

Abstract
Mycolic acids are essential components of the mycobacterial cell envelope, and their biosynthetic pathway is one of the targets of first-line antituberculous drugs. This pathway contains a number of potential targets, including some that have been identified only recently and have yet to be explored. One such target, FadD32, is required for activation of the long meromycolic chain and is essential for mycobacterial growth. We report here an in-depth biochemical, biophysical, and structural characterization of four FadD32 orthologs, including the very homologous enzymes from Mycobacterium tuberculosis and Mycobacterium marinum. Determination of the structures of two complexes with alkyl adenylate inhibitors has provided direct information, with unprecedented detail, about the active site of the enzyme and the associated hydrophobic tunnel, shedding new light on structure-function relationships and inhibition mechanisms by alkyl adenylates and diarylated coumarins. This work should pave the way for the rational design of inhibitors of FadD32, a highly promising drug target.
Keywords: crystal structure, drug design, enzyme inhibitor, mycobacteria, tuberculosis, fatty acyl ACP-synthase, fatty acyl-AMP ligase
Introduction
The structural hallmark of the causal agent of tuberculosis (TB),4 Mycobacterium tuberculosis, and other mycobacteria is their characteristic cell envelope (1), which has a much higher lipid content than the envelopes of other Gram-positive and Gram-negative bacteria (2). The considerable diversity of lipids present contributes to the unique nature and structural complexity of the mycobacterial cell envelope but also underlies key features of mycobacterial physiology. These lipids also play an important role in virulence, pathogenicity, and resistance to antibiotics (3) and in the control of inflammation and immune mechanisms (4). Mycolic acids, 2-alkyl, 3-hydroxy long-chain fatty acids, are major specific components of the mycobacterial cell envelope (5). They may be linked to arabinogalactan or form esters of trehalose or glycerol in the so-called “mycomembrane,” which plays a crucial role in establishing the architecture and impermeability of the cell envelope. Mycolic acids are essential for the viability of mycobacteria, and their biosynthetic pathway is a proven target for anti-mycobacterial drugs that continues to attract considerable attention from the community working on TB (5). The mycolic acid biosynthesis is complex. Briefly, two fatty acid synthases are involved in the biosynthesis of long fatty acids, yielding the α-alkyl (C24–C26) branch on the one hand and, after further modifications, the so-called meromycolic chain (C42–C62), on the other hand. These fatty acids are activated before condensation can take place, finally leading, after reduction, to the characteristic mycolic motif. Several essential enzymes are involved in this penultimate step of mycolic acid biosynthesis. They include Pks13, a polyketide synthase that has been identified as the condensing enzyme (6), the cognate 4′-phosphopantetheinyl transferase PptT responsible for its activation (7), an AccD4-containing carboxylase complex required for activation of the α-branch (8, 9), and FadD32, which activates the meromycolic chain through the formation of acyl-AMP (9). The fadD32 gene is adjacent to pks13 and accD4 and is essential for mycobacterial viability (6, 9). It belongs to a large family of fadD genes in the M. tuberculosis genome (10). The corresponding FadD (fatty acid degradation) proteins in M. tuberculosis are of two types, 12 fatty acyl-AMP ligases (FAALs) and 22 fatty acyl-CoA ligases (FACLs) (11). FAALs and FACLs are involved in fatty acid activation and use ATP to produce common acyl adenylate intermediates. However, FACLs catalyze a second reaction in which acyl chains are transferred to coenzyme A (CoA), whereas FAALs transfer the activated acyl chains onto the acyl carrier protein (ACP) domains of their cognate polyketide synthase. The FAAL activity of FadD32 and the FadD32-assisted transfer of fatty acids to the N-terminal ACP domain of Pks13, defining its fatty acyl-ACP synthetase (FAAS) activity, have been demonstrated biochemically (12, 13). FACLs, FAALs, and other acyl-activating enzymes, such as the adenylation domains of non-ribosomal peptide synthetases, belong to the superfamily of adenylate-forming enzymes (AFEs) (14). The M. tuberculosis genome encodes more than 60 AFEs involved in numerous essential biochemical processes, which therefore constitute attractive targets for the development of new antituberculous drugs (15). FadD32 has been identified as an important susceptible (16) and potentially druggable (13, 17, 18) target. We report here the full biochemical and biophysical characterization of four mycobacterial FadD32 enzymes. We also show the first crystal structures of FadD32 from Mycobacterium marinum and Mycobacterium smegmatis in complex with long-chain alkyl adenylate substrate analogs. Based on its high level of sequence identity, FadD32 from M. marinum is an ideal surrogate for the M. tuberculosis enzyme and should be a useful tool for the rational design of inhibitors.
Experimental Procedures
Plasmids
The cloning of the fadD32 genes from M. tuberculosis, M. smegmatis, and Corynebacterium glutamicum has been described elsewhere (13, 17). The M. marinum fadD32 gene was cloned according to published procedures, by PCR amplification from MYCM53 total DNA with the following primers: MmfadD32, 5′ CTCGCATATGATGGCGTACCACAACCCGTTC; MmfadD32, 3′ AGCTCATATGCTTGTTGGCTTGCGCGTCCTC.
Protein Production and Purification
We transformed competent Escherichia coli BL21 Star (DE3) One Shot (Invitrogen) with pET15b-fadD32 constructs for the production of full-length FadD32 proteins. Expression was induced with auto-inducible medium, as described by Studier (19). The transformed cells were first grown overnight in Luria Broth medium supplemented with 50 μg/ml carbenicillin at 37 °C and then diluted in auto-induction medium. Cells cultured for 72 h at 20 °C were harvested by centrifugation (3,000 × g for 15 min) at 4 °C, washed in 50 mm HEPES, 200 mm NaCl, pH 7.5. The pellets were resuspended in lysis buffer consisting of 50 mm HEPES, 10% glycerol (v/v), 30 mm imidazole, 500 mm NaCl, pH 7.5, 0.75 mg/ml lysozyme, and 2 mm phenylmethanesulfonyl fluoride (PMSF, Sigma) and frozen at −80 °C. The frozen bacterial pellets were thawed at room temperature, disrupted by sonication (four intermittent pulses of 30 s) on a VibraCell (Fisher Bioblock Scientific, Illkirch, France), and centrifuged at 20,000 × g for 30 min at 4 °C. Native proteins were purified at 4 °C. The clarified lysates were loaded onto a HisTrap HP (1 ml) affinity column (GE Healthcare). Recombinant FadD32 proteins were eluted in 150 mm imidazole in 50 mm HEPES, 500 mm NaCl, pH 7.5. Whenever appropriate, the 20-residue-long His tags of the affinity-purified FadD32 were removed by thrombin cleavage (Novagen), as follows. The protein solution was diluted 5-fold to decrease the imidazole concentration to 30 mm, concentrated on a Vivaspin 20 column (Sartorius, Göttingen, Germany) to obtain an optical density of 1.0, and then subjected to cleavage by incubation with 0.28 units/ml thrombin for 3 h at room temperature. The cleaved proteins were then reloaded onto the HisTrap HP affinity column to eliminate the uncleaved fractions. The protein-containing flow-through fractions were concentrated to an optical density of 3.0 and purified by size exclusion chromatography on a HighLoad 16/60 Superdex 200 pg column (GE Healthcare) equilibrated with 50 mm HEPES, 500 mm NaCl, pH 7.5, 0.2 mm 4-(2-aminoethyl) benzenesulfonyl fluoride (Sigma). The purified proteins were checked by SDS-PAGE with Coomassie Blue staining and were then concentrated to the desired concentrations. Samples used for kinetic experiments were stored at −20 °C in 50% glycerol. Samples used for biophysical studies were stored at −80 °C without glycerol. Crystallization was attempted only with freshly prepared proteins.
Kinetic and Inhibition Experiments
FadD32 enzyme activity was measured as described previously (17). Briefly, the pyrophosphate (PPi) released during the reaction was hydrolyzed in a pyrophosphatase-coupled reaction, and the resulting inorganic phosphate (Pi) was quantified with the colorimetric PiColorLockTM gold assay kit (Innova Biosciences, Cambridge, UK), by reading the absorbance at 630 nm (A630) resulting from the formation of the phosphomolybdate complex. Reactions were conducted at room temperature, in 30 μl of assay mix containing 50 mm HEPES, pH 7.5, 8 mm MgCl2, 0.001% Brij®35, 1 mm DTT, 2 milliunits/ml pyrophosphatase (Sigma), 1–2 mm ATP, and 20–200 μm fatty acid (as indicated). Reactions were initiated by adding 15 μl of FadD32 diluted in 50 mm HEPES, pH 7.5, to 15 μl of 2× assay mix. The reaction was stopped after 40–60 min by adding 30 μl of cold reaction buffer and 15 μl of malachite green reagent. The A630 was read after 5 min of incubation at room temperature in a CLARIOstar plate reader (BMG LABTECH, Ortenberg, Germany). A reaction without enzyme (for specific activity experiments), or without substrate (for Km and Vmax determinations), was used as a blank in each experiment. The concentration of Pi was determined from a calibration curve plotted with known concentrations of Pi from 10 to 80 μm in each experiment, in accordance with the manufacturer's recommendations.
For specific activity experiments (Table 1), the enzyme was first subjected to serial dilution in HEPES pH 7.5 (with protein concentrations of 12.5 to 800 nm for MtFadD32, MmFadD32, and CgFadD32 and from 2.5 to 160 nm for MsFadD32) and added to the reaction mixture containing 20 μm lauric acid (C12) as substrate and 2 mm ATP. We determined the apparent kinetic parameters (Km, Vmax, and kcat), by measuring the initial velocity (Vi, in μm formed PPi per min) as a function of the substrate concentration studied and at fixed concentrations of substrate with an incubation time of 40–60 min. The kinetic parameters for ATP were determined by measuring the initial velocity at a fixed concentration of lauric acid (200 μm) and various concentrations of ATP (0.0625 to 4 mm); the enzyme concentrations used were 0.4 μm for MtFadD32 and MmFadD32, 0.04 μm for MsFadD32, and 2 μm for CgFadD32. Kinetic parameters for fatty acids (lauric acid or myristic acid) were determined at a fixed concentration of ATP (4 mm), with various concentrations of fatty acid (3.1–200 μm); the enzyme concentrations used were 0.4 μm for MtFadD32 and MmFadD32 and 0.04 μm for MsFadD32. The saturation curves were fitted by non-linear regression analysis with a Graphpad Prism 4.02 equation: Vi = Vmax × [S]/(Km + [S]), where Vi is the initial velocity; Vmax is the maximal velocity; [S] is the substrate concentration, and Km is the Michaelis-Menten constant. The catalytic constant (kcat) reflects the maximum rate of product formation and was calculated by dividing Vmax by the total enzyme concentration [ET]: kcat = Vmax/[ET].
TABLE 1.
Specific activity and kinetic parameters of FadD32 orthologs
| Protein | MtFadD32 | MmFadD32 | MsFadD32 | CgFadD32 |
| Identity (%) | 100 | 92 | 74 | 39 |
| Specific activity (nmol·min−1·mg−1) | 5.37 ± 0.16 | 3.85 ± 0.12 | 39.91 ± 0.73 | 0.46 ± 0.03 |
| Kinetic parameters | ||||
| ATP | ||||
| Km (μm) | 248 ± 27 | 902 ± 114 | 112 ± 12 | |
| Vmax (μm·min−1) | 0.14 ± 0.01 | 0.29 ± 0.01 | 0.17 ± 0.01 | |
| kcat (min−1) | 0.35 ± 0.01 | 0.72 ± 0.03 | 4.33 ± 0.01 | |
| kcat/Km (min−1·μm−1) | 0.0014 ± 0.0003 | 0.0008 ± 0.0003 | 0.0387 ± 0.0063 | |
| Lauric acid (C12) | ||||
| Km (μm) | 72.09 ± 5.75 | 103.2 ± 13.94 | 25.9 ± 1.6a | |
| Vmax (μm.min−1) | 0.82 ± 0.03 | 0.60 ± 0.39 | 0.45 ± 0.01 | |
| kcat (min−1) | 2.04 ± 0.07 | 1.51 ± 0.09 | 11.29 ± 0.22 | |
| kcat/Km (min−1·μm−1) | 0.028 ± 0.012 | 0.015 ± 0.007 | 0.438 ± 0.139 | |
| Myristic acid (C14) | ||||
| Km (μm) | 4.77 ± 0.65 | 5.76 ± 0.56 | 5.2 ± 0.5a | |
| Vmax (μm.min−1) | 0.39 ± 0.01 | 0.27 ± 0.01 | 0.17 ± 0.01 | |
| kcat (min−1) | 0.98 ± 0.03 | 0.69 ± 0.01 | 4.18 ± 0.09 | |
| kcat/Km (min−1·μm−1) | 0.206 ± 0.042 | 0.119 ± 0.025 | 0.800 ± 0.184 | |
a Data are from Ref. 17.
For inhibition studies, the concentrations of ATP and lauric acid were adjusted to 1.6 mm and 100 μm, respectively. The alkyl adenylate substrate analogs AMPC12 and AMPC20, chemically synthesized as described previously (13), were first diluted in DMSO, and the substrate mixture was added to various concentrations of the compounds (0.03 to 31.6 μm). The reactions were started by adding the enzyme (0.4 μm for MtFadD32 and MmFadD32 and 0.04 μm for MsFadD32). They were performed for 60 min at room temperature, in quadruplicate. The results are expressed as the percent inhibition. Curve fitting and IC50 (half-maximal inhibitory concentration) calculations were performed with the dose-response curve as the model equation: Y = bottom + (top − bottom)/(1 + 10∧((logIC50 − X) × Hill slope) (GraphPad Prism 4.02 Software, San Diego), where top and bottom are plateaus in the units of the y axis (maximal and minimal inhibition, respectively), and Hill slope describes the steepness of the family of curves (a Hill slope of a constant value of +1.0 was applied).
Differential Scanning Fluorimetry (DSF)
DSF was used to characterize the thermal stability of the enzyme in various buffer and pH conditions and in the presence of AMPC12. A mixture of enzyme (4 μm), SYPRO® Orange (5×) (Invitrogen), the appropriate buffer at a concentration of 100 mm, and 500 mm NaCl was subjected to a temperature gradient from 25 to 80 °C, with increments of 0.3 °C. All measurements were performed in triplicate, in 96-well plates (Bio-Rad, Marnes-la-Coquette, France). Thermal transitions were monitored with a real time PCR CFX96 System (Bio-Rad). The melting points (Tm) were identified by the inflection points of the curves in relative fluorescence units = f(T). For DSF experiments in the presence of AMPC12, the final concentration of alkyl adenylate was 20 μm.
Microscale Thermophoresis (MST)
For MST measurements, FadD32 orthologs at a concentration of 20 μm were labeled with the RED fluorescent dye NT-647. Labeling and the removal of free dye were performed within 45 min. We then titrated 200 nm NT-647-labeled FadD32 protein against various amounts of AMPC12 or AMPC20 (9 nm to 270 μm) in 50 mm HEPES, pH 7.5, 500 mm NaCl, 0.05% Tween 20, 10% DMSO. The samples were incubated at room temperature for 5 min and then loaded into hydrophilic glass capillaries for MST analysis with Monolith NT.115 (NanoTemper Technologies GmbH, Germany). We monitored the thermophoretic movement of labeled FadD32. Dissociation constants (KD) and associated errors were calculated with NanoTemper software.
Size Exclusion Chromatography and Multiangle Static Light Scattering
FadD32 protein samples were buffered in 25 mm HEPES, pH 7.5, 500 mm NaCl, 1 mm DTT. We loaded 20 μl of protein sample at a final concentration of 40 μm (2.8 mg/ml) onto a Shodex KW402.5–4F column (Wyatt Technology, France) equilibrated with a filter-sterilized (passed through a filter with 0.1-μm pores) buffer consisting of 150 mm sodium phosphate at pH 7.0, in an Agilent 1260 Infinity LC chromatographic system (Agilent Technology). Separation was performed at 15 °C, with a flow rate of 0.35 ml·min−1. Data were collected on a DAWN HELEOS 8+ (8-angle) and Optilab T-rEX refractive index detector (Wyatt Technology, Toulouse France). Results were analyzed with ASTRA 6.0.2.9 software (Wyatt Technology Corp.).
Crystallization
Purified untagged MmFadD32 and MsFadD32 proteins were concentrated in 50 mm HEPES, 500 mm NaCl, pH 7.5, to 72 μm (5.4 mg/ml) and 153 μm (11 mg/ml), respectively. AMPC12 and AMPC20 solubilized in DMSO were mixed with proteins at a molar ratio of 3:1, with final concentrations in DMSO of 3 and 8%, respectively. MmFadD32-AMPC12 crystals were obtained by mixing equal volumes of the protein/inhibitor solution and a reservoir solution composed of 28% PEG 6000, 100 mm Tris-HCl, pH 8.7. In these conditions, triangular crystals, typically measuring 100 × 100 × 40 μm, were obtained in seeding experiments with a single spontaneously crystallized drop. These crystals displayed diffraction to a resolution of 2.5 Å with a synchrotron radiation beam. They belonged to space group P212121, with two molecules per asymmetric unit and 55% solvent. Crystals of MsFadD32 with either AMPC12 or AMPC20 were grown by mixing equal volumes of protein/inhibitor solutions and reservoir solution composed of PEG 1000, in 100 mm Tris-HCl, pH 8.2 to 8.7. These crystals were ∼100 × 100 × 30 μm in size and displayed diffraction to a maximum resolution of 3.3 Å with synchrotron beams. They belonged to space group P4, with eight molecules per asymmetric unit and 50% solvent. All crystals were cryoprotected by soaking for 2 min in reservoir solution supplemented with 10% glycerol (w/v), frozen under a cryogenic nitrogen stream, and stored in liquid nitrogen before data collection at 100 K.
Data Collection and Structure Determination
Data for MmFadD32-AMPC12 were obtained with ESRF beamline ID14-1, to a resolution of 2.5 Å. Data sets corresponding to MsFadD32 with AMPC12 and AMPC20 were obtained with ESRF beamline ID23-2, at 3.5 Å resolution. X-ray images were processed with Mosflm (20), and diffraction intensities were scaled with SCALA (21) from the CCP4 software package (22). The structure of the MmFadD32 protein was solved with the Balbes molecular replacement server (23). Two molecules were found in the asymmetric unit, when the structure of the FAAL from Legionella pneumophila was used (24) (PDB code 3KXW). The Q factor for this model was 0.637, and final refinement with Balbes gave Rwork and Rfree values of 0.415 and 0.452, respectively. After removal of the C-terminal domain, this model was subjected to several cycles of automatic building and refinement with Buccaneer (25), in which 90% of the protein (i.e. 575 residues) could be traced, giving Rwork and Rfree values of 0.29 and 0.33, respectively. Models were then constructed with the graphics program Coot (26), and refinement was carried out with Buster software (27) and PHENIX (28). String files for AMPC12 and AMPC20 were generated from Open Babel (29), and geometric restraints were generated from grade (30). The refined model corresponds to Rwork and Rfree values of 0.185 and 0.238, respectively. This model contains 610 of the 632 amino acids found in the sequence of the untagged protein, for both molecules A and B of the asymmetric unit. The 44 missing residues had poorly defined electron densities and were located in the N and C termini and in loops exposed to solvent. The side chains of 33 residues with a low electron density or no electron density at all were truncated to Cβ atoms. In total, seven glycerol molecules and 538 water molecules were positioned in the electron density map. The structure of MmFadD32-AMPC12 was used to solve the structure of MsFadD32 in complex with AMPC20, by molecular replacement with PHASER software (31). Structure refinement led to final Rwork and Rfree values of 0.223 and 0.286, respectively. Eight molecules of MsFadD32 were found in the asymmetric unit, and the refined structures contained 592–606 of the 630 amino acids present in the sequence of the untagged protein. In addition, a large number of side chains could not be traced because they had little or no electron density, and it was not possible to add solvent molecules due to the limited resolution. All structures were checked with PROCHECK (32) and during the PDB deposition process were analyzed with PROMOTIF (33), as implemented on the EMBL-EBI server, and visualized with PyMOL (34). The sequence alignment was generated with ESPript 3 (35). Protein structure databases were searched, and structures were superimposed with Dali software (36, 37).
Small Angle X-ray Scattering (SAXS) Experiments
MsFadD32 and CgFadD32 in 50 mm HEPES, pH 7.5, 500 mm NaCl were concentrated to about 5 mg·ml−1 (i.e. about 70 and 92 μm, respectively). AMPC12 in DMSO was mixed with proteins at a molar ratio of 2:1, with a final DMSO concentration of 2–3%. All buffers used for SAXS experiments were either collected from the gel filtration column used for purification, after the equilibration step, or by overnight dialysis, to ensure buffer matching. We supplemented 50-μl protein samples with 2 mm DTT and centrifuged them for 10 min at 10,000 rpm to eliminate all aggregates before x-ray analysis. Concentrations were checked by measuring UV absorption at λ = 280 nm on a Thermo Scientific NanoDrop 1000 spectrophotometer. SAXS experiments were conducted on the SWING beamline at the SOLEIL synchrotron, Gif-sur-Yvette, France (λ = 1.033 Å). The detector was positioned to collect data with an exploitable Q-range of 0.008–0.4 Å−1, where Q = 4πsinθ/λ, with a scattering angle of 2θ. Samples obtained directly or from a Bio SEC-3 (300 Å pore size) HPLC column (Agilent) were injected into the SAXS flow-through capillary cell at a flow rate of 0.05 and 0.2 ml·min−1, respectively, and a temperature of 15 °C. When the sample was directly injected in the capillary, a sample volume of 40 μl was used, and a total of 75 frames of 0.5 s each were recorded. In HPLC mode, SAXS data were collected throughout the whole elution time, with a frame time of 1 s. Frames corresponding to the elution peak were checked for the stability of the associated radius of gyration, and the resulting selection of curves was averaged. Data were reduced with the custom-built Foxtrot application and analyzed with the ATSAS suite (38). Theoretical scattering curves corresponding to crystal structures were calculated with CRYSOL software (39), with a solvent density of 0.34 e·Å−3 to take the salt contribution into account.
The atomic coordinates and structure factors (codes 5EY8 and 5EY9) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ.
Results
Biochemical and Enzymatic Characterization of Mycobacterial FadD32 Enzymes
We previously described the biochemical characterization of FadD32 from M. tuberculosis (MtFadD32) (13), and the use of the orthologous enzymes from M. smegmatis (MsFadD32, 74% sequence identity) and C. glutamicum (CgFadD32, 39% sequence identity) for comparative studies and the development of a high throughput screening assay for FadD32 activity (17). In this study, the FadD32 enzyme from M. marinum (MmFadD32), which displays a much higher degree of sequence identity (92%) to the M. tuberculosis enzyme, was used as a third surrogate. The four FadD32 proteins were produced and purified according to improved versions of published protocols (see under “Experimental Procedures”). The activity of the purified FadD32 proteins was then determined with our published FadD32-pyrophosphatase coupled assay (17), and their ability to release PPi, using lauric acid as a substrate, was compared (Table 1 and Fig. 1). MsFadD32 had the highest specific activity, as reported previously (17). However, we found the difference in specific activity between MsFadD32 and MtFadD32 to be much smaller than previously reported (factor of 7 here versus 75 in Ref. 17, and corrigendum 2014). This smaller difference reflects improvements in MtFadD32 activity, probably due to optimization of the purification protocol and the assay conditions used. Despite the higher purification yield and thermal stability (Fig. 2), the specific activity of CgFadD32 was only about one-twelfth that of the M. tuberculosis enzyme, precluding further kinetic and inhibition studies (Table 1). The FadD32 enzymes from M. tuberculosis and M. marinum had equivalent specific activities and affinities for fatty acid substrates (Table 1). This, and the very high sequence identity of these two proteins, highlights the relevance of MmFadD32 as a surrogate for the M. tuberculosis protein.
FIGURE 1.
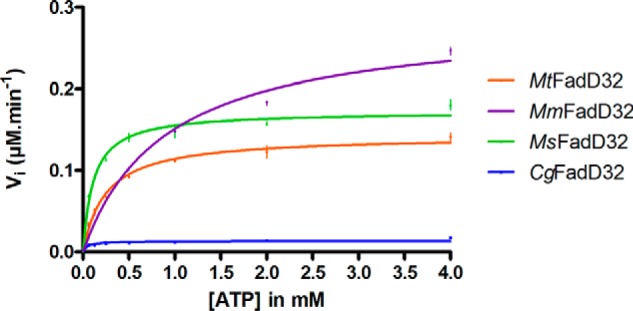
Michaelis-Menten curves for the determination of apparent Km and Vmax values for ATP. The specific activity of each FadD32 was deduced from the dose-response curve for the PiColorLock-based assay with lauric acid (200 μm) as a substrate (as described under “Experimental Procedures”). The initial velocity of the FadD32 reaction was assessed spectrophotometrically in the coupled assay. We determined the steady-state apparent parameters Km (Michaelis constant) and Vmax (maximum velocity) by using non-linear regression to fit the Michaelis-Menten equation to the data. Means and standard errors of the means of two replicates are shown.
FIGURE 2.
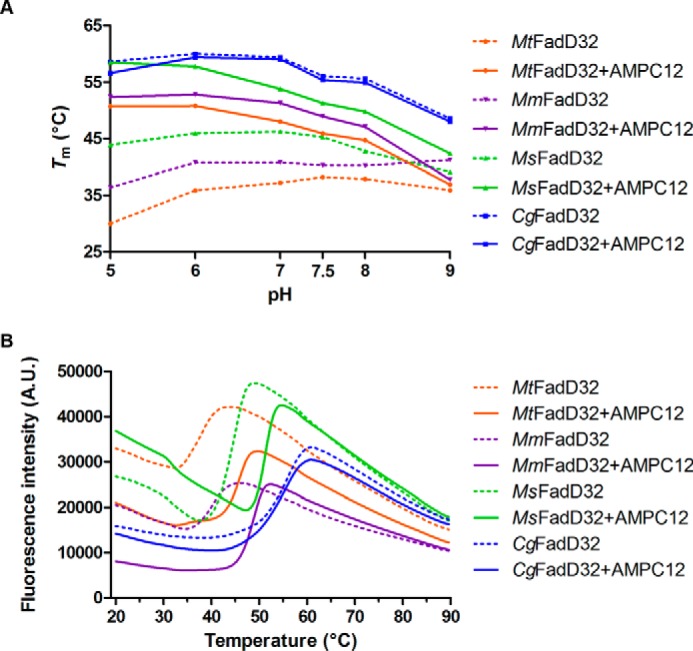
DSF analysis of FadD32. A, thermal stability of FadD32 orthologs was analyzed in various buffer conditions, in the absence (dashed lines) and in the presence (continuous lines) of AMPC12. Tm values were deduced from the curves for a temperature gradient from 25 to 80 °C. The means of three replicates are shown. Standard errors were found too low to be visualized and were not displayed. B, DSF (fluorescence versus temperature gradient) curves at pH 7.5. A.U., arbitrary units.
Inhibition of FadD32 Activity
We also previously reported the inhibition of MtFadD32 and MsFadD32 activity by the alkyl adenylate substrate analog AMPC12 (dodecyl-AMP) (13, 17). Here, we tested AMPC12 and the longer AMPC20 (eicosyl-AMP). We first investigated the ability of AMPC12 to induce a shift in the melting temperature (Tm) of the proteins, by DSF under various pH conditions (Fig. 2). The addition of AMPC12 to all proteins except CgFadD32 led to a significant thermal shift (ΔTm). For instance, at pH 7.5, the optimum pH for the M. tuberculosis enzyme, we obtained ΔTm values of +7.7 °C for MtFadD32, +8.6 °C for MmFadD32, and +6.0 °C for MsFadD32. These positive shifts indicated that the inhibitor bound the proteins, with this interaction probably stabilizing FadD32. Furthermore, AMPC12 and AMPC20 inhibited the three orthologs in a dose-dependent manner (Fig. 3). The calculated half-maximal inhibitory concentrations (IC50) of AMPC12 for the three orthologs were in the same range (1.5–2.75 μm), whereas the IC50 values for AMPC20 were lower by a factor of 2–3, suggesting stronger inhibition (Table 2), consistent with the long-chain substrate selectivity of FadD32 (Table 1) (13). Further characterization of the interaction between FadD32 and AMPC12 by MST yielded dissociation constants (KD) of 0.4 to 3.9 μm (Table 2), consistent with the IC50 values. The KD measurement by MST performed for MsFadD32 and AMPC20 gave a value of 0.24 ± 0.01 μm, in line with stronger binding and inhibition by longer substrates.
FIGURE 3.
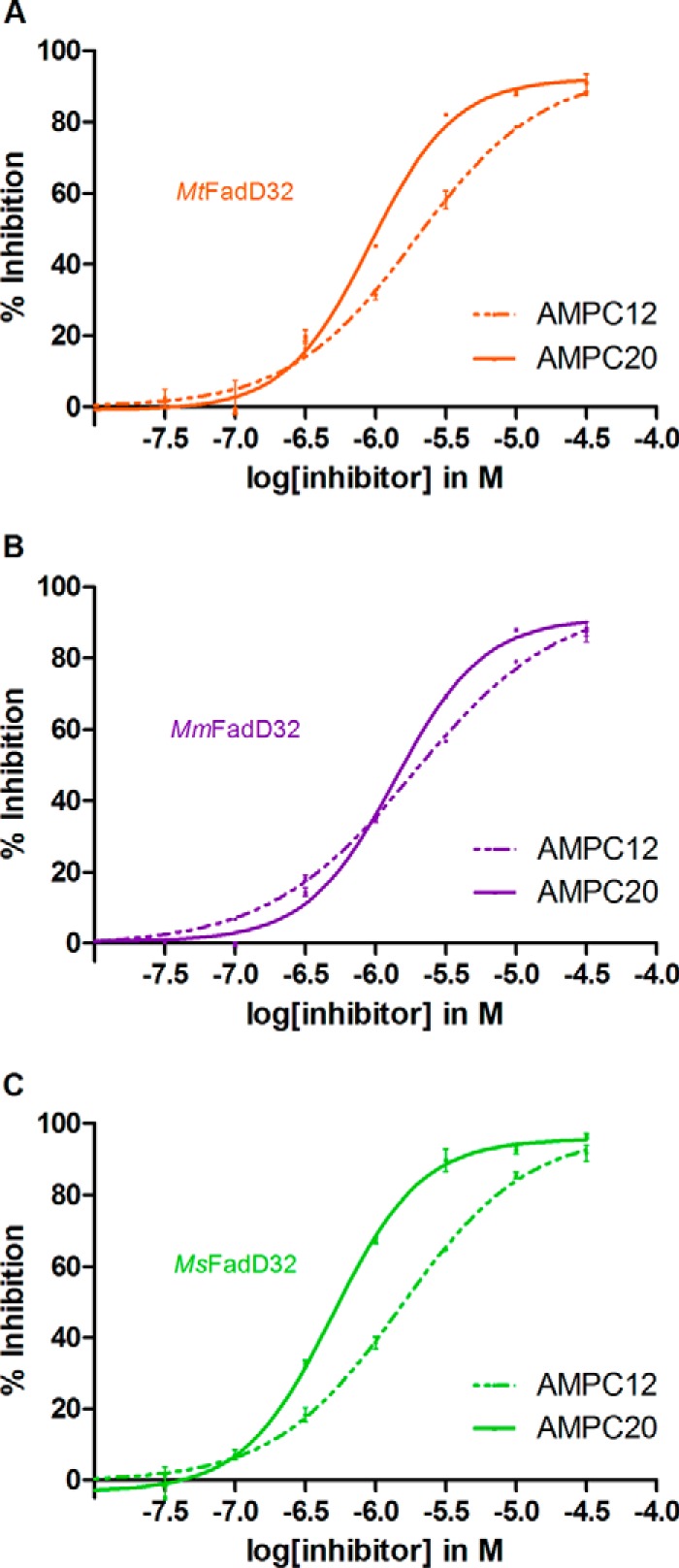
Curves for the inhibition of FadD32 activity by AMPC12 and AMPC20. Various concentrations (0.03–31.6 μm) of the compounds were added to the assay, and FadD32 activity was measured in the presence of 1.6 mm ATP and 100 μm lauric acid as substrates. Means and standard errors of the mean are shown for four replicates. IC50 (half-maximal inhibitory concentration) was calculated from the dose-response curve. A, MtFadD32. B, MmFadD32. C, MsFadD32.
TABLE 2.
Inhibition of FadD32 by AMPC12 and AMPC20
R = C12 (AMPC12) or C20 (AMPC20).
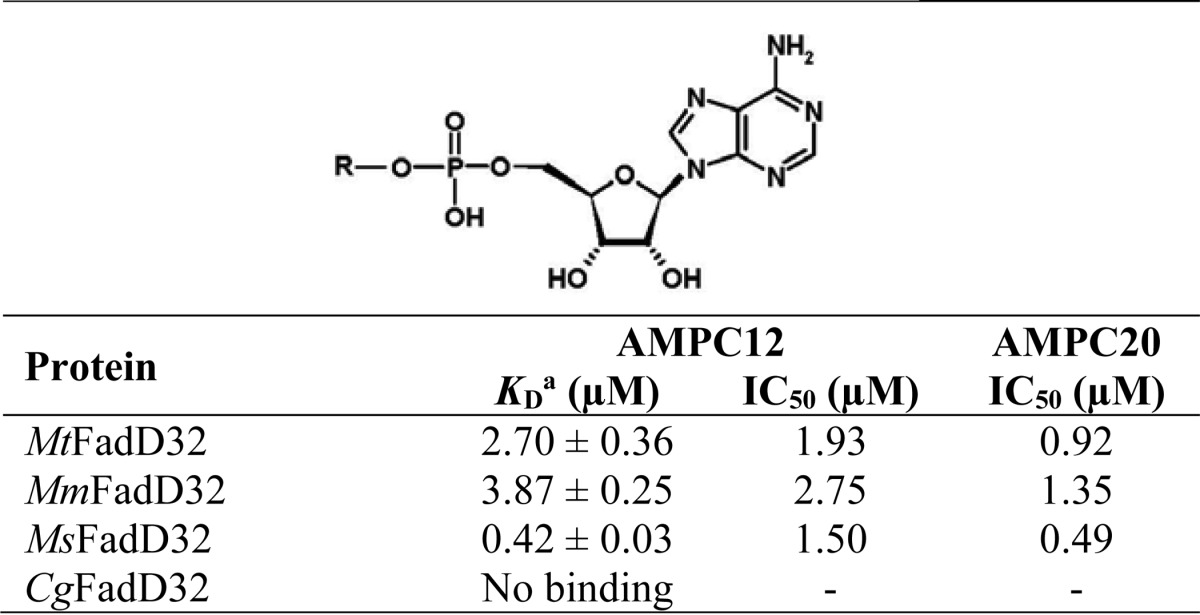
a Data were determined by microscale thermophoresis.
Overall Three-dimensional Structure of FadD32
Despite numerous attempts to crystallize apoenzymes and enzymes in the presence of substrate analogs, we were unable to obtain crystals of CgFadD32, and those obtained for MtFadD32 displayed only weak low resolution diffraction, making structural determination impossible. By contrast, crystals of MsFadD32 were obtained in the presence of AMPC12 or AMPC20, and the structure of the complex with AMPC20 was resolved at low resolution (i.e. 3.5 Å). The structure of MmFadD32-AMPC12 was determined at a much higher resolution (i.e. 2.5 Å) and will be used here as a reference (Table 3). Two molecules, denoted A and B, were present in the asymmetric unit of the orthorhombic MmFadD32 crystals. However, both crystal packing analysis and size exclusion chromatography multiangle static light scattering (Fig. 4) clearly indicated that the biologically active FadD32 unit was a monomer. This was confirmed by PISA calculations (40), which revealed no specific interactions likely to result in the formation of stable higher quaternary structures. The superimposition of molecules A and B yielded an r.m.s.d. value of 0.5 Å for 607 Cα carbons. Crystals of MsFadD32 in complex with either AMPC12 or AMPC20 belonged to the P4 tetragonal space group, and their asymmetric unit contained eight molecules. Consistent with the low resolution of diffraction, the corresponding structures lacked several residues and had many incomplete side chains, and only the structure in the presence of AMPC20 could be determined satisfactorily (Table 3). Cα-based pairwise comparison of the molecules constituting the asymmetric unit of MsFadD32 yielded r.m.s.d. values of 0.5 to 0.9 Å. Superimposition of the MmFadD32 and MsFadD32 structures yielded r.m.s.d. values of between 1.1 and 1.5 Å for about 590–600 common Cα atoms. FadD32 displays the classical two-domain fold observed in class I AFEs (Fig. 5) (14). The large N-terminal domain of MmFadD32 consists of amino acid residues 1–482 and adopts a complex α/β. The small C-terminal domain of MmFadD32 consists of residues 483–629. It extends from the last β-strand of the N-terminal domain through a four-amino acid linker and adopts a globular fold composed of two contiguous β-sheets surrounded by four α-helices and several long loops.
TABLE 3.
Crystallographic data collection and refinement statistics
| MmFadD32-AMPC12 | MsFadD32-AMPC20 | |||
|---|---|---|---|---|
| PDB code 5EY9 | PDB code 5EY8 | |||
| Data collection | ||||
| Beam line | ID14-1 | ID23-2 | ||
| Space group | P212121 | P4 | ||
| Unit cell a, b, c (Å) | 71.79, 100.15, 212.32 | 164.04, 164.04, 231.53 | ||
| Unit cell α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | ||
| Resolution (Å) | 2.50 (2.64–2.50)a | 3.50 (3.69–3.50) | ||
| No. of Unique | 53,766 (7,727) | 72,026 (10,689) | ||
| Rsym | 0.148 (0.484) | 0.232 (0.762) | ||
| Rpim | 0.074 (0.238) | 0.155 (0.515) | ||
| I/σI | 7.7 (2.9) | 4.6 (1.6) | ||
| Completeness (%) | 99.8 (100.0) | 93.9 (95.7) | ||
| Redundancy | 4.8 (5.0) | 3.0 (2.9) | ||
| Refinement | ||||
| Resolution range (Å) | 48.7–2.50 | 50.6–3.50 | ||
| No. of reflections | 53,613 | 72,012 | ||
| No. of molecules/AUb | 2 | 8 | ||
| Rwork/Rfree | 0.1847/0.2382 | 0.2227/0.2862 | ||
| Chain/No. of residues/missing residues | A/610/1–2,113–114,174–175,187–191,618–622,627–629 | A/598/1–10,189–191,558–562,617–630 | ||
| B/610/1–2,113–114,187–191,617–622,626–629 | B/594/1–11,46–48,112–113,547,556–561,618–630 | |||
| C/605/1–4,113–114,188–191,559–561,619–630 | ||||
| D/603/1–6,113–114,189,557–562,619–630 | ||||
| E/592/1–11,17–20,113–114,190–191,557–561,617–630 | ||||
| F/606/1–5,189–191,558–562,620–630 | ||||
| G/597/1–10,114,188–190, 268–269,408,558–562,620–630 | ||||
| H/595/1–9,16–19,36,113–114,190–191,241–242,275–276, 557–558,620–630 | ||||
| No. of atoms/average B-factors (Å2) | ||||
| All atoms | 10,027/36.0 | 33,378/62.0 | ||
| Protein atoms | 9,377/36.8 | 33,081/55.1 | ||
| Ligands, ions | 112/34.2 | 297/41.9 | ||
| Water molecules | 538/33.9 | −/− | ||
| r.m.s.d. | ||||
| Bond lengths (Å) | 0.003 | 0.005 | ||
| Bond angles (°) | 0.786 | 1.011 | ||
| Ramachandran plot (%) | ||||
| Allowed/disallowed | 2.1/0.1 | 3.5/0.5 | ||
| Most favored regions | 97.8 | 96.0 | ||
a Values in parentheses are for the highest resolution shell.
b AU is asymmetric unit.
FIGURE 4.
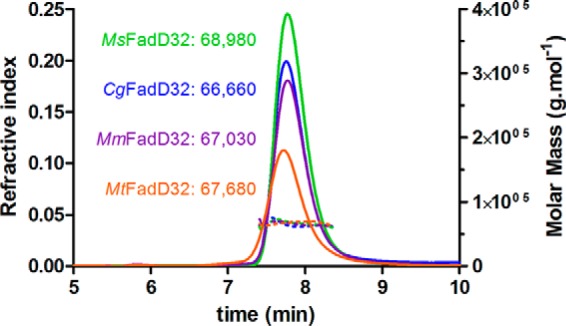
SEC-MALS experiments on FadD32 enzymes. Continuous lines represent the variation of refractive index against elution time from the size exclusion column for MtFadD32 (orange), MmFadD32 (purple), MsFadD32 (green), and CgFadD32 (blue). The experimentally measured molar mass distribution (dashed lines, same color code) and deduced mean molar mass obtained with ASTRA software (in g·mol−1) are indicated for each elution peak. Calculated molecular masses of untagged proteins used for experiments (in daltons): MtFadD32, 69,513; MmFadD32, 69,172; MsFadD32, 68,510; CgFadD32, 68,196. At the flow rate used (0.35 ml·min−1), the exclusion limit of the column corresponds to an elution time of 5.8 min.
FIGURE 5.

Overall structure of MmFadD32. A, ribbon representation. The various subdomains of the large N-terminal domain are shown in orange, cyan, and blue, whereas the smaller C-terminal domain is shown in yellow. The linker between the N- and C-terminal domains is shown in red. The six sequence insertions (SI1 to SI6, see text) of FadD32 are shown in magenta. Helices and β-strands are numbered sequentially. The bound AMPC12 molecule is shown as a stick representation, with carbon, nitrogen, oxygen, and phosphorus atoms in green, blue, red, and light gray, respectively. Top and bottom views are separated by a 180° rotation around a vertical axis. The same color scheme is used in all figures. B, topology diagram.
Distinctive Structural Features of Mycobacterial FadDs and Other AFEs
A structure-based sequence alignment was generated for the four FadD32 orthologs investigated here and other mycobacterial FadDs and AFEs of known structure (Fig. 6). Consistent with the low r.m.s.d. value obtained when their structures were superimposed, MmFadD32 and MsFadD32, which share 74% sequence identity, were found to have very similar structures. A single insertion of one residue and only a few differences in secondary structure were observed. The sequences of MmFadD32 and MtFadD32 are even more strongly conserved (92% sequence identity). Consistent with this high degree of sequence identity, a reliable three-dimensional homology model of MtFadD32 was constructed, based on the crystal structure of MmFadD32. Sequence differences mostly affect solvent-exposed residues, many of which are not involved in interactions. These residues are evenly distributed along the two protein sequences. Finally, comparison of the sequences of CgFadD32 and the other three orthologs revealed several insertions and deletions. The structures of three other FadD proteins from M. tuberculosis have been resolved and were included in the sequence alignment (Fig. 6 and Table 4). These structures included that of the N-terminal domain of the FAAL FadD28 (41) and those corresponding to the full-length FACL enzyme FadD13 and its N terminus (42, 43). As the orientations of the N- and C-terminal domains differ between these two proteins, the superimposition of FadD13 and MmFadD32 structures was based on the use of N- or C-terminal domains. The structure of the full-length FAAL enzyme FadD10 has also been determined (44). Again, the respective orientations of the N- and C-terminal domains differ in the structures of FadD10 and MmFadD32. Superimposition was therefore performed with these two domains separately. Thus, one key characteristic of AFEs is this well characterized flexibility of structural conformation between the N- and C-terminal domains. Indeed, searches for structural similarity based on either the N- or C-terminal domain of MmFadD32 identified a large (i.e. about 150) number of structural homologs. In searches based on the entire MmFadD32 structure, we were able to identify only a few homologous structures with the same orientation of N- and C-terminal domains as in MmFadD32: (i) E. coli FAAL (24); (ii) the benzoate CoA ligase from Burkholderia xenovorans (45); (iii) the malonyl CoA synthetase MatB from Rhodopseudomonas palustris (46); and (iv) the phenylalanine-activating domain PheA of gramicidin S synthetase 1 from Aneurinibacillus migulanus (Fig. 6 and Table 4) (47).
FIGURE 6.


Structure-based alignment of the sequences of mycobacterial FadDs with those of other adenylate-forming enzymes. Sequences were separated in two groups (upper group, selected mycobacterial FadDs; lower group, selected adenylate-forming enzymes). Within each group, sequence similarity is indicated by red letters, whereas sequence identity is indicated by white letters on a red background. Aligned and unaligned residues are displayed in uppercase and lowercase, respectively, taking MmFadD32 as reference. Residues 460–580 of FadD28, also in lowercase, are absent from the structure and were aligned manually. Secondary structure elements (arrows for β-strands and coils for α- and η-helices) of MmFadD32 are indicated at the top. Residues of MmFadD32 that are disordered in the crystal structure are also indicated at the top, by black bars. Sequence insertions (SI1 to SI6) in FadD32 are underlined in magenta. Residues important for alkyl adenylate binding are indicated by violet (adenine moiety) and orange (aliphatic chain) stars. Residues with side chains that undergo conformational changes to accommodate AMPC20 binding are indicated by red stars. Mutations that have been shown to confer resistance to coumarin inhibitors are indicated by blue stars.
TABLE 4.
Pairwise comparison of the structure of MmFadD32 with those of other adenylate-forming enzymes
| Protein name | Source | UniProtKB code | UniProtKB entry name | PDB code | Ref. | r.m.s.d./no. of aligned positions/sequence identity of aligned positions |
||
|---|---|---|---|---|---|---|---|---|
| N-terminal domain | C-terminal domain | Full length | ||||||
| FadD32 | M. marinum | B2HMK0 | FadD32_MYCMM | 5EY9 | This work | |||
| FadD32 | M. smegmatis | A0R618 | FadD32_MYCS2 | 5EY8 | This work | 1.0/466/74 | 0.6/128/79 | 1.1/595/75 |
| FadD28 | M. tuberculosis | P9WQ59 | FadD28_MYCTU | 3E53 | 41 | 1.7/417/38 | ||
| FadD13 | M. tuberculosis | P9WQ36 | FadD13_MYCTO | 3T5C | 43 | 2.4/377/20 | ||
| 3R44 | 42 | 2.7/380/20 | 2.0/99/22 | |||||
| FadD10 | M. tuberculosis | I6WXG2 | FadD10_MYCTU | 4IR7 | 44 | 2.7/362/18 | 2.3/104/20 | |
| Fatty acyl-AMP ligase | L. pneumophila | Q5ZTD3 | FAAL_LEGPH | 3KXW | 24 | 2.0/433/29 | 2.5 /102/24 | |
| Fatty acyl-AMP ligase | E. coli | Q8FDN4 | FAAL_ECOL6 | 3PBK | 24 | 2.4/421/30 | 1.8/103/33 | 2.5/524/ 30 |
| Benzoate-CoA ligase | B. xenovorans | Q13WK3 | BCL_BURXL | 2V7B | 45 | 2.5/375/18 | 1.8/94/19 | 2.8/469/19 |
| Malonyl-CoA synthetase MatB | R. palustris | Q6ND88 | MatB_RHOPA | 4FUT | 46 | 2.1/373/23 | 2.1/100/24 | 2.8/476/22 |
| Phenylalanine activating domain PheA of gramicidin S synthetase 1 | A. migulanus | P0C061 | GRSA_ANEMI | 1AMU | 47) | 2.4/382/19 | 2.2/97/14 | 3.1/481/18 |
The structure-based sequence alignment revealed the presence of several sequence insertion (SI) blocks in FadD32 (Figs. 5A and 6). Three of these blocks are specific to FadD32 (SI2, Arg-268–Gly-275; SI5, Leu-445–Gly-461; and SI6, Asn-523–Asp-543). One block (SI1, Phe-43–Asp-48) was also identified in FadD28, and two blocks (SI3, Pro-330–Thr-336, and SI4, Ile-363–Val-386) were found in FadD32 and FadD28 and in FAALs from E. coli and L. pneumophila. These six insertions interact with the rest of the protein, contributing to its globular fold (Fig. 5A). This finding is exemplified by the Ile-363–Val-386 SI4 segment, which bridges the N- and C-terminal domains. This motif is conserved among FAALs (24), and previous functional and structural studies of FadD28 have shown it to be a specific trait of FAAL homologs, sufficient to prevent the formation of acyl-CoA derivatives (41). Molecular modeling and further biochemical and structural studies on FadD13 have shown this insertion to be a prerequisite for FAAL activity, which requires a conserved hydrophobic patch between the insertion and the N-terminal domain (43). Two hydrophobic residues, phenylalanines 383 and 481, have been shown to be spatially adjacent in a molecular model of MtFadD32 derived from the structure of FadD28, and a F383A/F481A double mutant of MtFadD32 displays FACS activity (43). The insertion motif and its amino acid environment are highly conserved between M. tuberculosis, M. marinum and M. smegmatis sequences, and the structure of MmFadD32 confirms the existence of a hydrophobic core common to the insertion motif and the N-terminal domain, in which Phe-375 (add +8 for MtFadD32 numbering) occupies a central position (Fig. 7A). However, despite the close physical proximity of Phe-375 and Phe-473, the closest atoms of which are only 3.9 Å apart, no favorable interactions, such as π-stacking, were found between the two phenylalanine residues, the disruption of which may account for level of activity of the MtFadD32 F383A/F481A mutant. SI2 (Arg-268–Gly-275), the shortest of the three specific FadD32 sequence insertions, protrudes slightly from the surface of the protein. By contrast, SI5 (Leu-445–Gly-461) makes a long excursion and caps β-sheet E (Fig. 7B), and SI6 (Asn-523–Asp-543), which is not conserved in CgFadD32, also makes a long excursion at the surface of the protein and caps β-sheet G (Fig. 7C).
FIGURE 7.

Sequence insertions in MmFadD32. The three longest sequence insertions found in FadD32 and their interaction with the core of the protein are displayed and labeled. A, SI4 (Ile-363–Val-386), the trademark of FAALs bridging the N- and C-terminal domains. B, SI5 (Leu-445–Gly-461), which caps β-sheet E of the N-terminal domain. An enlarged view where all distances have been indicated is provided at the right. C, SI6 (Asn-523–Asp-543), which caps β-sheet G of the C-terminal domain.
Alkyl Adenylate Binding
The co-crystallization of MmFadD32 and MsFadD32 with AMPC12 and AMPC20, respectively, showed a well defined area of higher electron density in the active site of the enzymes (Fig. 8). This made it possible to position the alkyl adenylates unambiguously. An analysis of the topology of MmFadD32 revealed a deep, open, funnel-shaped cavity leading to the active site, and a long tunnel leaving the catalytic chamber and passing through the protein (Fig. 9). The mouth of the cavity is delimited by the N terminus of helix α9, the C-terminal tips of strands βA7 and βE5, helix η16, and the long βG3-α24 loop. The adenosine moiety resides in the vestibule of the cavity, and the C2′ exo-ribose and α-phosphate group are the most solvent-exposed parts of the ligand. By contrast, the adenine and the alkyl chain lie in the same plane and point toward the interior of the protein. The planar adenine sits in a small hydrophobic pocket, enclosed by the side chains of Pro-315, Tyr-342, and Ile-479 and the backbone atoms of Ser-313–Glu-314–Pro-315–Val-316 (Fig. 9A). In addition, hydrogen bonds are formed between endo/exocyclic nitrogen atoms (N1, N3, N6, and N7) and the main-chain atoms of serine residues 313 and 341 and water molecules (Fig. 9A). Five FadD32 residues seem to play a key role in anchoring the ribose-phosphate moiety, by establishing polar interactions with ribosyl hydroxyl groups and/or phosphate oxygen atoms as follows: Asp-468 with both O2′ and O3′; Arg-482 with O3′; Lys-600 with O5′ and O2P; Asp-231 with O1P; and His-230 with O2P (Figs. 8A and 9A). In MmFadD32, the dodecyl aliphatic chain of AMPC12 adopts an extended conformation and is buried in the hydrophobic tunnel. The 5–6 Å-wide and 16 Å-long slightly curved tunnel is delineated by protein segments exclusively from the N-terminal domain, comprising helix α8 (Val-210 and Leu-214), helix α9 (Met-232, Ile-235, Thr-236, and Leu-239), strand βA4 (Phe-247), strand βA5 (Phe-277, Ser-278, and Ala-279), strand βA6 (Leu-310 and Asn-311), part of the βA6-α14 loop (Gly-312 and Ser-313), strand βA7-helix η16 (Ser-341, Tyr-342, Gly-343, Leu-344, and Ala-345), and strand βA8 (Leu-349 and Phe-350). The electron density of the aliphatic chain is very well defined, but only one close contact is formed with protein residues (distance <3.5 Å, as shown in violet in Fig. 9A). AMPC12 binding upon co-crystallization with MmFadD32 is reminiscent, in terms of both ligand conformation and chemical environment, of previous observations for other structures, such as the long-chain FACS of Thermus thermophilus obtained after soaking crystals of the AMP-PNP complex in a myristate solution (48), the FAALs of E. coli and L. pneumophila co-purified with dodecanoyl/myristoyl adenylates (24), and M. tuberculosis FadD10 with dodecanoyl adenylate prepared by incubating the protein in a reaction mixture containing ATP, MgCl2, and lauric acid (44). Moreover, the structural features of the MsFadD32-AMPC20 complex were identical to those of MmFadD32-AMPC12 (Figs. 8 and 9), consistent with the 74% identity between the amino acid sequences of the two proteins. Conservation was found to be even stronger for residues described above as involved in interactions with the ribose-phosphate moiety (100% identity over 12 residues) or defining the hydrophobic tunnel (90% identity over 21 residues) (Fig. 6). One remarkable difference between the two structures was identified at the tunnel exit, which is closed in MmFadD32-AMPC12 but open in MsFadD32-AMPC20. This difference in the open/closed conformation of the tunnel is not due to differences in the local fold of the proteins. Instead, it is dependent simply on differences in the side-chain conformations of three residues as follows: Leu-239/Leu-240 (Δχ1 = 72°); His-245/His-246 (Δχ1 = 83°); and Phe-247/Phe-248 (Δχ1 = 100°) (Fig. 9). In addition, or as a result of this conformational change, the side chains of three residues at the very tip of the hydrophobic tunnel (Ile-212, Glu-219, and Ile-244) were found disordered in MsFadD32, whereas their counterparts in MmFadD32 (Leu-211, Glu-218, and Ile-243) could readily be assigned (Fig. 9). As a consequence of tunnel opening, up to 16 of the 20 carbons of AMPC20 were visible on the electron density map for the MsFadD32 structure. The remaining four carbon atoms were more disordered, probably because they were not shielded from the solvent.
FIGURE 8.
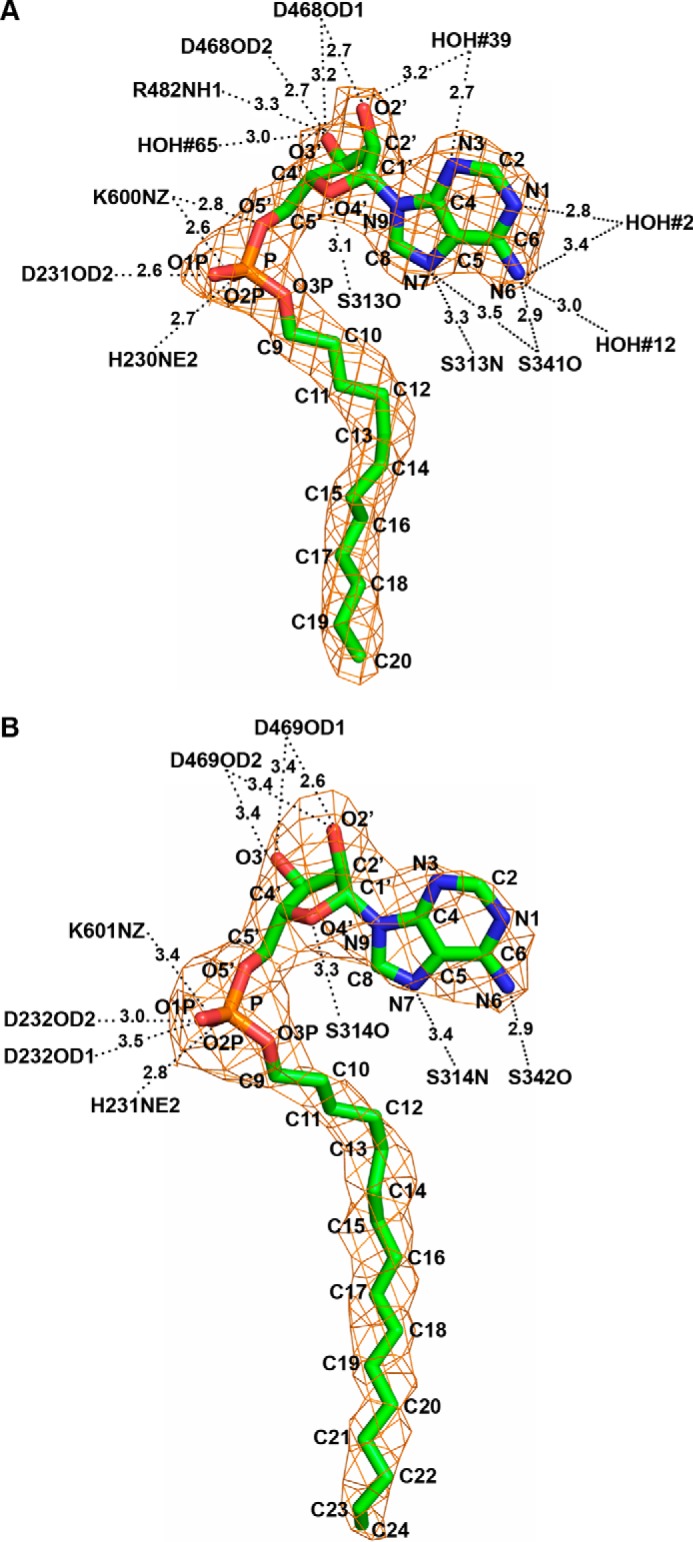
Structure of alkyl adenylates bound to FadD32. A, full-length AMPC12 bound to MmFadD32 (chain A). B, AMPC20 bound to MsFadD32 (chain D) for which 16 of the 20 carbons in the alkyl chain were assigned in the electron density map. The final σA-weighted 2Fo − Fc maps contoured at 1.0 σ are shown in orange. Ligand atoms are labeled. Polar interactions between protein atoms and alkyl adenylates with distances <3.5 Å are also indicated.
FIGURE 9.
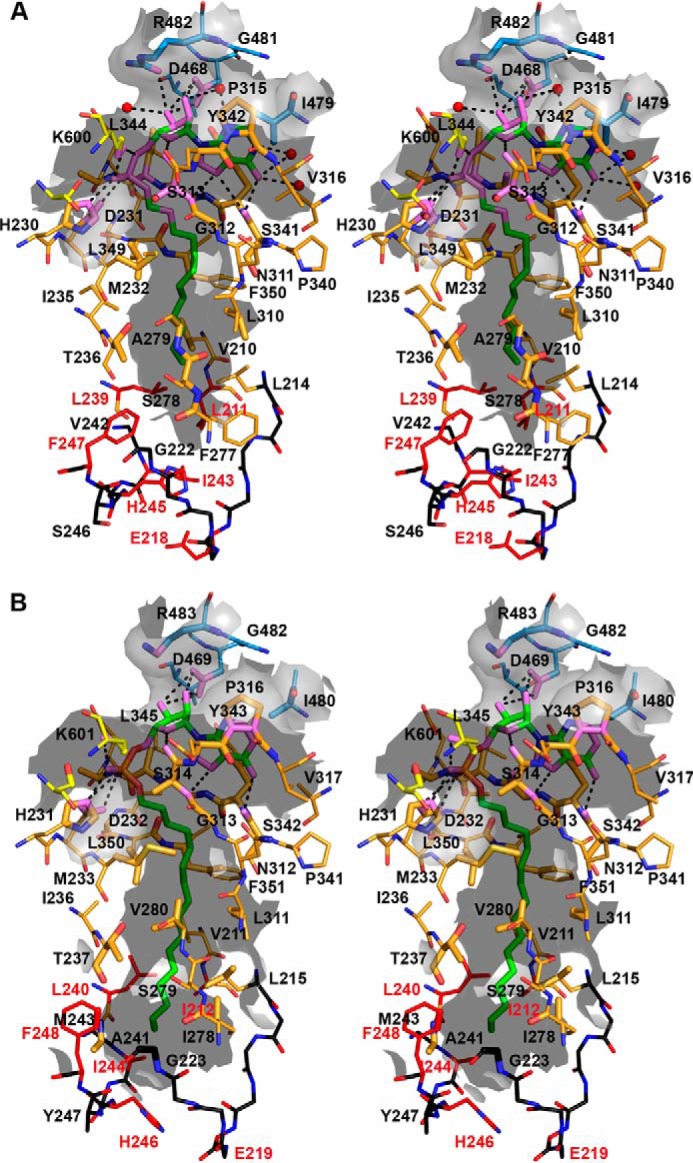
Stereoview of the alkyl adenylate-binding site of FadD32. Residues involved in alkyl adenylate binding are represented as sticks and labeled. Atoms found within 5 Å of the ligand are shown as enlarged sticks and depict the tunnel surface, shown as the gray semitransparent surface. Protein (or ligand) atoms found within 3.5 Å of ligand (or protein) atoms are shown in violet. Polar interactions are represented by black dotted lines. The backbone atoms of segments found at the very tip of the hydrophobic tunnel are shown in black, and those of side chains involved in the closed/open state are shown in red. A, AMPC12 bound to MmFadD32 (chain A). B, AMPC20 bound to MsFadD32 (chain D). Because their side chains were not visible on the electron density map, residues Ile-212, Glu-219, and Ile-244 of MsFadD32 were truncated as alanine.
SAXS Analysis
Free and ligand-bound forms of FadD32 were characterized at low resolution, by SAXS. These experiments were performed with CgFadD32 and MsFadD32, the only alternative to MtFadD32 available to us at the time. For CgFadD32, the SAXS curves obtained in the presence and absence of AMPC12 were similar (Fig. 10A). By contrast, for MsFadD32, there was a small but reproducible difference in SAXS profiles, particularly around Q = 0.15 Å−1 (Fig. 10B). To go further, the experimental curve obtained with unbound MsFadD32 was compared with the theoretical scattering patterns calculated for structures of the representative conformations of AFEs. The best fit (χ value of 1.4) was obtained with the adenylate-forming conformation as observed in FadD32 (Fig. 10, C and D).
FIGURE 10.

SAXS analysis of FadD32. A, experimental SAXS data for CgFadD32 in the unbound state (blue line) and in the presence of AMPC12 (red line). B, experimental SAXS data for MsFadD32 in the unbound state (blue line) and in the presence of AMPC12 (red line). C, comparison of the experimental SAXS data for unbound MsFadD32 (blue line) and theoretical scattering patterns computed from structures of representative conformations for AFEs: MsFadD32 (this work, PDB code 5EY8, cyan line), the long-chain fatty acyl-CoA synthetase from T. thermophilus (TtLC-FACS, PDB code 1ULT, green line), the fatty acyl-AMP ligase from L. pneumophila (LpFAAL, PDB code 3KXW, brown line), FadD13 from M. tuberculosis (MtFadD13, PDB code 3R44, orange line), and FadD10 from M. tuberculosis (MtFadD13, PDB code 4IR7, magenta line). D, surface representation of the structures used in C. The structures were superimposed on the basis of the N-terminal domain (marine) and are shown in the same orientation. The C-terminal domain, which adopts a different conformation, is shown in orange. The conserved FAAL motif of FadD32 is shown in magenta. Information is provided about the name of the enzyme, the type of conformation observed, and the χ value indicative of the quality of fit between the experimental diffusion curve and the theoretical curve computed from the corresponding structure.
Discussion
TB remains a major public health problem, as one of the leading causes of death due to a single infectious agent worldwide. It is very difficult to fight TB, due to a combination of correlated deleterious factors in this deadly disease, including drug treatments that are usually effective but difficult to cope with and drug resistance. Antibiotic resistance remains a major challenge, and the development of new drugs, together with effective vaccines and diagnostics, will be essential if we are to eliminate TB (49). The mycolic acid biosynthesis pathway and all the enzymes essential to this pathway have been validated as pertinent targets in the fight against this disease. FadD32, which is essential for mycolic acid biosynthesis (9), has been validated in both target-to-drug (17) and drug-to-target (18) approaches. The work described here constitutes a continuation of our efforts to characterize FadD32 enzymes fully (13, 17). We report here extensive biochemical and biophysical studies of four FadD32 orthologs and the characterization of their structure. Not only is such structural information important to our understanding of the structure-function relationships of enzymes essential for mycobacterial viability, it should also prove instrumental when the hits identified in chemical screens enter the target-to-drug pipeline.
It proved difficult to determine the structure of FadD32. Following massive unsuccessful efforts to obtain crystals of the M. tuberculosis enzyme displaying sufficiently high levels of diffraction for analysis, we extended our studies to orthologs from M. marinum, M. smegmatis, and C. glutamicum. All four enzymes were purified to high levels and biochemically characterized, to build on our previous work (13, 17). CgFadD32 had an unexpectedly low specific activity, contrasting with its high thermal stability on DSF analysis. Indeed, the Tm value of the protein was close to 60 °C, whereas values of 45, 40, and 38 °C were obtained for MsFadD32, MmFadD32, and MtFadD32. Furthermore, it was not modified by treatment with dodecyl-AMP (AMPC12), a characterized FadD32 inhibitor (13), whereas this treatment increased the Tm values of the other three proteins by at least 6.0 °C. Positive shifts of Tm are generally associated with protein stabilization through ligand interaction. It therefore seems likely that AMPC12 either cannot bind CgFadD32 or cannot stabilize it upon binding. The results of MST and SAXS experiments were consistent with a binding defect, because no complex of CgFadD32 with AMPC12 was detected. AMPC12 binding and inhibition clearly occurred for all three mycobacterial FadD32 proteins, yielding IC50 and KD values in the micromolar range. Consistent with these results, alkyl adenylate was required for protein crystallization, except for CgFadD32, which was unable to crystallize in any of the conditions tested. We were able to resolve the crystal structures of MmFadD32-AMPC12 and MsFadD32-AMPC20 at resolutions of 2.5 and 3.5 Å, respectively. These proteins adopt monomeric structures and display the classical fold of the AFE superfamily. However, although many AFE structures within the PDB were identified as displaying high levels of structural similarity to either the N- or C-terminal domain of FadD32, only four structures could be superimposed on the entire FadD32 protein. Versatility in the orientation of the N- and C-terminal domains is typical of AFEs, and it has been suggested that a rotation of the C-terminal domain by about 140°, in the domain alternation mechanism, would allow enzymes with CoA ligase activity to perform the second thioester-forming half-reaction upon completion of the initial adenylation reaction (50, 51). Indeed, close scrutiny of the PDB showed that most AFE structures fell into two classes in terms of the respective orientations of their N- and C-terminal domains as follows: those like FadD32 that are in the adenylate-forming conformation, and those in the thioester-forming conformation. Interestingly, we found that the linker between the N- and C-terminal domains systematically adopted a helical conformation (helix η19 in FadD32) in the adenylate-forming conformation, although it formed an open turn in the thioester-forming conformation. Moreover, some enzymes, such as M. tuberculosis FadD13 (42) adopted an intermediate conformation, with a smaller amplitude of C-terminal domain rotation, whereas others adopted a totally different conformation, such as the single open conformation observed for both the native and ligand-bound states of M. tuberculosis FadD10 (44). In the cases where the structures of AFEs were determined in both the native and ligand-bound adenylate-forming conformations, very few differences were found between the two forms (50, 52). We were unable to crystallize native FadD32, but based on SAXS experiments we hypothesize that the unbound structure should only slightly differ from the conformation trapped during FadD32 crystallization in the presence of alkyl adenylates. The role of FadD32 in the transfer of fatty acids to the N-terminal ACP domain of Pks13 raises questions about the need for a conformational change to facilitate FAAS activity and the nature of that change. This conformational change, if indeed such a change occurs, has yet to be characterized. The PA1221 non-ribosomal peptide synthetase protein of Pseudomonas aeruginosa, which contains adenylation and peptidyl carrier protein (PCP) domains, adopts the canonical AFE thioester-forming conformation (53). In PA1221, which lacks the conserved FAAL motif, the PCP domain (equivalent to polyketide synthase ACP) interacts with both the N- and C-terminal regions of the adenylation domain.
The alkyl adenylates used to solve the FadD32 structures adopt a U-shaped conformation and mode of binding similar to those for other complexes with substrate analogs. Asp-468 and Lys-600 are among the catalytic pocket residues involved in anchoring the inhibitor. Both are strictly conserved in the protein sequences featured in Fig. 6. Lys-600, His-230, and Asp-231 display strong chemical similarities with the active site alignment described for class I AFEs in which the positively charged lysine and histidine residues stabilize the pentavalent negatively charged phosphorus atom present in the transition state (14). The FadD32 structures also show how the long meromycolic chain can be accommodated in a tunnel with an adaptive mechanism based on the modification of a single side-chain rotamer of three residues. Insertions-deletions between the sequences of CgFadD32 and the mycobacterial orthologs are remote from the alkyl adenylate-binding site and do not seem to play a direct role in the lack of affinity of the C. glutamicum enzyme for AMPC12 and AMPC20. It also appears that all MmFadD32 residues involved in adenine and ribose/phosphate binding are strictly conserved in CgFadD32. By contrast, the degree of conservation of residues delineating the hydrophobic tunnel is lower, and changes in four residues that line the bottom of the tunnel (Leu-239 → Phe-262, Phe-277 → Tyr-301, Ala-279 → Val-303, and Leu-310 → Ile-333) might induce steric hindrance that could impede fully burying the aliphatic chains of the alkyl adenylate inhibitors.
Several strategies have been developed for identifying and designing potent and selective mycobacterial AFE inhibitors (15). Bisubstrate inhibitors mimicking acyl adenylate, such as acylsulfamoyl adenosine and alkyl adenylate analogs (13, 41), appear to be good starting materials, but the rationale for their improvement is not straightforward. Other approaches make use of high throughput screening. For instance, five novel classes of inhibitors were identified by screening a dedicated 10,000-compound library with a biochemical assay and MsFadD32 as a surrogate (17), whereas a 4,6-diaryl-5,7-dimethyl coumarin series was identified by screening about 20,500 small molecules for the ability to inhibit M. tuberculosis growth in a whole-cell GFP-based assay (18). The most potent substituted coumarin analog was as active as isoniazid and killed M. tuberculosis by directly inhibiting the FAAS activity of MtFadD32 (18). Three mutations (E120A, E120V, and F291L) in MtFadD32 have been shown to confer resistance to these coumarin inhibitors. The corresponding MmFadD32 residues (Glu-112 and Phe-283) belong to the N-terminal domain. Glu-112 is found in a solvent-exposed and partly disordered loop between strand βB2 and helix α4. Phe-283 is located at the N terminus of helix α13, at the interface with the C-terminal domain, with its side chain closely packed with the Tyr-514, Gly-599, Arg-595, and Phe-625 residues, which are also strictly conserved in MtFadD32. Phe-283 would be involved in the interface with the cognate Pks13 ACP domain if Pks13 ACP adopts the same configuration as reported for PCP in the structure of PA1221. Ser-553, the phosphopantetheine attachment site of PCP, is located 9.5 Å away from Phe-283. These observations are consistent with the proposed mode of action of 4,6-diaryl-5,7-dimethyl coumarins against FadD32 FAAS activity. Fragment-based drug discovery is another promising as yet unexplored approach that could be applied to the discovery of FadD32 inhibitors. In addition to providing fundamental details about the structure-function relationships of mycobacterial FadD enzymes, we hope that this work will facilitate the design and improvement of FadD32 inhibitors. This is particularly true for MmFadD32, which should be a useful surrogate for the tuberculosis enzyme, given the high degree of sequence identity between the two enzymes.
During completion of the writing of this manuscript, a report was published on the structures of the N-terminal domain of MsFadD32 in the unbound form and of the full-length protein in the ATP-bound state (54). This report provides support for some of the observations and conclusions drawn in this more extensive study.
Author Contributions
V. G. and H. M. conceived and designed experiments; V. G., S. G., S. L., V. M., C. B., N. E., and H. M. performed research; V. G., S. G., C. B., H. M., L. Maveyraud, and L. Mourey analyzed data; L. Mourey wrote the paper, with contributions from all the authors, and created the figures; M.D. and L. Mourey supervised the project.
Acknowledgments
We thank the scientific staff of the European Synchrotron Radiation Facility (Grenoble, France), SOLEIL (Gif sur Yvette, France), and ALBA (Barcelona, Spain) for the use of their excellent data collection facilities. We particularly thank the staff of beamlines ID14-1 and ID23-2 at the European Synchrotron Radiation Facility, where the crystallographic data used in this article were collected, and the staff of beamline SWING at SOLEIL where SAXS experiments were conducted. We thank NanoTemper Technologies, GmbH, for making the microscale thermophoresis equipment available to us. We also thank Dr. Patrice Gouet for the valuable help with ESPript software. The differential scanning fluorimetry and macromolecular crystallography equipment used in this study are part of the Integrated Screening Platform of Toulouse (PICT, IBiSA).
This work was supported by European Community New Medicines for Tuberculosis Program Grant LSHP-CT-2005-018923. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5EY8 and 5EY9) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TB
- tuberculosis
- ACP
- acyl carrier protein
- AFE
- adenylate-forming enzyme
- DSF
- differential scanning fluorimetry
- FAAL
- fatty acyl-AMP ligase
- FAAS
- fatty acyl-ACP synthetase
- FACL
- fatty acyl-CoA ligases
- MST
- microscale thermophoresis
- PCP
- peptidyl carrier protein
- SAXS
- small-angle x-ray scattering
- PDB
- Protein Data Bank
- SI
- sequence insertion
- r.m.s.d.
- root mean square deviation
- AMP-PNP
- 5′-adenylyl-β,γ-imidodiphosphate.
References
- 1.Daffé M., and Zuber B. (2014) in Bacterial Membranes: Structural and Molecular Biology (Remau H., and Fronzes R., eds) pp. 179–192, Caister Academic Press, Norfolk, UK [Google Scholar]
- 2.Daffé M. (2015) The cell envelope of tubercle bacilli. Tuberculosis 95, S155–S158 [DOI] [PubMed] [Google Scholar]
- 3.Daffé M., and Draper P. (1998) The envelope layers of mycobacteria with reference to their pathogenicity. Adv. Microb Physiol. 39, 131–203 [DOI] [PubMed] [Google Scholar]
- 4.Neyrolles O., and Guilhot C. (2011) Recent advances in deciphering the contribution of Mycobacterium tuberculosis lipids to pathogenesis. Tuberculosis 91, 187–195 [DOI] [PubMed] [Google Scholar]
- 5.Marrakchi H., Lanéelle M. A., and Daffé M. (2014) Mycolic acids: structures, biosynthesis, and beyond. Chem. Biol. 21, 67–85 [DOI] [PubMed] [Google Scholar]
- 6.Portevin D., De Sousa-D'Auria C., Houssin C., Grimaldi C., Chami M., Daffé M., and Guilhot C. (2004) A polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms. Proc. Natl. Acad. Sci. U.S.A. 101, 314–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chalut C., Botella L., de Sousa-D'Auria C., Houssin C., and Guilhot C. (2006) The nonredundant roles of two 4′-phosphopantetheinyl transferases in vital processes of mycobacteria. Proc. Natl. Acad. Sci. U.S.A. 103, 8511–8516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gande R., Gibson K. J., Brown A. K., Krumbach K., Dover L. G., Sahm H., Shioyama S., Oikawa T., Besra G. S., and Eggeling L. (2004) Acyl-CoA carboxylases (accD2 and accD3), together with a unique polyketide synthase (Cg-pks), are key to mycolic acid biosynthesis in Corynebacterianeae such as Corynebacterium glutamicum and Mycobacterium tuberculosis. J. Biol. Chem. 279, 44847–44857 [DOI] [PubMed] [Google Scholar]
- 9.Portevin D., de Sousa-D'Auria C., Montrozier H., Houssin C., Stella A., Lanéelle M. A., Bardou F., Guilhot C., and Daffé M. (2005) The acyl-AMP ligase FadD32 and AccD4-containing acyl-CoA carboxylase are required for the synthesis of mycolic acids and essential for mycobacterial growth: identification of the carboxylation product and determination of the acyl-CoA carboxylase components. J. Biol. Chem. 280, 8862–8874 [DOI] [PubMed] [Google Scholar]
- 10.Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E. 3rd, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., et al. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 [DOI] [PubMed] [Google Scholar]
- 11.Trivedi O. A., Arora P., Sridharan V., Tickoo R., Mohanty D., and Gokhale R. S. (2004) Enzymic activation and transfer of fatty acids as acyl-adenylates in mycobacteria. Nature 428, 441–445 [DOI] [PubMed] [Google Scholar]
- 12.Gavalda S., Léger M., van der Rest B., Stella A., Bardou F., Montrozier H., Chalut C., Burlet-Schiltz O., Marrakchi H., Daffé M., and Quémard A. (2009) The Pks13/FadD32 crosstalk for the biosynthesis of mycolic acids in Mycobacterium tuberculosis. J. Biol. Chem. 284, 19255–19264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Léger M., Gavalda S., Guillet V., van der Rest B., Slama N., Montrozier H., Mourey L., Quémard A., Daffé M., and Marrakchi H. (2009) The dual function of the Mycobacterium tuberculosis FadD32 required for mycolic acid biosynthesis. Chem. Biol. 16, 510–519 [DOI] [PubMed] [Google Scholar]
- 14.Schmelz S., and Naismith J. H. (2009) Adenylate-forming enzymes. Curr. Opin. Struct. Biol. 19, 666–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duckworth B. P., Nelson K. M., and Aldrich C. C. (2012) Adenylating enzymes in Mycobacterium tuberculosis as drug targets. Curr. Top. Med. Chem. 12, 766–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carroll P., Faray-Kele M. C., and Parish T. (2011) Identifying vulnerable pathways in Mycobacterium tuberculosis by using a knockdown approach. Appl. Environ. Microbiol. 77, 5040–5043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galandrin S., Guillet V., Rane R. S., Léger M., Eynard N., Das K., Balganesh T. S., Mourey L., Daffé M., and Marrakchi H. (2013) Assay development for identifying inhibitors of the mycobacterial FadD32 activity. J. Biomol. Screen. 18, 576–587 [DOI] [PubMed] [Google Scholar]
- 18.Stanley S. A., Kawate T., Iwase N., Shimizu M., Clatworthy A. E., Kazyanskaya E., Sacchettini J. C., Ioerger T. R., Siddiqi N. A., Minami S., Aquadro J. A., Schmidt Grant S., Rubin E. J., Hung D. T., Grant S. S., et al. (2013) Diarylcoumarins inhibit mycolic acid biosynthesis and kill Mycobacterium tuberculosis by targeting FadD32. Proc. Natl. Acad. Sci. U.S.A. 110, 11565–11570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 20.Leslie A. G., and Powell H. R. (2007) in Evolving Methods for Macromolecular Crystallography (Read R. J., and Sussman J. L., eds) pp. 41–51, Springer, Dordrecht, The Netherlands [Google Scholar]
- 21.Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 22.Collaborative Computational Project No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 23.Long F., Vagin A. A., Young P., and Murshudov G. N. (2008) BALBES: a molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 64, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z., Zhou R., Sauder J. M., Tonge P. J., Burley S. K., and Swaminathan S. (2011) Structural and functional studies of fatty acyl adenylate ligases from E. coli and L. pneumophila. J. Mol. Biol. 406, 313–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 26.Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 27.Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O. S., Vonrhein C., and Womack T. O. (2011) BUSTER, Version 2.10.0 Ed., Global Phasing Ltd, Cambridge, UK [Google Scholar]
- 28.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Boyle N. M., Banck M., James C. A., Morley C., Vandermeersch T., and Hutchison G. R. (2011) Open Babel: An open chemical toolbox. J. Cheminformatics 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smart O. S., Womack T. O., Sharff A., Flensburg C., Keller P., Paciorek W., Vonrhein C., and Bricogne G. (2011) grade, version 1.1.1, Global Phasing Ltd., Cambridge, United Kingdom [Google Scholar]
- 31.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laskowski R. A., MacArthur M. W. Moss D. S., and Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 33.Hutchinson E. G., and Thornton J. M. (1996) PROMOTIF–a program to identify and analyze structural motifs in proteins. Protein Sci. 5, 212–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeLano W. L. (2002) The PyMOL Molecular Graphics System, Version 1.7.5.0, Schrödinger, LCC, New York [Google Scholar]
- 35.Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holm L., and Park J. (2000) DaliLite workbench for protein structure comparison. Bioinformatics 16, 566–567 [DOI] [PubMed] [Google Scholar]
- 37.Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., and Svergun D. I. (2012) New developments in the program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Svergun D. I., Barberato C., and Koch M. H. J. (1995) CRYSOL–a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 40.Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 41.Arora P., Goyal A., Natarajan V. T., Rajakumara E., Verma P., Gupta R., Yousuf M., Trivedi O. A., Mohanty D., Tyagi A., Sankaranarayanan R., and Gokhale R. S. (2009) Mechanistic and functional insights into fatty acid activation in Mycobacterium tuberculosis. Nat. Chem. Biol. 5, 166–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andersson C. S., Lundgren C. A., Magnúsdóttir A., Ge C., Wieslander A., Martinez Molina D., and Högbom M. (2012) The Mycobacterium tuberculosis very-long-chain fatty acyl-CoA synthetase: structural basis for housing lipid substrates longer than the enzyme. Structure 20, 1062–1070 [DOI] [PubMed] [Google Scholar]
- 43.Goyal A., Verma P., Anandhakrishnan M., Gokhale R. S., and Sankaranarayanan R. (2012) Molecular basis of the functional divergence of fatty acyl-AMP ligase biosynthetic enzymes of Mycobacterium tuberculosis. J. Mol. Biol. 416, 221–238 [DOI] [PubMed] [Google Scholar]
- 44.Liu Z., Ioerger T. R., Wang F., and Sacchettini J. C. (2013) Structures of Mycobacterium tuberculosis FadD10 protein reveal a new type of adenylate-forming enzyme. J. Biol. Chem. 288, 18473–18483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bains J., and Boulanger M. J. (2007) Biochemical and structural characterization of the paralogous benzoate CoA ligases from Burkholderia xenovorans LB400: defining the entry point into the novel benzoate oxidation (box) pathway. J. Mol. Biol. 373, 965–977 [DOI] [PubMed] [Google Scholar]
- 46.Crosby H. A., Rank K. C., Rayment I., and Escalante-Semerena J. C. (2012) Structure-guided expansion of the substrate range of methylmalonyl coenzyme A synthetase (MatB) of Rhodopseudomonas palustris. Appl. Environ. Microbiol. 78, 6619–6629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Conti E., Stachelhaus T., Marahiel M. A., and Brick P. (1997) Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J. 16, 4174–4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hisanaga Y., Ago H., Nakagawa N., Hamada K., Ida K., Yamamoto M., Hori T., Arii Y., Sugahara M., Kuramitsu S., Yokoyama S., and Miyano M. (2004) Structural basis of the substrate-specific two-step catalysis of long chain fatty acyl-CoA synthetase dimer. J. Biol. Chem. 279, 31717–31726 [DOI] [PubMed] [Google Scholar]
- 49.Raviglione M., Marais B., Floyd K., Lönnroth K., Getahun H., Migliori G. B., Harries A. D., Nunn P., Lienhardt C., Graham S., Chakaya J., Weyer K., Cole S., Kaufmann S. H., and Zumla A. (2012) Scaling up interventions to achieve global tuberculosis control: progress and new developments. Lancet 379, 1902–1913 [DOI] [PubMed] [Google Scholar]
- 50.Gulick A. M., Lu X., and Dunaway-Mariano D. (2004) Crystal structure of 4-chlorobenzoate:CoA ligase/synthetase in the unliganded and aryl substrate-bound states. Biochemistry 43, 8670–8679 [DOI] [PubMed] [Google Scholar]
- 51.Reger A. S., Wu R., Dunaway-Mariano D., and Gulick A. M. (2008) Structural characterization of a 140 degrees domain movement in the two-step reaction catalyzed by 4-chlorobenzoate:CoA ligase. Biochemistry 47, 8016–8025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.May J. J., Kessler N., Marahiel M. A., and Stubbs M. T. (2002) Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc. Natl. Acad. Sci. U.S.A. 99, 12120–12125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mitchell C. A., Shi C., Aldrich C. C., and Gulick A. M. (2012) Structure of PA1221, a nonribosomal peptide synthetase containing adenylation and peptidyl carrier protein domains. Biochemistry 51, 3252–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li W., Gu S., Fleming J., and Bi L. (2015) Crystal structure of FadD32, an enzyme essential for mycolic acid biosynthesis in mycobacteria. Sci. Rep. 5, 15493. [DOI] [PMC free article] [PubMed] [Google Scholar]