of these residues in glucose binding has also been demonstrated by site-directed mutagenesis and biochemical studies, which are discussed in a later section. Despite the presence of similar residues at the sugar binding pocket of ppGBP and oaGBP, structural comparison has revealed two significant differences. Lys92 in ppGBP-glucose complex is replaced by Leu95 in the oaGBP-glucose complex structure. The water-mediated hydrogen bond between Asn380 and glucose at the sugar binding pocket of ppGBP-glucose complex is absent in oaGBP-glucose complex as Asn380 is replaced by Gly381 in the latter protein.
FIGURE 4.

Sugar binding pocket of ppGBP. A, interactions of glucose with ppGBP. Carbon atoms of glucose and amino acid residues are shown as yellow and light brown, respectively. Water 2 is shown as a red sphere. B, interactions of galactose with ppGBP. Carbon atoms of galactose and amino acid residues are shown as cyan and light brown, respectively. Water 2 is shown as a red sphere. C, superposition of ppGBP sugar binding pocket complexed with glucose (yellow) and galactose (cyan). Carbon atoms of the residues from glucose- and galactose-bound complexes are colored gray and cyan, respectively. Water 1 that displaces Glu41 in galactose-complexed ppGBP and water 2 involved in forming a water-mediated hydrogen bond are shown as orange spheres. Hydrogen bonds are presented as dotted lines in A, B, and C.
TABLE 3.
Hydrogen bond distances between the ppGBP sugar binding pocket residues and the hydroxyl groups of glucose/galactose
The residues shown in boldface depict significant differences in the polar interactions of glucose- and galactose-complexed ppGBP.
| Atom |
Distance (Å) |
||
|---|---|---|---|
| Protein | Sugar | Glucose | Galactose |
| His379/Nδ1 | O1 | 2.8 | 2.9 |
| Lys92/Nζ | O1 | 2.9 | 3.1 |
| Asn301/Nδ2 | O2 | 2.8 | 3.0 |
| Asn301/Oδ1 | O2 | 3.5 | 3.8 |
| Lys92/Nζ | O2 | 2.9 | 3.1 |
| Asp303/Oδ1 | O2 | 2.6 | 2.5 |
| Asp303/Oδ2 | O2 | 3.4 | 3.5 |
| Asp303/Oδ1 | O3 | 3.3 | 3.3 |
| Asp303/Oδ2 | O3 | 2.6 | 2.7 |
| Lys339/Nζ | O3 | 3.0 | 2.8 |
| Trp36/Nϵ1 | O3 | 2.9 | 3.0 |
| Lys339/Nζ | O4 | 2.9 | 4.0 |
| Glu41/Oϵ1 | O4 | 2.6 | 3.1 |
| Glu41/Oϵ2 | O4 | 4.7 | 2.9 |
| Trp36/Nϵ1 | O4 | 4.2 | 3.4 |
| Trp35/Nϵ1 | O5 | 3.2 | 3.2 |
| Trp35/Nϵ1 | O6 | 3.3 | 3.3 |
| Glu41/Oϵ2 | O6 | 2.8 | 3.9 |
Galactose is also bound in a full chair conformation (Figs. 1B and 4B) in the sugar binding pocket of ppGBP and has similar polar and hydrophobic interactions (Table 3) as observed in ppGBP-glucose complex. The total number of hydrogen bonds with the sugar in ppGBP-glucose complex is greater than those present in ppGBP-galactose complex. In the ppGBP-galactose structure, the C4 hydroxyl group is in an axial position, and therefore it cannot form a hydrogen bond with the Lys339 side chain but forms a hydrogen bond only with the Glu41 side chain. In addition, the side chain of Glu41 in ppGBP-galactose complex has moved closer to Lys339, and a water molecule occupies the same space (Fig. 4C). In ppGBP-galactose complex, both Glu41 carboxyl oxygen atoms form hydrogen bonds with the C4 hydroxyl group of galactose, whereas in the glucose-bound complex the Glu41 side chain forms hydrogen bonds with C4 and C6 hydroxyl groups. Because of the difference in the configuration of the hydroxyl group at C4 in glucose and galactose, the binding of these sugars must be associated to some conformational changes of residues in the binding pocket. ppGBP-galactose complex structure clearly shows the conformational change in Glu41 upon galactose binding to the protein (Fig. 4C). Although the corresponding glutamate residue (Glu13) is reported to be involved in galactose binding in ttGBP, the conformational change was not observed. A hydrogen bond between the C2 hydroxyl group of galactose and side chain of Asn301 is missing in ppGBP-galactose complex as compared with the glucose-bound form. This analysis indicates that there is a loss of polar interactions in the ppGBP-galactose as compared with the ppGBP-glucose complex. Lower affinity of ttGBP toward galactose has also been proposed because of a loss of polar interactions (1).
The comparison of the orientations of sugar molecules in the binding pocket of SBPs has not been reported earlier. Although sugar binding pockets have been reported to be similar for cluster D SBPs from T. thermophilus (1), O. anthropi, and Actinoplanes sp. (42) as well as for cluster B SBP from E. coli (15), careful comparison and analysis (Fig. 5) show that orientations of the sugar molecules in these proteins are different. In the six subunits of the ttGBP structure (Protein Data Bank code 2B3B), two glucose molecules are in full chair and four are in half-chair conformations. In the oaGBP structure (Protein Data Bank code 4R2B), the glucose molecules are in half-chair conformation. The crystal structures of ppGBP are of high resolution where clear electron density is visible for the sugar molecules. ppGBP-sugar complex structures depict the full chair β-conformation of the sugars and accurate orientation of the hydroxyl groups of the sugar molecule at the binding pocket. The orientation of glucose molecule in the ppGBP structure is equatorial like that observed in two molecules (of six in an asymmetric unit) of the ttGBP structure. However, the orientations of sugars present in SBPs of Actinoplanes sp. and E. coli are perpendicular and angular to the axis, respectively.
FIGURE 5.
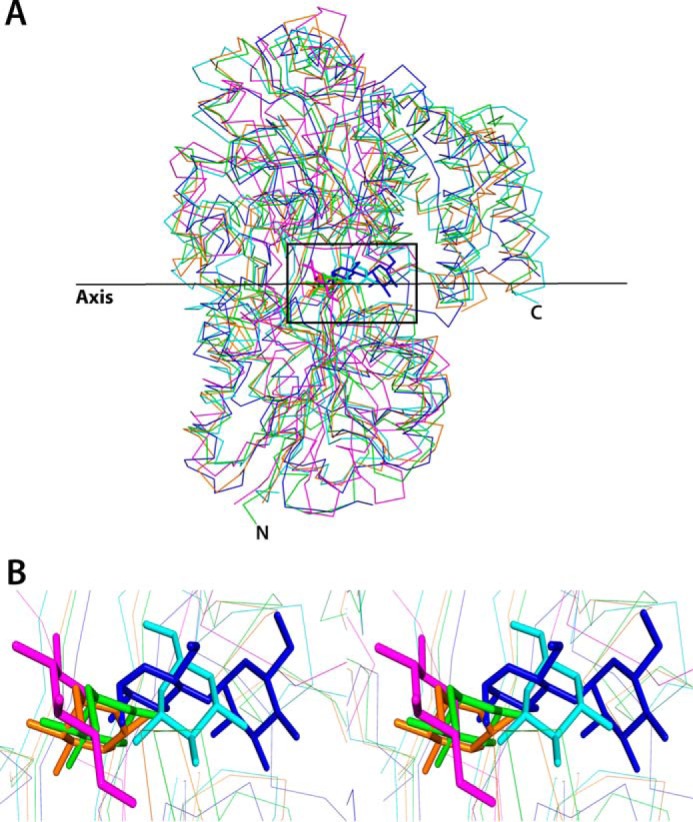
Orientations of sugars at the binding pocket. A, structural superposition ecMBP-maltose (blue), ecGGBP-glucose (magenta), AcbH-galactose (cyan), ttGBP-glucose (orange), and ppGBP-glucose (green) complexes showing the orientations of the bound sugars with respect to an imaginary axis. Sugars are shown as sticks, and the proteins are shown as lines. B, zoomed-in stereoimage of A, inset, showing the orientations of sugars at the binding pocket.
Sugar-induced Conformational Changes in ppGBP
The unliganded ppGBP was crystallized under the same condition as its complexes with different unit cell dimensions. Two almost identical (r.m.s.d. = 0.34 Å) molecules of unliganded-ppGBP are present in the asymmetric unit. Although most of the residues are well defined in the electron density, loop regions consisting of residues 64–69 and 64–70 could not be built in A and B subunits, respectively. The overall structural superposition of ppGBP-glucose complex with the unliganded structure (r.m.s.d. = 1.7 Å) reveals that the unliganded ppGBP has an open conformation (Fig. 6, A and B). The distance between the Cα atoms of Gly39 (N-terminal domain) and Asn251 (C-terminal domain) in unliganded and complexed ppGBP are 17.10 and 8.25 Å, respectively (Fig. 6B). The least square superposition of N- (residues 28–141, 303–337, and 343–352) and C-terminal domains (residues 144–300, 338–342, and 353–419) of both unliganded and glucose-bound structures showed r.m.s.d. values of 1.4 and 0.4 Å, respectively. These results indicate that the N-terminal domain has significant conformational changes, whereas the C-terminal domain remains almost unchanged. DynDom software (55, 56) was used to understand the conformational change in ppGBP structures. The analysis (Fig. 6, A and B) of unliganded and glucose-bound structures confirmed that the conformational change is mainly due to torsion angle changes in the three hinge regions (hinge one, 141–144; hinge two, 298–301; hinge three, 342–343) and a connecting loop (residues 352–358) between the two domains. The result also confirmed that the unliganded structure is in an open conformation compared with the liganded closed form (Fig. 6B) in which the two domains are closer to each other after a 19° rotation of one of the domains. In ecGGBP belonging to cluster B SBPs, domain closure upon glucose binding was reported (15) where the domains behaved as rigid bodies with conformational changes in the hinge region. However, the asymmetric conformational changes in the two domains of ppGBP upon sugar binding have not been reported for sugar-binding SBPs. Although many substrate-bound SBP structures are available, only a few structures of unliganded sugar SBPs (Protein Data Bank codes 2FW0, 5BRA, 4KQ9, 3UOR, 3KJT, and 1GCG) are reported demonstrating a Venus fly trap mechanism upon sugar binding (12). ecGGBP is the only representative of monosaccharide-binding SBPs where both unliganded (Protein Data Bank code 2FW0) and glucose-complexed (Protein Data Bank code 2FVY) structures have been reported. The poor representation of unliganded SBPs in the Protein Data Bank might be due to difficulties in crystallization of unliganded SBPs. In ppGBP, the unliganded and sugar-complexed structures indicate that the two domains move closer to each other upon sugar binding. Residues from both domains form hydrogen bonds with the sugar molecule. The torsional changes of the Cα backbone in the hinge region and conformational changes in the N-terminal domain of ppGBP are responsible for the domain closure. The observed domain movement is asymmetric, supporting a Venus fly trap mechanism. Such domain movements have also been observed in other SBPs and can be negligible (6°) as in TroA or can be large (60°) as in LivJ (6). Despite the availability of many structures of SBPs in the Protein Data Bank, there are only three reported structures (from Actinoplanes sp., O. anthropi, and T. thermophilus) of proteins that bind only monosaccharide but have folds similar to disaccharide-binding cluster D SBPs. These available structures are complexed with sugar; however, the corresponding apo structures are not reported. Although the structure of sugar-bound oaGBP is available in the Protein Data Bank, its structural and functional properties have not been reported. In this study, the complete structural features of apo- and sugar-complexed ppGBP describe the sugar-induced conformational change; this is the first report for the monosaccharide-binding SBPs belonging to cluster D.
FIGURE 6.

Domain movement in ppGBP upon sugar binding. A, superposition of the C-terminal domains of glucose-bound (green) and unliganded (magenta) ppGBP structures. The bound glucose molecule is shown as a stick. Positions of Cα atoms of Gly39 and Asn251 are shown as spheres. B, schematic depicting conformational changes in ppGBP upon sugar binding.
Structural Adaptations in ppGBP to Facilitate Monosaccharide Binding
According to the structural classification of SBPs (6), ppGBP belongs to cluster D disaccharide-binding SBPs (12); however, it binds to monosaccharides only (20, 21). Comparison of the ppGBP binding pocket with ecMBP (Fig. 7) reveals structural adaptations by ppGBP that made the sugar binding pocket smaller to accommodate monosaccharide. The binding pocket that accommodates maltose in ecMBP is wide enough to accommodate disaccharides; however, in ppGBP, it is filled with two loops (loop 1, residues 64–70; loop 2, 376–381) and α-helix (residues 189–204), making the pocket just sufficient to bind to monosaccharide. Residue Trp191 of α-helix in ppGBP obstructs the binding of a larger ligand like maltose. The side chains of His379 and Asn380 in loop 2 of ppGBP occupy the pocket where maltose binds in ecMBP. His379 from occluding loop 2 interacts with the C1 hydroxyl group of glucose/galactose through a hydrogen bond. Loop 1 of ppGBP is longer compared with ecMBP, and thus the spatial arrangement of this loop may be responsible for blocking disaccharide binding. Because of insertion of these secondary structural elements, the sugar binding pocket in ppGBP has moved toward helix 1 (residues 44–58). Our analysis by CASTp (57) reveals that the volume of the sugar binding pocket in ppGBP (203 Å3) is much smaller as compared with that of ecMBP (1247 Å3). Further analysis revealed the volumes of the sugar binding pockets of ttGBP, oaGBP, and Actinoplanes sp. galactose-binding protein to be 196, 497, and 436 Å3, respectively. The volumes of sugar binding pockets in ppGBP and ttGBP appear to be similar. The structure of ppGBP revealed insights into the structural adaptations at the sugar binding pocket to accommodate monosaccharide like glucose or galactose. Members of the SBP superfamily have been reported to modulate the sugar binding pocket by means of loops (58), bulky side chains (59), or cofactors (60). The cluster B SBPs, which bind to monosaccharide, have comparatively smaller binding pockets than cluster D (2, 21). These proteins are known to evolve from CheY-like ancestors by domain duplication and swapping (61), giving rise to five β-sheets in the core and one additional crossover between the two domains (2). The topology of ppGBP is similar to that observed in cluster D SBPs; however, functionally this protein is similar to cluster B SBPs. The volume of the binding pocket in ppGBP is smaller than that of ecGGBP, but the β-core sheet topology of ppGBP comprises five strands, and the fourth strand is antiparallel to the other strands. This type of topology is known to have evolved from cluster B SBPs due to domain dislocation, giving rise to the hypothetical progenitor of cluster D SBPs (2). Thus, the β-core sheet topology and volume of binding pocket of ppGBP indicate that it must have evolved from the cluster B SBPs. Our structure-based evolutionary analysis shows that ppGBP is related to thermophilic SBPs, especially to the well characterized ttGBP. The volumes of the sugar binding pockets in ppGBP and ttGBP are almost identical, indicating that these two proteins must have evolved from a common ancestor.
FIGURE 7.
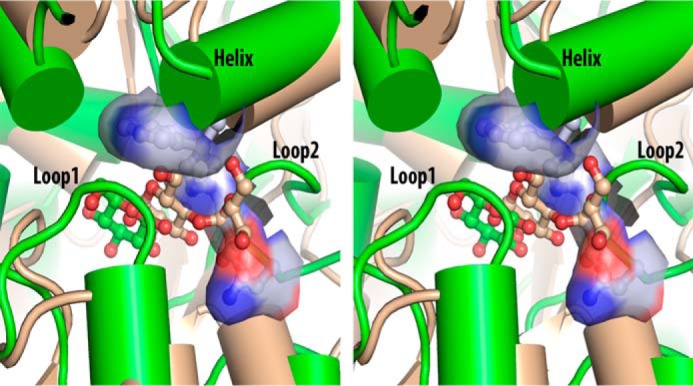
Structural adaptation for monosaccharide binding. A stereoimage comparing the sugar binding pockets of ppGBP-glucose (green) and ecMBP-maltose (light brown) complexes is shown. Sugars are presented as ball and stick. Secondary structural elements in ppGBP responsible for filling the disaccharide binding pocket are labeled. Three important residues (Trp191, His379, and Asn380) of ppGBP involved in making the monosaccharide binding pocket narrower are also presented as ball and stick inside the surface representation, showing the unavailability of space in the binding pocket for disaccharide binding.
Sugar Specificity of ppGBP
The crystal structures of ppGBP-sugar complexes showed involvement of 12 residues at the sugar binding pocket. Based on docking and mutational studies, involvement of Trp35, Trp36, Glu41, Lys92, Lys339, and His379 in glucose binding was proposed earlier (21). Five alanine mutants of Gln90, Trp250, Trp270, Asn301, and Asp303 were generated and confirmed by sequencing. The [14C]glucose binding activity of these purified mutants showed significant reduction in the glucose binding activity (Fig. 8A). The far-UV CD spectra of wild-type and mutant proteins were similar (data not shown). Therefore, the decrease in the activity can be attributed to the involvement of these residues in the glucose binding.
FIGURE 8.

Biochemical properties of ppGBP. A, [14C]glucose binding activity of ppGBP and mutants. Error bars represent S.E. B, glucose saturation profile of ppGBP as monitored by SPR spectroscopy. The inset represents the substrate saturation profile obtained by [14C]glucose binding assay. Error bars represent S.E. C, thermal denaturation profiles of ppGBP in the absence (●) and presence of 1 mm glucose (■) or galactose (▴).
The glucose binding constant of ppGBP using SPR was measured, and saturation was achieved at 6.25 μm (Fig. 8B). The binding constant, Kd, for glucose was calculated to be 0.39 μm, which was found to be similar to that measured (0.3 μm) using a [14C]glucose binding assay (Fig. 8B, inset). With galactose as a ligand, binding saturation was not observed even at 800 μm concentration. The temperature-dependent changes in the far-UV CD spectra for ppGBP-glucose complex showed a Tm value of 62 ± 0.3 °C (Fig. 8C). The melting temperature profiles of unliganded and galactose-bound ppGBP were similar with a Tm value of 55 ± 0.1 °C (Fig. 8C). These results indicate that thermostability of this protein increases with glucose binding. The structural analysis of the sugar binding pocket reveals that ppGBP-glucose complex has two additional strong hydrogen bonds as compared with the ppGBP-galactose complex. This may be the reason for the low affinity of the protein toward galactose, and hence the binding constant could not be determined. The ability of sugar-binding SBPs to bind glucose and galactose with different affinities has been observed (1, 6). The changes in the direct hydrogen bonding interactions at the substrate binding pocket probably attribute to the observed changes in the affinities. The insignificant change (2-fold) in affinity for glucose and galactose in ecGGBP is due to an equal number of hydrogen bonds at the binding pocket (15). The affinity of ttGBP for glucose is 12-fold higher than for galactose due to the presence of an additional hydrogen bond in the glucose complex compared with galactose (1).
Conclusion
In this study, we have determined high resolution crystal structures of ppGBP. This is the first structure-function study of a periplasmic component of a sugar ABC transporter from Pseudomonas sp. Among the available structures of SBPs belonging to cluster D, so far this is the highest resolution (1.25 Å) structure. The structural fold of ppGBP is similar to disaccharide-binding SBPs. However, the biochemical studies show that ppGBP binds to monosaccharides only with high specificity toward glucose. The structural analysis of ppGBP complexed with glucose or galactose reveals that changes in the hydrogen bonding interactions at the sugar binding pocket contribute to its high glucose affinity. The structure-based evolutionary studies indicate that ppGBP is structurally more similar to glucose/galactose-binding protein (ttGBP) of thermophilic bacteria.
Author Contributions
S. P., A. M., P. S. P., and P. B. conceived and coordinated the study. S. P., A. M., P. S. P., and P. B. designed and analyzed the experiments. S. P. and A. M. performed the experiments. S. P., A. M., P. S. P., and P. B. wrote the paper.
Acknowledgments
We thank the European Molecular Biology (EMBL) Laboratory staff Dr. Hassan Belrhali and Dr. Babu A. Manjasetty for providing support on the beamline as well as the EMBL and Department of Biotechnology for providing access to the BM14 beamline at the European Synchrotron Radiation Facility. We express our gratitude toward Ulka U. Sawant and Dr. Ashok K. Varma from the Advanced Centre for Treatment, Research and Education in Cancer (Navi Mumbai, India) for providing access to the x-ray diffractometer. We thank Dr. Vineeta Shah for the SPR studies and Dr. Kiran Kondabagil from the Indian Institute of Technology Bombay for input in the evolutionary aspect of this study. We also thank the reviewers for valuable comments.
This work was supported in part by a Ramalingaswami re-entry fellowship and research grant (to P. B.) from the Department of Biotechnology, Ministry of Science and Technology, India. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5DVF, 5DVI, and 5DVJ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
A. Modak and P. S. Phale, unpublished results.
- SBP
- substrate-binding protein
- ABC
- ATP-binding cassette
- ppGBP
- P. putida CSV86 glucose-binding protein
- ecGGBP
- E. coli glucose/galactose-binding protein
- ttGBP
- T. thermophilus glucose-binding protein
- ecMBP
- E. coli maltose-binding protein
- oaGBP
- O. anthropi glucose-binding protein
- tlMBP
- T. litoralis trehalose/maltose-binding protein
- TLS
- translation-libration-screw
- SPR
- surface plasmon resonance
- r.m.s.d.
- root mean square deviation.
References
- 1.Cuneo M. J., Changela A., Warren J. J., Beese L. S., and Hellinga H. W. (2006) The crystal structure of a thermophilic glucose binding protein reveals adaptations that interconvert mono and di-saccharide binding sites. J. Mol. Biol. 362, 259–270 [DOI] [PubMed] [Google Scholar]
- 2.Wilkinson T., Verschueren K. H. G., Wilkinson T., and Verschueren K. H. G. (2003) Crystal structures of periplasmic solute binding proteins in ABC transport complexes illuminate their function in ABC proteins in ABC Proteins: From Bacteria to Man (Higgins C. F., and Higgins C. F., eds) pp. 187–208, Academic Press, London [Google Scholar]
- 3.Miller D. M. 3rd, Olson J. S., and Quiocho F. A. (1980) The mechanism of sugar binding to the periplasmic receptor for galactose chemotaxis and transport in Escherichia coli. J. Biol. Chem. 255, 2465–2471 [PubMed] [Google Scholar]
- 4.Chen J., Sharma S., Quiocho F. A., and Davidson A. L. (2001) Trapping the transition state of an ATP-binding cassette transporter: evidence for a concerted mechanism of maltose transport. Proc. Natl. Acad. Sci. 98, 1525–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prossnitz E., Nikaido K., Ulbrich S. J., and Ames G. F. (1988) Formaldehyde and photoactivatable cross-linking of the periplasmic binding protein to a membrane component of the histidine transport system of Salmonella typhimurium. J. Biol. Chem. 263, 17917–17920 [PubMed] [Google Scholar]
- 6.Berntsson R. P., Smits S. H., Schmitt L., Slotboom D.-J., and Poolman B. (2010) A structural classification of substrate-binding proteins. FEBS Lett. 584, 2606–2617 [DOI] [PubMed] [Google Scholar]
- 7.de Lorimier R. M., Smith J. J., Dwyer M. A., Looger L. L., Sali K. M., Paavola C. D., Rizk S. S., Sadigov S., Conrad D. W., Loew L., and Hellinga H. W. (2002) Construction of a fluorescent biosensor family. Protein Sci. 11, 2655–2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lager I., Fehr M., Frommer W. B., and Lalonde S. (2003) Development of a fluorescent nanosensor for ribose. FEBS Lett. 553, 85–89 [DOI] [PubMed] [Google Scholar]
- 9.Looger L. L., Dwyer M. A., Smith J. J., and Hellinga H. W. (2003) Computational design of receptor and sensor proteins with novel functions. Nature 423, 185–190 [DOI] [PubMed] [Google Scholar]
- 10.Benson D. E., Haddy A. E., and Hellinga H. W. (2002) Converting a maltose receptor into a nascent binuclear copper oxygenase by computational design. Biochemistry 41, 3262–3269 [DOI] [PubMed] [Google Scholar]
- 11.Quiocho F. A., and Ledvina P. S. (1996) Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: variation of common themes. Mol. Microbiol. 20, 17–25 [DOI] [PubMed] [Google Scholar]
- 12.Fukami-Kobayashi K., Tateno Y., and Nishikawa K. (1999) Domain dislocation: a change of core structure in periplasmic binding proteins in their evolutionary history. J. Mol. Biol. 286, 279–290 [DOI] [PubMed] [Google Scholar]
- 13.Lee Y. H., Deka R. K., Norgard M. V., Radolf J. D., and Hasemann C. A. (1999) Treponema pallidum TroA is a periplasmic zinc-binding protein with a helical backbone. Nat. Struct. Biol. 6, 628–633 [DOI] [PubMed] [Google Scholar]
- 14.Mao B., Pear M., McCammon J. A., and Quiocho F. A. (1982) Hinge-bending in l-arabinose-binding protein. The “Venus's-flytrap” model. J. Biol. Chem. 257, 1131–1133 [PubMed] [Google Scholar]
- 15.Borrok M. J., Kiessling L. L., and Forest K. T. (2007) Conformational changes of glucose/galactose-binding protein illuminated by open, unliganded, and ultra-high-resolution ligand-bound structures. Protein Sci. 16, 1032–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Basu A., Apte S. K., and Phale P. S. (2006) Preferential utilization of aromatic compounds over glucose by Pseudomonas putida CSV86. Appl. Environ. Microbiol. 72, 2226–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shrivastava R., Basu B., Godbole A., Mathew M. K., Apte S. K., and Phale P. S. (2011) Repression of the glucose-inducible outer-membrane protein OprB during utilization of aromatic compounds and organic acids in Pseudomonas putida CSV86. Microbiology 157, 1531–1540 [DOI] [PubMed] [Google Scholar]
- 18.Phale P. S., Paliwal V., Raju S. C., Modak A., and Purohit H. J. (2013) Genome sequence of naphthalene-degrading soil bacterium Pseudomonas putida CSV86. Genome Announc. 1, e00234–e00212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paliwal V., Raju S. C., Modak A., Phale P. S., and Purohit H. J. (2014) Pseudomonas putida CSV86: a candidate genome for genetic bioaugmentation. PLoS One 9, e84000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Basu A., Shrivastava R., Basu B., Apte S. K., and Phale P. S. (2007) Modulation of glucose transport causes preferential utilization of aromatic compounds in Pseudomonas putida CSV86. J. Bacteriol. 189, 7556–7562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Modak A., Bhaumik P., and Phale P. S. (2014) Periplasmic glucose-binding protein from Pseudomonas putida CSV86—identification of the glucose-binding pocket by homology-model-guided site-specific mutagenesis. FEBS J. 281, 365–375 [DOI] [PubMed] [Google Scholar]
- 22.Adewoye L. O., and Worobec E. A. (2000) Identification and characterization of the gltK gene encoding a membrane-associated glucose transport protein of Pseudomonas aeruginosa. Gene 253, 323–330 [DOI] [PubMed] [Google Scholar]
- 23.Pandey S., Modak A., Phale P. S., and Bhaumik P. (2015) Cloning, purification, crystallization and preliminary x-ray diffraction studies of periplasmic glucose binding protein of Pseudomonas putida CSV86. Adv. Biosci. Biotechnol. 6, 164–171 10.4236/abb.2015.63016 [DOI] [Google Scholar]
- 24.Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 25.Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 26.Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., and Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stein N. (2008) CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J. Appl. Crystallogr. 41, 641–643 10.1107/S0021889808006985 [DOI] [Google Scholar]
- 29.Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 30.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 32.Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 33.Painter J., and Merritt E. A. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62, 439–450 [DOI] [PubMed] [Google Scholar]
- 34.Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 35.Schellman J. A. (1987) The thermodynamic stability of proteins. Annu. Rev. Biophys. Biophys. Chem. 16, 115–137 [DOI] [PubMed] [Google Scholar]
- 36.Hsieh H. V., Pfeiffer Z. A., Amiss T. J., Sherman D. B., and Pitner J. B. (2004) Direct detection of glucose by surface plasmon resonance with bacterial glucose/galactose-binding protein. Biosens. Bioelectron. 19, 653–660 [DOI] [PubMed] [Google Scholar]
- 37.Gestwicki J. E., Hsieh H. V., and Pitner J. B. (2001) Using receptor conformational change to detect low molecular weight analytes by surface plasmon resonance. Anal. Chem. 73, 5732–5737 [DOI] [PubMed] [Google Scholar]
- 38.Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 [DOI] [PubMed] [Google Scholar]
- 40.Owen R. L., Rudiño-Piñera E., and Garman E. F. (2006) Experimental determination of the radiation dose limit for cryocooled protein crystals. Proc. Natl. Acad. Sci. U.S.A. 103, 4912–4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spurlino J. C., Lu G. Y., and Quiocho F. (1991) The 2.3 Å resolution structure of the maltose-or maltodextrin-binding protein, a primary receptor of bacterial active transport and chemotaxis. J. Biol. Chem. 266, 5202–5219 [DOI] [PubMed] [Google Scholar]
- 42.Licht A., Bulut H., Scheffel F., Daumke O., Wehmeier U. F., Saenger W., Schneider E., and Vahedi-Faridi A. (2011) Crystal structures of the bacterial solute receptor AcbH displaying an exclusive substrate preference for β-d-galactopyranose. J. Mol. Biol. 406, 92–105 [DOI] [PubMed] [Google Scholar]
- 43.Diez J., Diederichs K., Greller G., Horlacher R., Boos W., and Welte W. (2001) The crystal structure of a liganded trehalose/maltose-binding protein from the hyperthermophilic archaeon Thermococcus litoralis at 1.85 Å. J. Mol. Biol. 305, 905–915 [DOI] [PubMed] [Google Scholar]
- 44.Han Y., Agarwal V., Dodd D., Kim J., Bae B., Mackie R. I., Nair S. K., and Cann I. K. (2012) Biochemical and structural insights into xylan utilization by the thermophilic bacterium Caldanaerobius polysaccharolyticus. J. Biol. Chem. 287, 34946–34960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Telmer P. G., and Shilton B. H. (2003) Insights into the conformational equilibria of maltose-binding protein by analysis of high affinity mutants. J. Biol. Chem. 278, 34555–34567 [DOI] [PubMed] [Google Scholar]
- 46.Schäfer K., Magnusson U., Scheffel F., Schiefner A., Sandgren M. O., Diederichs K., Welte W., Hülsmann A., Schneider E., and Mowbray S. L. (2004) X-ray structures of the maltose-maltodextrin-binding protein of the thermoacidophilic bacterium Alicyclobacillus acidocaldarius provide insight into acid stability of proteins. J. Mol. Biol. 335, 261–274 [DOI] [PubMed] [Google Scholar]
- 47.Ejby M., Fredslund F., Vujicic-Zagar A., Svensson B., Slotboom D. J., and Abou Hachem M. (2013) Structural basis for arabinoxylo-oligosaccharide capture by the probiotic Bifidobacterium animalis subsp. lactis Bl-04. Mol. Microbiol. 90, 1100–1112 [DOI] [PubMed] [Google Scholar]
- 48.Chekan J. R., Kwon I. H., Agarwal V., Dodd D., Revindran V., Mackie R. I., Cann I., and Nair S. K. (2014) Structural and biochemical basis for mannan utilization by Caldanaerobius polysaccharolyticus strain ATCC BAA-17. J. Biol. Chem. 289, 34965–34977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsumoto N., Yamada M., Kurakata Y., Yoshida H., Kamitori S., Nishikawa A., and Tonozuka T. (2009) Crystal structures of open and closed forms of cyclo/maltodextrin-binding protein. FEBS J. 276, 3008–3019 [DOI] [PubMed] [Google Scholar]
- 50.Higgins M. A., Abbott D. W., Boulanger M. J., and Boraston A. B. (2009) Blood group antigen recognition by a solute-binding protein from a serotype 3 strain of Streptococcus pneumoniae. J. Mol. Biol. 388, 299–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vahedi-Faridi A., Licht A., Bulut H., Scheffel F., Keller S., Wehmeier U. F., Saenger W., and Schneider E. (2010) Crystal structures of the solute receptor GacH of Streptomyces glaucescens in complex with acarbose and an acarbose homolog: comparison with the acarbose-loaded maltose-binding protein of Salmonella typhimurium. J. Mol. Biol. 397, 709–723 [DOI] [PubMed] [Google Scholar]
- 52.Chaudhuri B. N., Ko J., Park C., Jones T. A., and Mowbray S. L. (1999) Structure of D-allose binding protein from Escherichia coli bound to D-allose at 1.8 Å resolution. J. Mol. Biol. 286, 1519–1531 [DOI] [PubMed] [Google Scholar]
- 53.Cuneo M. J., Beese L. S., and Hellinga H. W. (2008) Ligand-induced conformational changes in a thermophilic ribose-binding protein. BMC Struct. Biol. 8, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Flocco M. M., and Mowbray S. L. (1994) The 1.9 Å x-ray structure of a closed unliganded form of the periplasmic glucose/galactose receptor from Salmonella typhimurium. J. Biol. Chem. 269, 8931–8936 [PubMed] [Google Scholar]
- 55.Hayward S., and Berendsen H. J. (1998) Systematic analysis of domain motions in proteins from conformational change: new results on citrate synthase and T4 lysozyme. Proteins 30, 144–154 [PubMed] [Google Scholar]
- 56.Taylor D., Cawley G., and Hayward S. (2014) Quantitative method for the assignment of hinge and shear mechanism in protein domain movements. Bioinformatics 30, 3189–3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y., and Liang J. (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34, W116–W118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heddle J., Scott D. J., Unzai S., Park S.-Y., and Tame J. R. (2003) Crystal structures of the liganded and unliganded nickel-binding protein NikA from Escherichia coli. J. Biol. Chem. 278, 50322–50329 [DOI] [PubMed] [Google Scholar]
- 59.Quiocho F. A., Spurlino J. C., and Rodseth L. E. (1997) Extensive features of tight oligosaccharide binding revealed in high-resolution structures of the maltodextrin transport/chemosensory receptor. Structure 5, 997–1015 [DOI] [PubMed] [Google Scholar]
- 60.Guo M., Harvey I., Yang W., Coghill L., Campopiano D. J., Parkinson J. A., MacGillivray R. T., Harris W. R., and Sadler P. J. (2003) Synergistic anion and metal binding to the ferric ion-binding protein from Neisseria gonorrhoeae. J. Biol. Chem. 278, 2490–2502 [DOI] [PubMed] [Google Scholar]
- 61.Stock A. M., Mottonen J. M., Stock J. B., and Schutt C. E. (1989) Three-dimensional structure of CheY, the response regulator of bacterial chemotaxis. Nature 337, 745–749 [DOI] [PubMed] [Google Scholar]
- 62.Thompson J. D., Higgins D. G., and Gibson T. J. (1994) ClustalW. Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Modak A. (2014) Studies on periplasmic GBP-dependent glucose ABC transporter from Pseudomonas putida CSV86. Ph.D. thesis, Indian Institute of Technology Bombay, India [Google Scholar]