

Abstract
NusA is an essential protein that binds to RNA polymerase and also to the nascent RNA and influences transcription by inducing pausing and facilitating the process of transcription termination/antitermination. Its participation in Rho-dependent transcription termination has been perceived, but the molecular nature of this involvement is not known. We hypothesized that, because both Rho and NusA are RNA-binding proteins and have the potential to target the same RNA, the latter is likely to influence the global pattern of the Rho-dependent termination. Analyses of the nascent RNA binding properties and consequent effects on the Rho-dependent termination functions of specific NusA-RNA binding domain mutants revealed an existence of Rho-NusA direct competition for the overlapping nut (NusA-binding site) and rut (Rho-binding site) sites on the RNA. This leads to delayed entry of Rho at the rut site that inhibits the latter's RNA release process. High density tiling microarray profiles of these NusA mutants revealed that a significant number of genes, together with transcripts from intergenic regions, are up-regulated. Interestingly, the majority of these genes were also up-regulated when the Rho function was compromised. These results provide strong evidence for the existence of NusA-binding sites in different operons that are also the targets of Rho-dependent terminations. Our data strongly argue in favor of a direct competition between NusA and Rho for the access of specific sites on the nascent transcripts in different parts of the genome. We propose that this competition enables NusA to function as a global antagonist of the Rho function, which is unlike its role as a facilitator of hairpin-dependent termination.
Keywords: Escherichia coli (E. coli), gene regulation, microarray, RNA polymerase, transcription termination, NusA, Rho, nut site, rut site
Introduction
The bacterial transcription terminator, Rho, functions as a hexameric RNA-dependent ATPase that is capable of translocating along the RNA. It dislodges the elongating RNA polymerase (RNAP)4 once it is loaded onto the nascent RNA (1, 2). Its termination function is facilitated upon interaction with the transcription elongation factor, NusG (3–5). Recent genomics studies have revealed that Rho-dependent termination occurs at more than one-third of the operons of a dividing Escherichia coli (6, 7). This mode of transcription termination is involved in many physiological processes, like control of translation (2), riboswitch formation (8), inhibition of unwanted antisense transcription, etc. (7). Rho is capable of loading onto unstructured stretches of RNA (the rut ([R]ho [ut]ilization) sites), and once it is bound to the RNA, it translocates in a processive manner along the 5′to 3′ direction, which may induce unwanted termination of transcription. Interestingly, in vivo negative regulation by cellular factors or any control switch of the Rho function has not been documented. In principle, any RNA-binding protein that has overlapping binding sites with the rut sites on the nascent RNA could influence the Rho activity by a direct competition for occupancy of the same sites.
NusA, a ubiquitous transcription elongation factor of bacteria, interacts with nascent RNA upon binding to the elongating RNAP (9). The NusA N-terminal domain interacts with the β-flap domain of the RNAP, whereas its S1-KH1-KH2 (SKK) domain interacts with the nascent RNA (Fig. 1A). The AR1 and AR2 domains interact with the antiterminator protein N and the α-subunit of RNAP, respectively (9). Nut (N utilization) sites of lambdoid phages and the sequences in the antitermination box (AT-box) found in the rRNA operons are the few characterized high affinity NusA binding sites on the RNAs (10) (Fig. 1B). On the AT-box, NusA together with NusB, NusE, and other ribosomal proteins form an antitermination complex that increases the rate of transcription elongation in the rrn operons, which is believed to decrease the kinetic window for the Rho protein to act (11). In general, NusA induces pauses during the transcription elongation. In particular, the hairpin-dependent pauses are enhanced by NusA via stabilization of the hairpin-RNAP interaction (12). NusA improves the termination efficiency at many hairpin-dependent terminators by facilitating the hairpin folding (13) and also by stabilizing the latter's interactions in the RNA exit channel of the transcription elongation complex (14).
FIGURE 1.

Descriptions of the NusA protein and the nut and rut sites. A, various functional domains of E. coli NusA as indicated. The boundaries of each domain are shown by amino acid numbers. Crystal structures of each of the domains are also shown (Protein Data Bank entries 1L2F, 1WCL, and 1WCN) (29). B, sequences of some bona fide NusA binding sites in E. coli. rrn AT, rrn operon antitermination box; nut L and R, N utilization sites from λ phage. C, depiction of overlapping rut and nut sites in the λtR1 terminator sequence. Rut sites and boxA and boxB elements of the nut site are indicated. The termination zone is indicated by an underline.
Previously, different studies have indicated the involvement of NusA in the Rho-dependent termination process (15–17). A NusA mutant, nusA1, was shown to suppress defects of a Rho mutant (16). In in vitro Rho-dependent termination assays, the presence of NusA delayed the Rho-dependent termination (18). Genome-wide gene expression patterns of NusA-deleted strains showed same pattern as that of the NusG-deleted ones, where the latter is a bona fide partner of Rho (6). More recently, a NusA mutant was observed to produce a termination defect at a Rho-dependent terminator, H-19B tR1 (17).
NusA, by virtue of its RNA and RNAP binding properties, could affect Rho-dependent terminations in different ways (Fig. 1A). NusA and Rho may compete for the same or overlapping binding sites on the exiting mRNA; thereby, the former could act as an antagonist to the latter (Fig. 1C describes this case). NusA-induced pausing of the transcription elongation complex (EC) may facilitate the Rho-dependent termination. Moreover, NusA may also modulate the access of the EC exit channel to Rho by interacting with the exit channel-forming domain, the β-subunit flap.
Here, we isolated and characterized two NusA mutants, located in the SKK domain, that exhibited inhibition of Rho-dependent termination strictly in a nut site-dependent manner. Higher affinity of these mutants for the nut site helps them to compete with the RNA loading step(s) of Rho. A genome-wide gene expression pattern revealed that a significant number of genes/operons were up-regulated when these two NusA mutants were expressed. Gene expressions of the majority of these up-regulated operons were also under the control of Rho-dependent termination. This indicated that NusA-induced inhibition of the Rho-dependent termination is widespread throughout the genome. We propose that NusA, contrary to its role as a facilitator of hairpin-dependent termination, functions as a general antagonist of Rho-dependent termination, which helps bacteria to avoid unwanted transcription termination of the latter kind.
Experimental Procedures
Materials
NTPs for in vitro transcription reactions were from GE Healthcare. [γ-32P]ATP (3000 Ci/mmol) and [α-32P]CTP (3000 Ci/mmol) were from Jonaki, Board of Radiation and Isotope Technology (Hyderabad, India). Antibiotics, isopropyl 1-thio-β-d-galactopyranoside, lysozyme, DTT, and BSA were purchased from Affymetrix. Primers for PCR were obtained from either MWG or Xcelri. Restriction endonucleases, polynucleotide kinase, and T4 DNA ligase were obtained from New England Biolabs. WT E. coli RNAP holoenzyme was purchased from Epicenter Biotechnologies. Taq DNA polymerases were either from Roche Applied Science or from Sigma or Invitrogen. Nickel-nitrilotriacetic acid-agarose beads were from Qiagen. Streptavidin-coated magnetic beads were from Promega. The bacterial RNA purification kit was from Qiagen. RNAlaterTM used for storing RNA samples used in microarray experiments was from Ambion. All of the bacterial growth media were from Difco.
Isolation of NusA Mutants
The nusA mutants defective for rho-dependent termination were isolated from the E. coli MC4100 strain (RS734) having a single terminator (tR1) containing reporter cassette Plac-H19B tR1-lacZYA, residing as a λRS45 lysogen. A random mutagenized library of the nusA cloned in a low copy (∼5) pCL1920 plasmid (pRS1472) was prepared by passing it through the XL-1 Red mutator strain (Stratagene) (19). The mutagenized library obtained from the mutator strain was then electroporated into RS734, following which the genomic copy of the nusA was deleted by P1 (ΔnusA:KanR) transduction, and the transductants were screened for pink colonies on MacConkey-lactose plates. The plasmids from pink colonies were isolated, retransformed to confirm the phenotype, and sequenced to identify the mutations. All of the strains used are listed in Table 1.
TABLE 1.
Strains, plasmids, and phages
| Description | Source/Reference | |
|---|---|---|
| Strains | ||
| GJ3161 | MC4100 galEp3 | Ref. 49 |
| MG1655 | ||
| GJ5147 (RS734) | MC4100 galEp3, Plac H-19B nutR-tR1- lacZYA | J. Gowrishankar |
| RS1019 | MC4100 galEp3, λRS45 lysogen carrying Plac-−λnutR-λtR1 −lacZYA | Ref. 22 |
| RS1038 | MC4100 galEp3, λRS45 lysogen carrying Plac- trpt′-lacZYA | Ref. 22 |
| RS1428 | MC4100 galEp3, λRS45 lysogen carrying PRM −racR/tRAC- lacZYA | Ref. 21 |
| RS1471 | MC4100 galEp3, λRS45 lysogen carrying PLac −AT-H19B nutR-tR1- lacZYA | |
| XL1-Red | endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac mutD5 mutS mutT Tn10 (TetR) | Stratagene |
| Phage | ||
| λRS45 | J. Gowrishankar | |
| Plasmids | ||
| pHyd3011 | ||
| pRS22 | pTL61 T with pT7A1-H-19B nutR-TR′T1T2-lacZYA, AmpR | Ref. 50 |
| pRS106 | pT7A1-trp t′-lacZ, AmpR | Ref. 43 |
| pRS431 | pTL61T with Plac − lacZYA by deletion of H-19B nutR-tR1 between HindIII and BamHI sites from pK8628, AmpR | Ref. 20 |
| pRS604 | T7A1-λTR1 fragment cloned at HindIII site of pRS22. | Ref. 41 |
| pRS649 | pCL1920 with rho (WT) with its own promoter cloned at HindIII and SacI sites. | Ref. 22 |
| pRS703 | nusA (WT-FL) cloned at NdeI/SalI sites of pHYD3011 vector, AmpR | Ref. 25 |
| pRS1165 | nusA (R258C-FL) cloned at NdeI/SalI sites of pHYD3011 vector, AmpR | This study |
| pRS1251 | nusA (R258C) with nusA promoter cloned at BamHI/SalI sites of pCL1920 vector, SpecR, StrepR | This study |
| pRS1252 | nusA (G181D) with nusA promoter cloned at BamHI/SalI sites of pCL1920 vector, SpecR, StrepR | This study |
| pRS1396 | nusA (WT-SKK) cloned at NdeI/SalI sites of pHYD3011 vector, AmpR | This study |
| pRS1447 | pTL61T with PLac- rrnG AT-H-19B nutR-tR1 -lac ZYA, AmpR | This study |
| pRS1472 | nusA (WT) with nusA promoter cloned at BamHI/SalI sites of pCL1920 vector, SpecR, StrepR | This study |
| pRS1520 | nusA (G181D-SKK) cloned at NdeI/SalI sites of pHYD3011 vector, AmpR | This study |
| pRS1521 | nusA (R258C-SKK) cloned at NdeI/SalI sites of pHYD3011 vector, AmpR | This study |
| pRS1522 | nusA (G181D-FL) cloned at NdeI/SalI sites of pHYD3011 vector, AmpR | This study |
| pRS1530 | pTL61T with PT7A1− rrnG AT-H-19B nutR-tR1 -lac ZYA, AmpR | This study |
| pRS1609 | pT7A1-AT-trp t′-lacZ, AmpR | This study |
| pRS1695 | nusA (WT-SKK) His-tagged, cloned at NdeI/SalI sites of pHYD3011 vector, AmpR | This study |
| pRS1696 | nusA (G181D-SKK) His-tagged, cloned at NdeI/SalI sites of pHYD3011 vector, AmpR | This study |
| pRS1697 | nusA (R258C-SKK) His-tagged, cloned at NdeI/SalI sites of pHYD3011 vector, AmpR | This study |
In Vivo Termination Assays
The in vivo termination assays were performed in E. coli MC4100 rac− strain. Strains having the reporters Plac-H19BtR1-lacZYA (RS734), Plac-λtR1-lacZYA (RS1019), Plac-trpt′-lacZYA (RS1038), and PRM-trac-lacZYA (RS1428) were used to measure the β-galactosidase activities, which in turn gave the in vivo termination efficiency at the terminators present upstream of the lacZ gene. These strains were transformed with pCL1920 plasmids expressing WT (pRS1472), G181D (pRS1252), and R258C (pRS1251) NusA proteins. The genomic copy of the nusA of these strains was deleted by P1 (nusA:KanR) transduction.
To study whether overexpression of different varieties of either full-length NusA or its RNA-binding domain (SKK) alone is capable of producing the Rho inhibition effect, we expressed them from a modified pBAD vector, pHYD3011, in the presence of chromosomal nusA. The strains RS734 and RS1428 were transformed with the pHYD3011 plasmids expressing WT NusA (pRS703) and its mutant derivatives, G181D (pRS1522) and R258C (pRS1165). Similarly, different SKK fragments having WT or G181D or R258C mutations were also expressed from the plasmids pRS1396, pRS1520, and pRS1521, respectively. The strains were cultured overnight in the presence of 0.1% glucose to suppress the nusA expressions from these plasmids, which was followed by subculturing them in the absence of glucose to express the genes, and subsequently the Rho-dependent termination efficiencies were measured as described above.
All of the measurements of β-galactosidase activities were performed at 37 °C using a SpectramaxTM Plus plate reader following the published procedures (20).
Preparations of Different NusA Mutants and Fragments
The NusA mutants, R258C and G181D, were constructed in pCL1920 by site-directed mutagenesis (Stratagene). These mutant derivatives and the SKK fragments of NusA were also cloned in the pHYD3011 plasmid to be used in in vivo competition assays. For in vitro experiments, full-length and SKK fragment of R258C NusA were made in a pET28b plasmid to introduce a C-terminal His tag, whereas to introduce a heart muscle kinase tag in the N terminus, these mutants were also cloned in pET33b plasmid. This tag was used for end-labeling with 32P. To check the in vivo levels of NusA by Western blotting, His-tagged derivatives of the SKK fragment having WT, G181D, and R258C sequences were also cloned in pHYD3011 plasmids; these constructs have an N-terminal hexahistidine tag. Purification of these NusA derivatives was performed using nickel-nitrilotriacetic acid beads (Qiagen). Labeling of the heart muscle kinase-tagged R258C NusA was performed using protein kinase A and [γ-32P]ATP (3000 Ci/mmol).
DNA Template Preparations
Linear DNA templates for in vitro transcription assays were made by PCR using the plasmids pRS604 (T7A1-λtR1; oligonucleotide pairs RS83/RS147, RS83/RS333, and RS83/994), pRS106 (pT7A1-trpt′; oligonucleotide pairs RS83/RSRK-1, RS83/RS303, and RS83/177), pRS649 (rho gene with its own promoter; oligonucleotide pairs RS323/RS555 and RS1116/RS1142), and pRS1609 (pT7A1-AT-trpt′-lacZ; oligonucleotide pairs RS83/RSRK-1). These DNA fragments were gel-purified. When required, a lac operator sequence was inserted after the terminator regions using a downstream primer having the operator sequence. In order to immobilize the DNA templates to the streptavidin-coated magnetic beads, a biotin group at the 5′-end of the templates was incorporated by using the biotinylated primer RS83. In all of these templates, transcription was initiated from a T7A1 promoter unless mentioned otherwise. All of the plasmids used to make the DNA templates are listed in Table 1.
In Vitro Termination Assays
For the transcription using strong T7A1 promoter, we followed the regime of single round transcription. However, due to the poor yield from Prho promoter, multiple-round transcription was employed by omitting rifampicin from the reaction mixture. Reactions were carried out in the transcription buffer (20 mm Tris-Cl, pH 8.0, 10 mm MgCl, 50 mm KCl, 1 mm DTT, and 100 μg/ml BSA) at 37 °C. For single round transcription, the reactions were initiated with 175 μm ApU, 5 μm GTP, 5 μm ATP, 2.5 μm CTP, and [α-32P]CTP (3000 Ci/mmol) to make a 23-mer EC (EC23). The EC23 was then chased with 250 μm each of ATP, GTP, CTP, and UTP. For multiple-round transcription, the RNAP-promoter complex was chased with 250 μm each of ATP, GTP, and UTP. In these reactions, concentrations of DNA template, RNAP, NusA, and Rho were 5, 25, 200, and 50 nm, respectively. The concentration of CTP was 50 μm, and the RNA was labeled with [α-32P]CTP (3000 Ci/mmol). The reactions were stopped by extraction with phenol followed by ethanol precipitation. Samples were loaded onto a 6% sequencing gel and analyzed using an FLA 9000 phosphor imager (Fuji).
RNA Release Kinetics
For measuring the RNA release kinetics from the stalled ECs, transcription reactions were performed in the presence of 100 nm lac repressor so that the ECs are roadblocked at the lacO site present downstream of the terminator region. The DNA templates were immobilized on streptavidin magnetic beads (Promega) through streptavidin-biotin interaction. The transcription reactions were continued for 2 min to form the stalled EC, and the excess NTPs were removed by thoroughly washing the beads, following which 50 nm Rho plus 1 mm ATP was added. 10 μl of samples were removed at different time points and separated into supernatant (S) and pellet (P) fractions by keeping the microcentrifuge tubes on magnetic stands. Half of the supernatant (S) was directly mixed with an equal volume of formaldehyde loading buffer. The rest of the sample (S + P) was phenol-extracted and mixed with dye. Fractions of released RNA ((2S)/(S) + (S + P)) were plotted against time, and the curves were fitted to exponential rise equations either of the form, y = a(1 − exp−bx) or y = y0 + a(1 − exp−bx), where b denotes the rate, a is maximum RNA release amount, and y0 is the residual amount of RNA release at the 0 time point.
NusA Dissociation Assays in the Presence of Cold Competitors
For measuring the binding strength of WT and mutant derivatives of NusA to the nut site in the presence of a stalled EC, transcription reactions were performed in the presence of 100 nm lac repressor to form a roadblocked EC at the lacO site present downstream of various terminators. The lac repressor-bound DNA templates were immobilized on streptavidin-coated magnetic beads (Promega) through streptavidin-biotin interaction at their 5′-end. The roadblocked ECs were formed by chasing EC23 with 250 μm NTPs along with a 20 nm concentration of either 32P-labeled WT or R258C NusA for 2 min. Excess NTPs were then removed by thoroughly washing the beads, following which varying concentrations (10–200 nm) of unlabeled (cold) WT and R258C NusA proteins were added as the competitors. 30 μl of samples were removed at different time points and fractionated into supernatant and pellet fractions by holding the tubes on the magnetic stands. Half of the supernatant (S) and the rest of the sample (S + P, denoted as P in the figures) were mixed with an equal volume of SDS loading buffer. Fractions of NusA dissociated ((2S)/(S) + (S + P)) were plotted against time, and the curves were fitted to a three-parameter hyperbolic binding equation of the form, y = y0 + ((a*x)/(b + x)), where b denotes the amount of competitor at 50% dissociation, and a is the amplitude of the dissociation curves.
Fluorescence Assays to Estimate the Kd of the NusA-RNA Interaction
To measure the binding constants (Kd) of the NusA-RNA interactions in isolation, we have monitored the tryptophan fluorescence changes of the WT and R258C derivatives of the NusA-SKK fragments. This fragment has two tryptophan residues. Fluorescence experiments were performed in a Hitachi FL7000 spectrometer at 25 °C. The excitation wavelength was 295 nm, and the emission range was 310–400 nm. Fluorescence intensities at 340 nm were plotted against increasing concentrations of an HPLC-pure 20-mer RNA having the rrn AT-box sequence. The Kd values were calculated by fitting the binding isotherms to the equation, y = yo + (ab/(b + x)), where b denotes the Kd.
NusA-Rho Competition Assays for the Same or Overlapping Binding Site(s)
Stalled ECs were formed, in the same way as described earlier, either downstream of the AT-box (PCR-amplified from pRS1530 using the RS83/1127 oligonucleotide pair) or in the terminator zone of the rho gene (PCR-amplified from the chromosomal DNA using the RS1116/RS1142 oligonucleotide pair). A C-terminal heart muscle kinase-tagged WT Rho was used to radiolabel at its C-terminal. The presence of this tag did not affect its activity (5). (i) In the experiments where Rho was added prior to the competitor, 5 nm 32P-labeled WT Rho together with 1 mm AMP-PNP were added to the stalled EC for 5 min. The beads were washed, followed by the addition of 200 nm cold competitors, and incubation was continued for another 5 min, following which supernatant and pellet fractions were collected. (ii) In the experiments where Rho and the competitors were added together, 5 nm 32P-labeled WT Rho (+1 mm AMP-PNP) and a 200 nm concentration of the competitors were added together to the stalled EC. Supernatant and pellet fractions were collected after 5 min. The fractions of Rho dissociated from the EC were calculated as described in the above mentioned dissociation assays. (iii) In the experiments where competitors were added before Rho, 200 nm competitors were added to the stalled EC, following which 5 nm radiolabeled Rho was added to the mixture. Other experimental conditions were the same as above.
Nascent RNA-mediated Rho ATPase Assays on the EC Stalled Downstream of the AT-box
The stalled ECs were formed in a similar way as in the RNA release kinetics experiments but without using immobilized template. To obtain a sufficient amount of RNA for the ATPase assays, the concentrations of DNA template and RNAP were increased 4-fold compared with that used in the in vitro transcription assays. Rho + 1 mm ATP was mixed with [γ-32P]ATP (3000 Ci/mmol), was then added to the stalled ECs, both in the absence and presence of WT/R258C NusA, and the inorganic phosphate (Pi) release was observed on polyethyleneimine thin layer chromatography sheets (Merck) under 0.75 m KH2PO4 (pH 3.5) as the mobile phase. The composition of the assay mixture was 25 mm Tris-HCl (pH 8.0), 50 mm KCl, and 5 mm MgCl2. The reactions at the indicated time points were stopped with 1.5 m formic acid. thin layer chromatography sheets were analyzed by Fuji FLA 9000. The amounts of [γ-32P]ATP and the released Pi were calculated from the intensities of the spots of these products using ImageQuantTM software.
Microarray
E. coli MG1655 was transformed with pCL1920 vector expressing WT and different NusA mutants (WT/G181D/R258C). The genomic copy of the nusA was deleted by P1 transduction (nusA:KanR). Two colonies from each strain were grown in LB until the A600 reached ∼0.4. The cell pellets were dissolved in 1.5 ml of RNAlaterTM (Ambion). The RNA isolation, microarray experiments, and gene expression analysis were performed by Genotypic Technologies Pvt. Ltd. (Bangalore, India). Two types of Agilent arrays were used as described below.
In the low density arrays, Agilent E. coli K12 microarray slides in an 8000 × 15,000 format along with Agilent's one-color (cyanine 3-labeled targets) microarray-based gene expression analysis were used. The total number of coding regions available on the chip was 4294. The average number of probes per coding region sequence was 3, and the total number of probes designed was 10,828. The total number of probes used for non-coding region was 4380. For the Agilent high density tiling microarray, the specifications were as follows: format, 2 × 400,000; total number of genes covered, 4515; total number of probes, 416,505, of which 249,903 probes correspond to the genic region and 166,602 probes correspond to the non-genic regions. Two independent biological replicates were used for each strain. Only those genes showing a change of ≥1.5-fold compared with the WT strain were considered. Microarray data for the Rho mutants and those obtained in the presence of bicyclomycin were reported earlier (21, 32). These were used to compare with the data obtained for the NusA mutants.
RT-PCR and qRT-PCR Assays
To validate the microarray data, RT-PCR and qRT-PCR for selected genes were carried out with the same strains as were used in the microarray experiments. Two colonies from each strain were grown until A600 of ∼0.4. RNA was isolated from the bacterial pellets using the RNeasy RNA isolation kit (Qiagen). In brief, 3 μg of total RNA was used for DNase I treatment (for 15 min at 37 °C) and was used for cDNA synthesis using the SuperScript III first strand synthesis system (Invitrogen). 1 μl of cDNA was used as template for PCR in a 20-μl reaction. The product from rpoC was used as an internal control because it is expressed constitutively. The RT-PCR products were run on 1.5% TAE-agarose gels.
The qRT-PCR was performed in an Applied Biosystem 7500 PCR system using Cyber Green dye. The amount of product formed was calculated by determining the Ct value (the midpoint of the sigmoidal curve obtained by plotting the fluorescence intensity against the number of PCR cycles) for each sample. The 2−ΔΔCt method was used for calculating the -fold change (2−ΔΔCt) in the RNA levels in the mutants with respect to the WT, where Ct = number of threshold cycle; ΔCt = (Ct of target gene) − (Ct of internal control); ΔΔCt = ΔCt of the mutant − ΔCt of the WT. The RNA level of rpoC was used as an internal control. The central part of each gene was used for designing oligonucleotide pairs having comparable Tm producing cDNA products of length 200–300 nt.
Western Blotting
Western blotting was employed to check the levels of different derivatives of full-length NusA and NusA SKK domain cloned in pCL1920 and pHYD3011 plasmids. For full-length NusA, monoclonal antibody of NusA was used (Neoclone), whereas for SKK fragments, an anti-His monoclonal antibody (Qiagen) was used. Western blotting was also performed to measure the in vivo concentrations of Rho in the presence of WT and different nusA mutants using polyclonal antibodies specific for Rho (see Fig. 10). In general, E. coli MG1655 was transformed with pCL1920 and pHYD3011 plasmids having either nusA derivatives (WT/G181D/R258C) or rho. In cases of measurements of the in vivo level of Rho (see Fig. 10) or different NusA proteins (Fig. 2), after transformations with the pCL1920 NusA plasmids, the genomic copy of nusA was deleted by P1 (nusA:KanR) transduction. Each strain was grown until A600 of ∼0.4. The bacterial pellets were suspended in 100 μl of SDS dye (0.5 m Tris, pH 6.8, 50% glycerol, 10% SDS, β2-mercaptoethanol, bromphenol blue) and boiled at 100 °C for 10 min to obtain the cell lysates. Proteins in these lysates were resolved on SDS-PAGE (12%), and subsequently the Western blotting was performed by following standard procedures using polyclonal antibody of the Rho protein. The blots were viewed in a chemiluminescent detection system FluorChemTM E (ProteinSimple).
FIGURE 10.

Rho-NusA competition at the leader region of the rho operon. A, schematic showing the organization and the relevant sequences of the rho operon. Oligonucleotides RS323, RS1116, and RS1142 (also see Table 1) were used to amplify the DNA template, used in transcription assays, from the genomic DNA. Untranslated region is shown in italic type, and the underlined portion within this has a high G/C ratio of 3. The boldface GA sequence is the possible transcription start site, whereas the boldface ATG is the translation start site. B, autoradiogram showing the in vitro transcription assays on the above template under different conditions. The termination zones are indicated by dashed lines together with the amount of RO products. RNA length markers are shown on the left of the gel. C, schematic showing the stalled EC in the termination zone using LacI as a roadblock. Lac operator sequence was introduced at the end of the template using RS1142 having the operator site, and the site is 135 bp downstream of the translation start site of rho. Competition between Rho and NusA fragments for the overlapping binding site(s) in the untranslated region is indicated. D and E, autoradiograms showing the distribution of radiolabeled Rho in different fractions (half of the supernatant (S) or pellet plus the rest of the supernatant, denoted as P) upon challenge by the indicated competitors. Competitors were added together with Rho in D and after the addition of Rho in E. Fractions ([2S]/([S] + [S + P])) of dissociated Rho are indicated below the lanes. S.D. values (error bars) were obtained from 2–3 experiments. F, left, a schematic depicting the feedback regulation in the rho operon. Right, Western blot to estimate the amount of in vivo concentrations of Rho in the presence of WT and mutant NusA. The blot for the β′-subunit of RNAP is used as a loading control.
FIGURE 2.

Effects of NusA mutants on the in vivo and in vitro transcriptions. A, Western blots showing the in vivo level of WT and mutant NusA proteins expressed either from chromosome or from pCl1920 plasmids. A monoclonal antibody (Neoclone) of NusA was used. A blot for the β′-subunit of RNAP was used as a loading control. Anti-β′ polyclonal antibody was used for this purpose. Signal intensity of chromosomal NusA (lane 1) was set as 1, and amounts of different NusA proteins in other lanes (lanes 2–4) were expressed with respect to that. In lanes 2–4, MG1655 strain was transformed with the indicated plasmids, following which the chromosomal nusA was deleted by P1 transduction. Equal amounts of cells, as judged by A600, were loaded in each of the lanes. S.D. values (error bars) were obtained from three measurements. Prestained protein molecular weight markers are shown beside the gels to identify the protein size. Note that NusA migrates as a higher molecular weight protein. B, bar diagrams showing the β-galactosidase activities obtained from the lacZ reporter cassettes fused downstream of the different (as indicated) Rho-dependent terminators, trpt′, trac, λtR1, and H-19B tR1. Bars are grouped according to the nusA alleles, as stated. The enhancements in activities in the mutant alleles are indicated by upward arrows. These enhancements indicate termination defects at the Rho-dependent terminators in the presence of mutant nusA alleles. β-Galactosidase activities are expressed in arbitrary units (A.U.). The error bars were obtained from five independent experiments. C and D, autoradiograms showing the in vitro transcription assays performed on indicated linear DNA templates, where transcripts were initiated from the T7A1 promoter. Templates carry the Rho-dependent terminators, nutR/tR1 (C) and trpt′ (D). Triangles, increasing concentrations of NusA ranging from 5 to 200 nm, as indicated. The termination zone and the RO products are marked. The 0 NusA lane represents the Rho alone condition. Amounts of RO in the presence of specific concentrations of NusA are indicated below the gels. RNA length markers are indicated beside the gels. These values were calculated using the equation, [RO] = [RO]/([RO] + intensities of all of the bands in the termination zone). E, schematic showing the locations of the point mutations in the structure of NusA (Protein Data Bank entry 1L2F) as spheres. F, changes in fluorescence intensities of the tryptophan residues of the SKK domain upon the addition of increasing concentrations of 20-mer oligo-RNA having AT-box sequence. F and F0, final and initial intensities at 340 nm, respectively. The average of three independent titration profiles is plotted. The average Kd values indicated in the plots are obtained by fitting the points to a hyperbolic curve.
Bioinformatics
The operonic arrangements of the genes whose expressions were affected by both NusA and Rho mutants were organized using information from the EcoCyc database. Gaps between the genes were defined as intergenic regions, which could be targets of Rho binding. To represent the common genes as a subset of the two nusA mutants (G181D and R258C), as well as subsets between a nusA mutant (G181D/R258C) and the three rho mutants (N340S, G324D, and P279S), respectively, different Venn diagrams were drawn. The criterion of an identical gene or another gene from the same operon being up-regulated in the two mutants (rho/nusA) was chosen to denote the overlap.
Results
Nut Site-dependent Inhibition of Rho Function by NusA Mutants
Using a genetic screen as described under “Experimental Procedures,” we isolated a nusA mutant, G181D, that is defective for Rho-dependent termination. We also constructed an earlier reported nusA mutant, R258C, having a similar defect (17). We have chosen E. coli MC4100 strains, each carrying one of the four lacZ reporter cassettes Plac-H19B tR1-lacZYA (16), Plac-λtR1-lacZYA (22), PRM-trac-lacZYA (21), and Plac-trpt′-lacZYA (22) to measure the in vivo termination defects of the nusA mutants. In these reporter cassettes, Rho-dependent terminators tR1 (obtained either from H-19B or from λ phages), trac, and trpt′ were fused upstream to the lacZYA operon. The higher the amount of β-galactosidase activity from each of the reporters, the greater is the defect in the Rho-dependent termination. WT and the mutant derivatives of NusA were expressed from their own promoter cloned in a low copy plasmid (pCL1920). The in vivo expression levels of NusA proteins from this plasmid were close to the physiological level (Fig. 2A). It should be mentioned that in these strains, the chromosomal deletion of nusA leaves behind the N-terminal fragment of NusA (7). However, this fragment does not have any function in vivo because these strains were not viable in the absence of the WT nusA supplied from the plasmid (data not shown).
We made the following observations. (i) NusA mutants, G181D and R258C, caused 3–5-fold enhancement in the β-galactosidase activities compared with their WT counterpart from the reporters having tR1 terminators (from the H-19B and λ phages; Fig. 2B). The effect of R258C NusA was more pronounced. (ii) This enhancement was not observed from the reporters having the trpt′ and trac terminators (Fig. 2B). Among these terminators, only the tR1 terminators have an overlapping NusA binding site, nutR (10) (Fig. 1C); hence, the termination defects caused by these two NusA mutants were only observed with the terminators having nut sites.
Next we studied the anti-Rho dependent termination effects of WT and R258C NusA proteins in a purified system. We performed in vitro transcription assays using a DNA template where λ nutR/tR1 and trpt′ terminators were fused downstream of a strong T7A1 promoter (Fig. 2, C and D). In the in vitro termination assays, Rho terminates transcription over a region called a termination zone. We observed that in the presence of both WT and the R258C NusA, this termination zone moved toward the longer transcript side, indicating a delay in the termination process (Fig. 2C). This effect of NusA was not observed at the trpt′ terminator that does not have an overlapping nut site (Fig. 2D). The termination at λtR1 was further delayed by R258C NusA compared with WT. In addition to this, a significant fraction of the transcript escaped the termination and reached the end of the template (run-off (RO); compare the amounts of RO product indicated below the lanes) when R258C NusA was used. This suggests that NusA on its own is capable of delaying Rho-dependent termination in a nut site-dependent manner, and this inhibitory effect is enhanced by the R258C mutations in NusA. Because nut and rut sites overlap (see Fig. 1C), it was not possible to make mutations in the nut site without affecting the rut site; hence, we used a nut-less Rho-dependent terminator, trpt′, to understand the nut dependence of the NusA.
We localized these two NusA mutations in its SKK domain that confers the RNA binding properties to NusA (Fig. 2E) (23). Because they are in the RNA binding domain, it is possible that enhanced inhibitory activities of these two mutants were due to their tighter RNA binding to the nut site.
To test this, we determined the affinity of the WT and R258C NusA-SKK domains for an RNA oligomer (20-mer) having the nut site from the rrn AT-box (Fig. 1C). Isolated full-length NusA does not bind to RNA due to the inhibitory effect of the NusA N-terminal domain (10), so we used only the RNA-binding fragment of the NusA. We measured the binding constants (Kd) of these two NusA SKK domains by monitoring the changes in their tryptophan (Trp-276 and Trp-334) fluorescence at 340 nm upon interaction with the increasing concentrations of the AT-box RNA oligomer (Fig. 2F). We observed that the isolated SKK domain with R258C mutation has ∼4-fold higher affinity than its WT counterpart. This indicates that the intrinsic affinity of the NusA RNA binding domain for the nut site increased due to the R258C mutation. The obtained binding constant of the WT SKK domain is consistent with what has been reported earlier (10).
The above data indicate that the binding constant of the free NusA for an RNA oligonucleotide is in the micromolar range, which is quite moderate for an RNA-binding protein, and the functional relevance of this low affinity NusA-RNA interaction outside the EC is questionable. Due to the increase in local concentrations, this affinity is likely to be enhanced when NusA is bound to the EC, and also the interaction becomes more functionally relevant; hence, below, we decided to estimate the RNA binding properties of the WT and mutant derivatives of NusA when bound to the EC.
Higher Concentrations of the Full-length and the SKK Domain Fragment of WT NusA Produce the Same Inhibitory Effects as the Two SKK Domain Mutants
If the enhanced inhibitory effects of the NusA mutants are due to their higher affinity for the nut sites, it is expected that the elevated concentrations of WT NusA would also elicit similar responses as the NusA mutants. We increased the in vivo concentrations of WT NusA artificially by expressing it from the PBAD promoter cloned in a high copy number (∼20–25) plasmid, pHYD3011 (see Table 1). This is a modified PBAD plasmid having a strong ribosome binding site from a pET vector, which enables it to express a very high level of any protein upon derepression. Due to these modifications, it also expresses proteins to a significant level even in the presence of the repressor. The expression was induced by removing glucose, a repressor of PBAD, from the medium. These strains were not maintained under uninduced conditions to avoid any harmful effect, if any, due to the elevated concentrations of NusA. We confirmed by Western blotting that from these constructs, the NusA level was elevated ∼3-fold upon induction (data not shown). We measured the β-galactosidase activities in the same way as described above. We observed that at elevated concentrations, WT NusA was capable of inhibiting the Rho-dependent termination at the tR1 terminator to the same extent as the two NusA mutants (Fig. 3A). The β-galactosidase activity, which is the measure of the read-through at a terminator, was observed to increase ∼3-fold. This concentration-dependent inhibition was also nut site-specific because it was not observed at the trac terminator (21), which is devoid of this site (Fig. 3B).
FIGURE 3.

Effects of overexpression of full-length and SKK domain of NusA on the in vivo transcription. β-Galactosidase activities from the lacZ reporter fused downstream of Rho-dependent terminators, H-19B tR1 (A and C) and trac (B and D). The effects of overexpression of different derivatives of full-length (FL) NusA (A and B) and its SKK fragment (C and D) are shown. Concentrations of these two species were varied by adding (+) or omitting (−) 0.1% glucose (Glu) as they were expressed from the PBAD promoter. In vivo levels of full-length NusA and SKK fragments were verified by Western blotting (data not shown). Because the in vivo levels of different NusA derivatives were similar (∼3-fold enhancement upon induction), we did not attempt to normalize the β-galactosidase activities. S.D. values (error bars) were obtained from five independent measurements. vector, empty plasmid without the nusA. A.U., arbitrary units.
If NusA binding to the nut site is the only requirement to inhibit Rho action, then higher concentrations of the RNA binding domain of NusA, the SKK domain fragment (23.8 kDa, amino acids 113–348; see Fig. 1A), should also exhibit the same effect as the full-length NusA. We expressed the SKK domains each having either the WT or the two mutations in the same way as stated above from the inducible PBAD promoter. Higher levels of expression (∼4-fold) of these NusA fragments were also confirmed by Western blotting (data not shown). We observed a ∼5-fold increase in β-galactosidase activities at the tR1 terminator in the presence of elevated concentrations of all of the three derivatives of the SKK domain (Fig. 3C). This increase was not observed at the trac terminator (Fig. 3D). Therefore, binding of the isolated SKK domain in trans to the nut site is sufficient for the Rho inhibition.
In Fig. 3, A and C, we observed a nonspecific increase of β-galactosidase activities in the presence of the vector itself. We think that this arose due to the titration of the Rho molecules by the high level of untranslated transcripts initiated from the strong PBAD promoter under induced conditions.
The aforementioned results indicated that (i) the NusA-mediated inhibition of Rho occurred through the nut site binding of its SKK domain, and (ii) Rho-inhibitory activity of R258C and G181D NusA is probably due to their enhanced affinity for the nut site. In this regard, it should be mentioned that the enhanced inhibitory effect of these two mutants suggests that the WT NusA has an intrinsic Rho-inhibitory activity (also see Fig. 2C, WT NusA panel); hence, these mutants with enhanced inhibitory activities could be used as tools to understand the mechanism of this inhibition.
Estimating the Higher Affinity of the NusA Mutant When Bound to a Stalled EC
To measure the higher affinity of R258C NusA mutant for the nut site as well as the stability of the NusA-EC complex, we employed competition assays with the purified components. We measured the dissociations of WT and mutant NusA proteins from the ECs stalled at the lac operator sites present either close to the nut site (∼60 nt downstream; Fig. 4A, left, Template I) or away from it (∼250 nt downstream; Fig. 4A, middle, Template II) or 63 nt downstream of a terminator that is devoid of the nut site (Fig. 4A, right, Template III with the trpt′ terminator). The ECs were stalled at the lacO sites on the different templates in the presence of lac repressor molecules. The NusA-nut interaction weakens as the EC moves away from the nut site, so it is likely that NusA dissociation would happen more readily from the stalled ECs formed on template I. Due to the absence of the nut site, the interaction would also be weaker on template III. It should be mentioned that the NusA N-terminal domain interacts with RNAP, which also contributes to the stability of the NusA-EC complex formation (24). However, NusA SKK domain-RNA interactions are the major component that contributes to stabilizing this complex formation, because very high concentrations of the free NusA N-terminal domain are required to observe the interaction of this fragment with EC in the in vitro experiments (24). Under our experimental conditions, ∼70% of the NusA molecules remained associated with the EC in the absence of any competitor (data not shown). In these experiments, both the WT and the R258C NusA proteins were radiolabeled with 32P at their heart muscle kinase tag (used to radiolabel proteins) sequence. The stalled ECs were immobilized on magnetic beads using a biotinylated DNA template so that the dissociation of [32P]NusA could be measured in the supernatant fractions. Due to the poor solubility of the G181D NusA protein, we did not attempt any in vitro experiments with it.
FIGURE 4.

Cold competition assays of radiolabeled NusA bound to stalled elongation complexes. A, schematics showing the stalled EC formed downstream of Rho-dependent terminators by the lac repressor (lacI). On templates I (left) and II (middle), ECs are stalled 60 and 250 nt downstream of the nut site of λ nutR, respectively. In template III (right), trpt′, lacking any nut site, is located 63 nt downstream of the terminator. All of the templates were immobilized to streptavidin-coated magnetic beads through a streptavidin-biotin bonding at their 5′-end. B, fractions of dissociated radiolabeled NusA ([32P]NusA) bound to the stalled EC on template I (left), template II (middle), and template III (right). Different derivatives of [32P]NusA bound to stalled ECs are indicated. C, plots obtained from the fraction of dissociated NusA in the presence of increasing concentrations of cold competitors, WT, and R258C NusA. Stalled ECs on template I (left), template II (middle) and template III (right) were bound to either WT (WT-EC) or R258C (R258C-EC) NusA. Data points were fitted to a hyperbolic equation. Concentrations of cold competitor corresponding to 50% change are indicated. In all of the experiments, S.D. values (error bars) were calculated from three independent experiments.
In these experiments, at first a 23-mer EC (EC23) was formed by omitting UTP from the reaction mix (see “Experimental Procedures”), which was subsequently chased with all of the NTPs together with either radiolabeled WT or R258C NusA proteins. NusA-bound stalled ECs (at the lac operator sites in the presence of lac repressor) formed on templates I (Fig. 4, B and C, left), II (Fig. 4, B and C, middle), and III (Fig. 4, B and C, right) were then competed out by adding different concentrations of either unlabeled (“cold”) WT or R258C NusA. The amounts of the competitors required to dissociate 50% of the EC-bound radiolabeled NusA proteins, indicated in the plots, gave quantitative measures of the stability of each of the NusA-EC complexes. The lower the concentration of the cold competitor required for competition, the lesser is the stability of the NusA-EC complex. We observed the following. (i) 50% dissociation of both the WT and R258C NusA proteins bound to the ECs stalled on template I was exerted by their respective cold counterparts at a concentration of ∼4 nm (Fig. 4C, left). This facile dissociation by the cold competitors also indicates that it is an equilibrium methodology to estimate the stability of the complexes. (ii) On the same template, EC-bound R258C NusA required ∼17-fold higher concentrations of cold WT NusA to obtain 50% dissociation (Fig. 4C, left). (iii) This cold competition was more facile when the labeled NusAs were part of EC stalled on the template II (Fig. 4C, middle), which indicates that the stability of NusA-EC as well as NusA-RNA interactions weakens when the EC moves away from the nut site. (iv) This stability was significantly reduced when the EC was stalled downstream of the trpt′ terminator sequence that is devoid of the nut site (template III; Fig. 4C, right). However, compared with template II, higher concentrations of cold competitors were required to induce dissociation of EC-bound NusA proteins on this template. Probably, the presence of some unknown “nut-like” sequences in the trpt′ terminator could have improved the affinity of NusA.
These data further support the results obtained in Fig. 2F, which suggest an enhanced Rho inhibition by R258C NusA due to its higher affinity for the nut site. However, the enhanced stability of this mutant for the EC was observed only when the EC was in the vicinity of the nut site.
NusA Binding to Nut Site(s) Delays the Loading of Rho at the Rut Site(s)
The aforementioned results strongly indicated that the NusA-mediated inhibition of Rho function is due to a direct interaction of the former with the nut site. Next, in order to identify the step(s) of Rho-dependent termination affected by this NusA-nut interaction, we measured the Rho-induced RNA release from an EC stalled downstream of either λtR1 or trpt′ terminator (Fig. 5A). The Rho action was measured in the presence of either WT or R258C NusA. In these experiments, the in vitro transcription was initiated from the T7A1 promoter, and the elongation occurred either through the λtR1 (Fig. 4, Template II) or the trpt′ (Fig. 4, Template III) terminator regions to reach the lac operator/repressor-mediated roadblock. DNA templates were immobilized to magnetic beads to measure the released RNA in the supernatant (Fig. 5, B and C).
FIGURE 5.

Assays to show direct NusA-Rho competition for the overlapping nut and rut sites. A, schematic showing the configuration of the stalled ECs at a LacI roadblock (RB) downstream of either λtR1 or trpt′ terminators. DNA templates were immobilized to magnetic beads via a streptavidin-biotin linkage. This immobilization enables us to measure the released RNA in the solution. Rut sites of each of the terminators are indicated. Autoradiograms show the time courses of RNA release from the ECs stalled downstream of either λtR1 (B) or trpt′ (C) terminators. These time courses were obtained either in the absence of NusA or in the presence of WT or R258C NusA. Inverted triangles denote the increasing time as indicated. S and P, one-half of the supernatant and one-half of the supernatant plus pellet fractions, respectively. D and E, plots of fractions of released RNA obtained in B and C, respectively, against time. The indicated rates of RNA release (k) for each curve were calculated from the exponential fitting of the curves. The fraction of released RNA was estimated from the expression, [2S]/([S] + [S + P]). S.D. values (error bars) were calculated from three independent measurements.
We observed that both the WT and R258C NusA proteins (Fig. 5, B and D) slowed down the Rho-induced rate of RNA release significantly when the ECs were stalled downstream of the λtR1 terminator. Similar effects were not observed from the ECs stalled downstream of the nut-less trpt′ terminator (Fig. 5, C and E).
These results indicated that there could be a direct competition between Rho and NusA for the nut/rut sites. Consistent with these observations, we have earlier shown that the nut and rut sites overlap in the spacer region of the nutR/tR1 sequence (see Fig. 5 of ref. 22 and Fig. 6 of Ref. 25). The presence of NusA delays the RNA loading as well as the RNA-dependent activation step(s) of Rho, which is reflected as a slow rate of RNA release by Rho. We also observed that R258C NusA did not induce enhanced pausing by the elongating RNAP, which could have been an alternative way of delaying the Rho-dependent termination (data not shown).
FIGURE 6.

Rho-NusA competition at the AT-box of an rRNA operon. A, schematic showing the AT-box sequence of E. coli rrnG operon together with the flanking regions. B, details of the T7A1-AT-box-trpt′ construct. The T7A1 promoter, AT-box sequence preceding the 16S RNA gene of rrnG operon, and the rut sites of the trpt′ terminator are indicated. C, autoradiograms showing in vitro transcription assays on T7A1-trpt′ and T7A1-AT-trpt′ templates under various indicated conditions. The termination zones (dashed lines) and the RO products are indicated. The amounts of RO product in each transcription reactions are shown below each lane. [RO] was calculated as [RO]/([RO] + total band intensities in the termination zone). RNA length markers are indicated. S.D. values (error bars) were calculated from 2–3 independent experiments. D, schematic showing an EC stalled 44 nt downstream of the AT-box sequence using LacI as a roadblock. The AT-box region is the target of Rho as well as NusA fragments (SKK and ΔAR1–2 NusA) having the RNA-binding domains. E–G, autoradiograms showing the amounts of radiolabeled Rho in supernatant (S; half of the supernatant) and pellet (P; half supernatant + pellet) fractions in the presence of different amounts of the competitors added either with Rho (E) or after adding Rho (F) or before adding Rho (G). Fractions of dissociated Rho in different cases are given below each gel. Fractions of dissociated Rho were calculated as [2S]/([S] + [P]). H, autoradiograms showing a similar competition experiment described in D except that the EC was stalled downstream of the trpt′ terminator, which lacks a bona fide nut site. This template has been described in Fig. 5A. S.D. values (error bars) were obtained from 2–3 independent measurements.
Competition between Rho and NusA for the AT-box of rRNA Operons
The AT-box in the untranslated ribosomal operons is responsible for the antitermination of primarily the Rho-dependent termination (26, 27). A complex of NusA, NusB, NusE, and other ribosomal proteins bound to the AT-box may prevent Rho loading onto the mRNA and thereby bring about the transcription antitermination in this operon (11). This AT-box comprises a NusA-binding site, the boxA sequence, and also an overlapping Rho-binding site (Figs. 1B and 6A (10)). The aforementioned results and the composition of the AT-box of the ribosomal operon led us to hypothesize that the NusA-mediated inhibition of Rho-dependent termination is likely to be a major component of the rRNA-antitermination event, and this inhibition occurs due to a direct NusA-Rho competition for the AT-box.
To test the hypothesis, we cloned a 22-base pair AT-box sequence of rrnG (29) (Fig. 6B) upstream of the trpt′ terminator that is devoid of any NusA-binding site. We measured the transcription termination at the trpt′ terminator that is fused downstream of the AT-box, both in the absence and presence of WT and R258C NusA (Fig. 6C). The transcription termination patterns in the presence (T7A1-AT-trpt′) and absence (T7A1-trpt′) of the AT-box have been compared side by side (Fig. 6C). Transcription assays were performed with immobilized DNA templates to assess the terminated transcripts in the supernatant fractions. We observed that the presence of the AT-box induced a significant delay in transcription termination in a NusA-dependent manner, and this delay increased the amounts of RO transcripts. The presence of R258C NusA produced a higher level of RO products but, unlike the tR1 terminator (Fig. 2B), did not produce more delayed termination zone compared with its WT counterpart. This could be because of fewer pausing sequences in this template, such that the EC could have escaped from the action of Rho. Similar to the case of λtR1 terminator, R258C NusA presumably has a higher affinity for the boxA sequence of the AT-box. These results suggest that even in the absence of other Nus factors (11), binding of NusA to the AT-box is sufficient to bring about Rho inhibition as well as the in vitro antitermination. We restricted our study to only the in vitro experiments because the role of NusA in rRNA antitermination would have been difficult to identify in vivo in the presence of all of the other essential Nus factors.
Next, we designed experiments to directly demonstrate the Rho-NusA competition for the AT-box of the nascent RNA. We stalled the EC, 44 nt downstream of the AT-box (Fig. 6D), by using the lac repressor. The template was immobilized in a similar way as described in the legend to Fig. 4. We performed three types of competitions. In the first case, both radiolabeled Rho and the different cold competitors (the SKK domain fragment with R258C mutation (R258C-SKK) and ΔAR1–2 NusA that is devoid of AR1 and AR2 domains) were mixed together and added to the stalled EC (Fig. 6E). In the second case, Rho was added to the EC prior to the competitors (Fig. 6F). In the third case, NusA derivatives were added to the EC prior to the Rho (Fig. 6G). Both the SKK domain and the NusA devoid of AR1–2 domains are capable of binding to RNA in trans (10), and the latter construct was used because the presence of AR1 and AR2 domains inhibits NusA from binding to the RNA (30). To avoid the translocase activity of Rho, instead of ATP, its non-hydrolyzable analogue, AMP-PNP, was used. In the presence of ATP or AMP-PNP, Rho forms a translocase-competent complex on the rut site (31). The amounts of cold competitors were a 150- and 600-fold molar excess of the radiolabeled Rho (also see “Experimental Procedures”). The amount of Rho dissociated was estimated from the supernatant fractions. The experiments described in the legend to Fig. 6E (Rho and competitor added together) were also repeated on a control template that was devoid of the AT-box (T7A1-trpt′) (Fig. 6H).
We observed that under two conditions, the radiolabeled Rho was competed out efficiently: when Rho was mixed together with the cold competitors before the addition to the EC (Fig. 6E) and when it was added after the competitors were added to the stalled EC (Fig. 6G). This competition was not evident on the template lacking the AT-box (Fig. 6H) and when the Rho was added to the EC prior to the competitors (Fig. 6F). Therefore, the specific Rho-NusA competition occurs at the recognition step of the AT-box, and the competition does not occur if either Rho-RNA or NusA-RNA complexes were formed first. These results also indicated that the presence of overlapping binding sites for Rho and NusA blocks the entry of one species in the prior presence of the other.
We also checked whether R258C NusA has a higher affinity for the AT-box when it is part of the EC (Fig. 7, A–C). Using the same stalled EC as described in the legend to Fig. 6D and following the same procedures as described in the legend to Fig. 4, we observed that, compared with WT, R258C NusA-bound EC required significantly higher concentrations of the WT NusA than the cold competitor, which indicated that this mutant NusA also has higher affinity for the AT-box, which is consistent with what we observed with the isolated AT-box RNA oligomer (Fig. 2F).
FIGURE 7.

Effects of R258C NusA at the AT-box. A and B, autoradiograms estimating the stability of the complexes formed between radiolabeled WT/R258C NusA and stalled EC. The same template as described in the legend to Fig. 6D was used to form the stalled ECs. The same procedures as described in Fig. 4 were used to perform the competition assays using “cold” WT NusA as competitor. The amounts of dissociated radiolabeled WT or R258C NusA proteins were estimated from the S (half of the supernatant, [S]) and P (pellet plus the rest of the supernatant, [S + P]) fractions. C, curves showing the fractions ([2S]/([S] + [S + P]) of labeled WT/R258C NusA dissociated from the stalled EC upon the addition of increasing concentrations of the cold competitors. Amounts of competitors required to bring about 50% dissociation of the radiolabeled NusA are also indicated. S.D. values (error bars) were obtained from 2–3 experiments. D, representatives thin layer chromatography plates showing the 32Pi release from [γ-32P]ATP due to the RNA-dependent ATPase activities of Rho both in the presence and absence of WT/R258C NusAs. In these experiments, Rho was added to the stalled ECs bound to either WT or R258C NusA, as described in the legend to Fig. 6D. Rho utilizes the nascent RNA of the stalled EC to hydrolyze ATP. E, fractions of ATP hydrolyzed as calculated from the signal intensities of the spots in the thin layer chromatography plates were plotted against time. Symbols are described in the key. Rates of ATP hydrolyzes calculated from the linear fittings of the points are indicated. S.D. values (error bars) were obtained from three sets of measurements.
If this competition for binding to the AT-box is functionally relevant, then NusA would reduce the rate of the nascent RNA-dependent ATPase activity of Rho. We used the same stalled EC described in the legend to Fig. 6D for measuring the ATPase activity of Rho using the nascent RNA exiting from the EC as the substrate, both in the absence and presence of WT and R258C NusA proteins (Fig. 7, D and E). We observed that the rate of ATP hydrolysis of Rho was reduced by ∼2–3-fold in the presence of both the WT and R258C NusA proteins. These assays strongly indicated that the Rho-NusA competition observed by the binding assays described above (Fig. 6, E–H) is also functionally significant.
Therefore, these results strongly suggest the existence of a direct Rho-NusA competition for the AT-box sequence under an in vitro setup, which could be an important component in preventing Rho-dependent termination of this untranslated operon.
Genome-wide Pattern of Rho-NusA Competition
Results so far have convincingly indicated that at certain well characterized terminators, NusA inhibits Rho function by a direct competition for the same or overlapping binding sites on the RNA. This raises a possibility of the existence of this type of competition in many other operons, which in turn would lead to a generalized negative regulation of Rho-dependent termination by NusA. To test the hypothesis of the existence of this genome-wide negative regulation of Rho, we performed both low density and high density (Agilent tiling arrays) microarray analyses of mid-log phase MG1655ΔnusA strains expressing WT or mutant nusA (G181D and R258C) genes from a low copy number pCL1920 plasmid.
We plotted the -fold changes in gene expression in different nusA mutants relative to that of the WT (signal intensity with respect to the WT; Fig. 8, A, B, D, and E). Because these two nusA mutants caused a defect in Rho-dependent termination (Fig. 2), we interpreted the up-regulations in different genes/operons as the consequences of defects in Rho-dependent termination (Fig. 8, A and B). This up-regulation is consistent with what was reported for the MDS42ΔnusA strain (6). We ignored the down-regulated genes because they might have arose from indirect effects. We observed that a significant number of genes were up-regulated in these two nusA mutants (Fig. 8, A and B). In addition to this, close to 100 different non-coding regions, mostly the intergenic ones, were also up-regulated (Fig. 8, D and E). The list of all of the up-regulated genes is given in supplemental Fig. S1. A significant number of up-regulated genes/operons (supplemental Fig. S1 and Fig. 8C) as well as intergenic regions (supplemental Fig. S2A and Fig. 8F) were common between the two nusA mutants, which indicates the reliability and robustness of the microarray data. It should be mentioned that in addition to its altered RNA binding properties, G181D mutation makes NusA conformationally unstable, which could have indirect influence on global gene expression patterns. We believe that this might have reduced the overlap between the microarray profiles of these two NusA mutants. It should be noted that the number of genes/operons affected by these two NusA mutants was not as high as that observed for the microarrays obtained in the presence of the Rho inhibitor bicyclomycin (6) or different Rho mutants (21, 32).
FIGURE 8.

Microarray profiles and their validations by q-PCR. A and B, show the plots of microarray profiles of the coding regions (genes) obtained from MG1655rac+ strains expressing different nusA mutants as indicated. The ratio of the hybridization intensities obtained from WT and different mutants gave the measure of the -fold changes that is expressed in log2 scale as per the convention. Up-regulated genes with ≥1.5-fold changes are shown. Each gene is covered by ∼60 probes, and the averages of the -fold change values obtained from each of the probes were assigned as the -fold change values of each gene. C, Venn diagram showing the number of overlapping genes and operons between the two nusA mutants. D and E, microarray profile plots for the non-coding regions obtained under the same conditions as in A and B. The coordinate at the midpoint of each of the intergenic regions was considered for plotting. F, Venn diagram showing the overlaps between the non-coding regions of the two NusA mutants. G, Venn diagrams showing the numbers of overlapping genes and operons between the NusA (R258C and G181D) and the Rho mutants (N340S, G324D, P279S) and those obtained in the presence of bicyclomycin (BCM). H, overlapping patterns of total number of up-regulated genes obtained in all of the NusA and Rho mutants together with those obtained in the presence of the BCM. I–K, validations of the microarray data of the indicated genes by qRT-PCR. In vivo levels of RNA of the up-regulated genes in the presence of the NusA mutants, G181D (H) and R258C (I), and the Rho mutant, N340S (J). mRNA levels are expressed as -fold changes of the CT values (threshold cycle) obtained from the qRT-PCR as per the convention, details of which are given under “Experimental Procedures.” S.D. values (error bars) were calculated from the two biological replicates. y axis breaks are used to accommodate the wide range of -fold change values for different genes.
If a direct competition between NusA and Rho occurs during the gene expressions from different parts of the genome, both the NusA and Rho mutants defective for Rho-dependent termination are likely to produce overlapping up-regulated gene expression profiles in the microarrays. We compared the NusA mutant profiles with that obtained earlier (21, 32) with the termination-defective mutants of Rho, N340S, G324D, and P279S, as well as in the presence of the Rho inhibitor bicyclomycin. We observed that either a high number of the same genes or the genes of the same operons were up-regulated when Rho function was impaired or in the presence of NusA mutants (Fig. 8, G and H; see supplemental Fig. S1 for the full list). About 60% of the up-regulated genes in the NusA mutants were also observed to be up-regulated when Rho function was affected (Fig. 8H). We observed that some of the up-regulated intergenic regions as well as their neighboring regions were also shared by both the NusA and Rho mutants (supplemental Fig. S2B). These results suggest that although the number of up-regulated genes is lower in the NusA mutants, the majority of them are also found to be up-regulated in the different Rho mutants as well as in the presence of bicyclomycin. This is indicative of a widespread Rho-NusA competition.
To validate the above microarray data, we measured the in vivo RNA level of some selected genes in NusA and Rho mutants by qRT-PCR (Fig. 8, H–J). The RNA levels for a few intergenic regions (Fig. 9A) were measured by RT-PCR (Fig. 9B). We have chosen the genes yhil, yjhQ, cysQ, tdcD, rbsR, gfcE, fimD, wecF, mgtA, racR (from the rac prophage), and rho for qRT-PCR, based on the following criteria: (i) highly up-regulated genes in NusA mutants, (ii) common up-regulated genes between the NusA and the Rho mutants, and (iii) other genes of the same operon where one gene is up-regulated by the NusA mutants. We observed that relative mRNA levels of all of these genes were significantly high in both the NusA and Rho mutant strains, thus validating the microarray data. Up-regulation of hundreds of genes together with intergenic regions in the NusA mutants and the overlap of a significant number among them with the microarray profiles of the Rho mutants (Fig. 8H and supplemental Figs. S1 and S2B) strongly indicate that NusA-mediated inhibition of Rho-dependent termination is a genome-wide phenomenon.
FIGURE 9.

Analyses of the microarray data of the non-coding regions. A, a ∼7.7-kb non-coding region due to the presence of many pseudogenes. This region has been highly up-regulated in both of the NusA mutants (Fig. 8, D and E). Underlined subregions are up-regulated in both the NusA and Rho mutants. Horizontal bars of different styles indicate different Rho and NusA mutants, as indicated. The values of the -fold changes in up-regulation are indicated against each mutant. B, mRNA levels from two of the non-coding regions, ydbA_2 and rhsE, estimated by RT-PCR. Products were made from the total RNA obtained from the strains expressing the indicated NusA and Rho mutants. A significant increase in the amount of RT-PCR signals in the mutant strains was observed. The rpoC product was an internal control. C, examples of highly up-regulated genes (in black) in NusA and Rho mutants that are part of operons with stretches of untranslated regions. Gaps between the genes in each of the operons are likely to be the untranslated regions.
The best characterized physiological functions that are influenced by the Rho-dependent termination are as follows: (i) prophage gene silencing (6) and maintaining their lysogenic states (33), (ii) regulation of small RNA expression (34), and (iii) riboswitch formations (8). We probed the up-regulation status of the genes involved in these functions in both the NusA and Rho mutants. Tables described in supplemental Fig. S2C revealed that many prophage genes are up-regulated in the NusA mutants, and a significant proportion of that or the neighboring genes from the same prophage are also up-regulated in the Rho mutants. An increase in the qRT-PCR signal (Fig. 8, I–K) of the rac prophage gene, ydaF, validates the microarray profile. We have observed a few small RNAs(supplemental Fig. S2D), and an Mg2+ sensor riboswitch-controlled gene, mgtA (Fig. 4, H–J, and supplemental Fig. S1, mgtA is highlighted in supplemental Fig. S1) was expressed at a high level in both the NusA and the Rho mutants. These results indicate that important physiological functions that are under the control of Rho-dependent termination are also the target of NusA antagonism. However, because both NusA and Rho binding sequences are quite ambiguous, we failed to predict and locate such sequences in these operons.
Among the genes tested, tdcD, gfcE, fimD, and wecF are part of very long operons (supplemental Fig. S2E), and transcriptions of those would be ideal targets for Rho-dependent termination. A closer look into the microarray data revealed that many of the genes of these operons are significantly up-regulated in the presence of both the NusA (G181D and R258C) and Rho mutants (N340S/G324D/P279S; in supplemental Fig. S2E, see the tables for the -fold changes; Rho data were obtained from Refs. 21 and 32). These data are suggestive of the existence of Rho-NusA competition in these long operons.
The classical role of Rho-dependent termination is to terminate transcription of the untranslated operons because the long stretches of unstructured naked RNAs are the targets of Rho (35). Fig. 8, D and E, revealed that close to 100 intergenic regions were up-regulated in the nusA mutants. Some of the same regions as well as several of their neighboring regions (up to 300-nt regions upstream or downstream) were also up-regulated in the Rho mutants (see supplemental Fig. S2B and Ref. 32). A closer look into the tiling array profile revealed that a ∼7.7-kb region (dotted boxes in Fig. 8, D and E, and bracketed in supplemental Fig. S2A), crowded with intergenic regions, is highly up-regulated in both of the nusA mutants (Figs. 8 (D and E) and 9A). The presence of several pseudogenes (as per the EcoCyc database; Fig. 9A) formed different subregions of the long stretches of untranslated regions that are up-regulated not only in NusA mutants but also in the different Rho mutants (indicated in Fig. 9A). We have validated the up-regulation of the microarray data of two of the untranslated regions corresponding to the pseudogenes by RT-PCR (Fig. 9B). We also noticed that genes that were highly up-regulated in the NusA and Rho mutants have many untranslated regions (Fig. 9C). These untranslated regions are likely to have Rho-binding site(s). Indeed, an RNA secondary structure prediction program, M-fold, predicted that many of these regions have the signature of the rut sites that are characterized by high C/G ratio with favorable free energy to remain unstructured (data not shown). The aforementioned detailed genome-wide analyses strongly suggest a pattern of overlapping existence of Rho targets and NusA binding sites on the same RNA of many operons that is likely to lead to functional competitions between these two factors and thereby support our proposed hypothesis of NusA-mediated global antagonism of Rho-dependent termination.
NusA-Rho Competition in the rho Operon
We observed the up-regulation of rho in the presence of NusA mutants (Fig. 8 (I and J) and supplemental Fig. S1), which indicates that expression of the rho operon itself could be under NusA competition. The rho gene is preceded by an untranslated region (rhoL), part of the sequence of which has a very favorable C/G ratio (∼3) for becoming a potential rut site (Fig. 10A). This Rho-binding site is responsible for the feedback regulation present in this operon (36). We probed the NusA-mediated inhibition of the Rho-dependent transcription termination in the rho operon.
We performed multiple-round transcriptions of a DNA fragment containing a part of the rho operon (Fig. 10A; see the schematic) having the rho promoter, Prho, from which the in vitro transcription was initiated (Fig. 10B). Rho-dependent termination occurred at the 5′-proximal region of the rho gene (indicated in Fig. 10B by dashed lines and also in Fig. 10A). Similar to that observed in Figs. 2 and 6, WT and R258C NusA delayed the termination zone, and in their presence a significant amount of transcript reached the end of the template (RO) without being terminated (Fig. 10B). The amount of RO product in the presence of R258C NusA was much higher, indicating its higher inhibitory effect on Rho. Thus, it is likely that the untranslated region preceding the rho ORF harbors a NusA binding site.
In order to provide further evidence for the existence of this NusA binding site, we monitored the dissociation of Rho from an EC stalled in the termination zone of the rho ORF using the lac repressor as a roadblock (Fig. 10C). The amount of dissociation of radiolabeled Rho from the EC was estimated in the presence of NusA SKK domain and ΔAR1–2 NusA as competitors. Competition experiments were performed essentially following the same procedures as described in the legend to Fig. 6. Assays were performed on immobilized templates so that the amount of released radiolabeled Rho could be measured in the supernatant fractions (Fig. 10, D and E). Similar to what is observed in Fig. 6, Rho was efficiently competed out on this template when it was added to the EC together with the two competitors (Fig. 10D), and this competition was negligible when Rho was added before the competitors (Fig. 10E). These results strongly suggest a Rho-NusA competition for the same or overlapping site(s) on the nascent RNA of the Rho leader region.
The Rho protein level increases when Rho-dependent termination is compromised (36). Therefore, it is expected that NusA-mediated inhibition of Rho-dependent termination in this operon should also increase the level of Rho, and this increase would be much higher in the presence of NusA mutants, G181D and R258C. We measured the in vivo level of Rho protein in the presence of both the WT and mutant NusA by Western blotting using a polyclonal antibody raised against Rho (Fig. 10F) and observed a significantly higher level of Rho expression in the presence of these two mutants. This further reinforced our proposal of the existence of NusA-mediated inhibition of Rho-dependent termination of the rho operon.
Discussion
Recent genomic studies (6, 7, 32) have revealed the presence of genome-wide targets of Rho-dependent termination, which are mostly required to prevent unwanted gene expressions. However, the inherent less specific nature of its RNA binding sites (rut sites) poses the danger of unwanted termination events, and not much is known about how to minimize this event. Once Rho is committed for translocation along the RNA, it may not be controlled because this mechanical process is irreversible. The rut site recognition step is likely to be the best point to control Rho. In principle, many single-stranded RNA binding proteins could block the rut recognition step by functioning as direct competitors. Here, we have provided the following evidence to establish that the transcription elongation factor NusA functions as a general antagonist of Rho by competing for the same or overlapping sites on the mRNA. (i) We have shown that the two NusA-SKK domain (RNA binding domain) mutants as well as the isolated SKK fragment are capable of inhibiting Rho-dependent termination both in vitro and in vivo, and this inhibition was nut site-dependent (Figs. 2 and 3). SKK domain mutations increase the affinity for the nut site (Fig. 2F). (ii) This inhibition originates from the direct competition between overlapping nut and rut sites (Fig. 4), which delays the Rho loading onto the rut sites (Fig. 5). (iii) The existence of direct competition between Rho and NusA was shown during the antitermination of Rho-dependent termination at the rRNA operon (Figs. 6 and 7) and as a part of the feedback mechanism of the rho expression (Fig. 10). (iv) Finally, high resolution tiling microarrays revealed the genome-wide presence of overlapping targets of NusA and Rho, indicating the existence of NusA-mediated antagonism of Rho in many operons as well as in different intergenic regions (Figs. 8 and 9 and supplemental Figs. S1 and S2). Based on all of this evidence, we propose that NusA, unlike a facilitator of hairpin-dependent termination, is an antagonist of Rho-dependent termination. This adds one more function to the multitasking abilities of NusA, in addition to its roles in transcription elongation, antitermination, and DNA repair (11). It should be noted that this model prescribes for the most effective antagonism by NusA in the vicinity of the nut/rut sites, because EC-bound NusA is likely to vacate this site with the progress of transcription elongation. However, on the sites where NusA-nut interaction is very strong, NusA may remain bound to these sites even after the EC has progressed farther away from the nut site. An alternative to the direct competition between NusA and Rho for the overlapping sites on RNA, in the cases where rut and nut sites do not overlap, it is possible that NusA binding to the nut site could remodel the nearby rut site RNA structure to block the entry of Rho.
Two kinds of crystal structures of Rho has been reported to date. When the primary RNA binding site of Rho was occupied by oligonucleotides, the structure was an open ring (37), whereas when both the primary and secondary RNA binding sites were occupied by DNA or RNA oligonucleotides, the structure appeared as a symmetrical hexameric closed ring (38). Based on these sequential occupations of the primary and secondary RNA binding sites, the Rho activation steps may be described by the following equilibria (21),
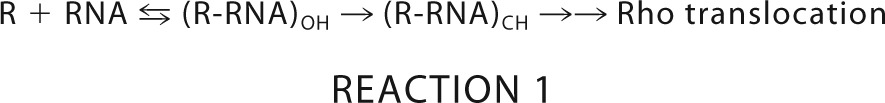 |
where R represents Rho.
Initially, Rho attains an open hexamer (OH) conformation upon recognizing the rut site, following which it undergoes an irreversible isomerization to form a closed hexameric (CH) state. As the CH formation is accompanied by the onset of the ATP hydrolysis, Rho becomes committed for the translocation. Due to this irreversibility, once Rho is committed for translocation, it is unlikely to be dissociated by any RNA-binding protein. Here, we observed that NusA could compete out Rho only when it was added together with it (Figs. 6 and 10), which indicates that the competition occurs at the (R-RNA)OH formation stage. We propose that the OH formation is the only step susceptible to the RNA-binding protein competitors, and thereby the Rho antagonism is likely to occur via this step.
NusA induces transcription elongation pausing (11); it especially enhances the RNA-hairpin dependent pauses (39). As per the kinetic coupling model of Rho-dependent termination (40), pausing of ECs should enhance the termination process. Hence, NusA should function as a stimulator of Rho-dependent termination at the hairpin-dependent pause sites, which may occur less frequently in vivo. However, the RNA hairpin-induced paused/arrested/backtracked ECs are not good substrates for Rho (41), and therefore, pause enhancement at these sites by NusA is not likely to stimulate the Rho function.
The antitermination complex that assembles on the AT-boxes of the rRNA operon consists of NusA, NusB, NusE, and NusG and primarily protects the transcription of this untranslated operon from Rho-dependent termination (26, 27). Assembly of these Nus factors and their subsequent interactions with the EC increase the transcription elongation rate, which is thought to be the major way of overcoming the “Rho chase” of the EC (42). Here, we show that in the in vitro setup, the presence of NusA alone is sufficient to prevent the loading of Rho to the AT-box region, thereby bringing about efficient antitermination (Fig. 6). In vitro this effect of NusA may be localized in the vicinity of the nut site, which only delays the Rho loading onto the rut site. However, under the in vivo conditions, assembly of the Nus factors further enhance the stability of their interactions at the AT-box, which helps them to occupy this site even when the EC has traversed far away to the downstream regions, possibly through a looped out RNA structure, further reducing the chances of Rho to load onto the nascent RNA. Hence, we propose that NusA-Rho competition for the AT-box is an important component of the antitermination event in addition to the enhanced elongation rate of the EC induced by the Nus factors. Because NusE (S10), a component of the antitermination machinery, can interact with NusG interaction (43), one can also speculate that this interaction would prevent Rho-NusG interaction, which in turn would reduce the efficiency of Rho-dependent termination at this operon.
The genome-wide antagonism of Rho-dependent termination arose from the observations of the up-regulation of a significant number of common genes/operons in the presence of termination-defective NusA or Rho mutants (Figs. 8 and 9 and supplemental Figs. S1 and S2). It is possible that this mode of down-regulation of Rho-dependent termination by NusA in non-rRNA operons is reminiscent of the antitermination mechanism described above. A remote possibility could also be the existence of an rRNA-like antitermination system in some other operons, which was manifested as the NusA-mediated inhibition in our experiments.
Bacteriophage P4 protein Psu (44, 45), E. coli Hfq (46), and YaeO (47, 48) proteins and the most recently discovered specific RNA sequence, RARE, in Salmonella (28) are the other inhibitors of the Rho function. They bind directly to Rho and inhibit its function. Hence, because of the presence of NusA and all of these other cis and trans factors, a negative control for Rho is always present in the cell as a default, which is necessary to prevent unwanted transcription termination.
Author Contributions
M. Z. Q. performed all of the in vitro and part of the in vivo experiments. D. D. performed some in vivo experiments. R. S. conceived the project and designed the experiments. M. Z. Q. and R. S. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank laboratory members Dr. Shweta Singh, Gairika Ghosh, and Richa Gupta for critically reading the manuscript.
This work was supported by Department of Biotechnology, Government of India, Grant BT/PR13297/BRB/10/746/2009 and the intramural grant of the Center for DNA Fingerprinting and Diagnostics. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1 and S2.
The microarray data have been deposited in the NCBI Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession numbers GSE77008, GSE77009, GSE77176, and GSE77177).
- RNAP
- RNA polymerase
- AT-box
- antitermination box
- EC
- transcription elongation complex
- AMP-PNP
- 5′-adenylyl-β,γ-imidodiphosphate
- qRT-PCR
- quantitative RT-PCR
- nt
- nucleotide(s)
- RO
- run-off
- OH and CH
- open and closed hexamer, respectively.
References
- 1.Banerjee S., Chalissery J., Bandey I., and Sen R. (2006) Rho-dependent transcription termination: more questions than answers. J. Microbiol. 44, 11–22 [PMC free article] [PubMed] [Google Scholar]
- 2.Peters J. M., Vangeloff A. D., and Landick R. (2011) Bacterial transcription terminators: the RNA 3′-end chronicles. J. Mol. Biol. 412, 793–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sullivan S. L., and Gottesman M. E. (1992) Requirement for E. coli NusG protein in factor-dependent transcription termination. Cell 68, 989–994 [DOI] [PubMed] [Google Scholar]
- 4.Li J., Mason S. W., and Greenblatt J. (1993) Elongation factor NusG interacts with termination factor rho to regulate termination and antitermination of transcription. Genes Dev. 7, 161–172 [DOI] [PubMed] [Google Scholar]
- 5.Chalissery J., Muteeb G., Kalarickal N. C., Mohan S., Jisha V., and Sen R. (2011) Interaction surface of the transcription terminator Rho required to form a complex with the C-terminal domain of the antiterminator NusG. J. Mol. Biol. 405, 49–64 [DOI] [PubMed] [Google Scholar]
- 6.Cardinale C. J., Washburn R. S., Tadigotla V. R., Brown L. M., Gottesman M. E., and Nudler E. (2008) Termination factor Rho and its cofactors NusA and NusG silence foreign DNA in E. coli. Science 320, 935–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters J. M., Mooney R. A., Grass J. A., Jessen E. D., Tran F., and Landick R. (2012) Rho and NusG suppress pervasive antisense transcription in Escherichia coli. Genes Dev. 26, 2621–2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hollands K., Proshkin S., Sklyarova S., Epshtein V., Mironov A., Nudler E., and Groisman E. A. (2012) Riboswitch control of Rho-dependent transcription termination. Proc. Natl. Acad. Sci. U.S.A. 109, 5376–5381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mooney R. A., Davis S. E., Peters J. M., Rowland J. L., Ansari A. Z., and Landick R. (2009) Regulator trafficking on bacterial transcription units in vivo. Mol. Cell 33, 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prasch S., Jurk M., Washburn R. S., Gottesman M. E., Wöhrl B. M., and Rösch P. (2009) RNA-binding specificity of E. coli NusA. Nucleic Acids Res. 37, 4736–4742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sen R., Chalissery J., Qayyum M., Vishalini V., and Muteeb G. (2014) Nus factors of Escherichia coli. EcoSal Plus 10.1128/ecosalplus.4.5.3.1 [DOI] [PubMed] [Google Scholar]
- 12.Toulokhonov I., Artsimovitch I., and Landick R. (2001) Allosteric control of RNA polymerase by a site that contacts nascent RNA hairpins. Science 292, 730–733 [DOI] [PubMed] [Google Scholar]
- 13.Gusarov I., and Nudler E. (2001) Control of intrinsic transcription termination by N and NusA: the basic mechanisms. Cell 107, 437–449 [DOI] [PubMed] [Google Scholar]
- 14.Kolb K. E., Hein P. P., and Landick R. (2014) Antisense oligonucleotide-stimulated transcriptional pausing reveals RNA exit channel specificity of RNA polymerase and mechanistic contributions of NusA and RfaH. J. Biol. Chem. 289, 1151–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt M. C., and Chamberlin M. J. (1987) nusA protein of Escherichia coli is an efficient transcription termination factor for certain terminator sites. J. Mol. Biol. 195, 809–818 [DOI] [PubMed] [Google Scholar]
- 16.Zheng C., and Friedman D. I. (1994) Reduced Rho-dependent transcription termination permits NusA-independent growth of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 91, 7543–7547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saxena S., and Gowrishankar J. (2011) Compromised factor-dependent transcription termination in a nusA mutant of Escherichia coli: spectrum of termination efficiencies generated by perturbations of Rho, NusG, NusA, and H-NS family proteins. J. Bacteriol. 193, 3842–3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burns C. M., Richardson L. V., and Richardson J. P. (1998) Combinatorial effects of NusA and NusG on transcription elongation and Rho-dependent termination in Escherichia coli. J. Mol. Biol. 278, 307–316 [DOI] [PubMed] [Google Scholar]
- 19.Muteeb G., and Sen R. (2010) Random mutagenesis using a mutator strain. Methods Mol. Biol. 634, 411–419 [DOI] [PubMed] [Google Scholar]
- 20.Chalissery J., Banerjee S., Bandey I., and Sen R. (2007) Transcription termination defective mutants of Rho: role of different functions of Rho in releasing RNA from the elongation complex. J. Mol. Biol. 371, 855–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shashni R., Qayyum M. Z., Vishalini V., Dey D., and Sen R. (2014) Redundancy of primary RNA-binding functions of the bacterial transcription terminator Rho. Nucleic Acids Res. 42, 9677–9690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muteeb G., Dey D., Mishra S., and Sen R. (2012) A multipronged strategy of an anti-terminator protein to overcome Rho-dependent transcription termination. Nucleic Acids Res. 40, 11213–11228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Worbs M., Bourenkov G. P., Bartunik H. D., Huber R., and Wahl M. C. (2001) An extended RNA binding surface through arrayed S1 and KH domains in transcription factor NusA. Mol. Cell 7, 1177–1189 [DOI] [PubMed] [Google Scholar]
- 24.Ha K. S., Toulokhonov I., Vassylyev D. G., and Landick R. (2010) The NusA N-terminal domain is necessary and sufficient for enhancement of transcriptional pausing via interaction with the RNA exit channel of RNA polymerase. J. Mol. Biol. 401, 708–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mishra S., Mohan S., Godavarthi S., and Sen R. (2013) The interaction surface of a bacterial transcription elongation factor required for complex formation with an antiterminator during transcription antitermination. J. Biol. Chem. 288, 28089–28103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Albrechtsen B., Squires C. L., Li S., and Squires C. (1990) Antitermination of characterized transcriptional terminators by the Escherichia coli rrnG leader region. J. Mol. Biol. 213, 123–134 [DOI] [PubMed] [Google Scholar]
- 27.Squires C. L., and Zaporojets D. (2000) Proteins shared by the transcription and translation machines. Annu. Rev. Microbiol. 54, 775–798 [DOI] [PubMed] [Google Scholar]
- 28.Sevostyanova A., Groisman E. A. (2015) An RNA motif advances transcription by preventing Rho-dependent termination. Proc. Natl. Acad. Sci. U.S.A. 112, E6835-E6843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Squires C. L., Greenblatt J., Li J., Condon C., and Squires C. L. (1993) Ribosomal RNA antitermination in vitro: requirement for Nus factors and one or more unidentified cellular components. Proc. Natl. Acad. Sci. U.S.A. 90, 970–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schweimer K., Prasch S., Sujatha P. S., Bubunenko M., Gottesman M. E., and Rösch P. (2011) NusA interaction with the α subunit of E. coli RNA polymerase is via the UP element site and releases autoinhibition. Structure 19, 945–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalyani B. S., Muteeb G., Qayyum M. Z., and Sen R. (2011) Interaction with the nascent RNA is a prerequisite for the recruitment of Rho to the transcription elongation complex in vitro. J. Mol. Biol. 413, 548–560 [DOI] [PubMed] [Google Scholar]
- 32.Shashni R., Mishra S., Kalayani B. S., and Sen R. (2012) Suppression of in vivo Rho-dependent transcription termination defects: evidence for kinetically controlled steps. Microbiology 158, 1468–1481 [DOI] [PubMed] [Google Scholar]
- 33.Menouni R., Champ S., Espinosa L., Boudvillain M., and Ansaldi M. (2013) Transcription termination controls prophage maintenance in Escherichia coli genomes. Proc. Natl. Acad. Sci. U.S.A. 110, 14414–14419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Figueroa-Bossi N., Valentini M., Malleret L., Fiorini F., and Bossi L. (2009) Caught at its own game: regulatory small RNA inactivated by an inducible transcript mimicking its target. Genes Dev. 23, 2004–2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richardson J. P. (2002) Rho-dependent termination and ATPases in transcript termination. Biochim. Biophys. Acta 1577, 251–260 [DOI] [PubMed] [Google Scholar]
- 36.Barik S., Bhattacharya P., and Das A. (1985) Autogenous regulation of transcription termination factor Rho. J. Mol. Biol. 182, 495–508 [DOI] [PubMed] [Google Scholar]
- 37.Skordalakes E., and Berger J. M. (2003) Structure of the Rho transcription terminator: mechanism of mRNA recognition and helicase loading. Cell 114, 135–146 [DOI] [PubMed] [Google Scholar]
- 38.Skordalakes E., and Berger J. M. (2006) Structural insights into RNA-dependent ring closure and ATPase activation by the Rho termination factor. Cell 127, 553–564 [DOI] [PubMed] [Google Scholar]
- 39.Artsimovitch I., and Landick R. (2000) Pausing by bacterial RNA polymerase is mediated by mechanistically distinct classes of signals. Proc. Natl. Acad. Sci. U.S.A. 97, 7090–7095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin D. J., Burgess R. R., Richardson J. P., and Gross C. A. (1992) Termination efficiency at rho-dependent terminators depends on kinetic coupling between RNA polymerase and rho. Proc. Natl. Acad. Sci. U.S.A. 89, 1453–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dutta D., Chalissery J., and Sen R. (2008) Transcription termination factor rho prefers catalytically active elongation complexes for releasing RNA. J. Biol. Chem. 283, 20243–20251 [DOI] [PubMed] [Google Scholar]
- 42.Paul B. J., Ross W., Gaal T., and Gourse R. L. (2004) rRNA transcription in Escherichia coli. Annu. Rev. Genet. 38, 749–770 [DOI] [PubMed] [Google Scholar]
- 43.Burmann B. M., Schweimer K., Luo X., Wahl M. C., Stitt B. L., Gottesman M. E., and Rösch P. (2010) A NusE:NusG complex links transcription and translation. Science 328, 501–504 [DOI] [PubMed] [Google Scholar]
- 44.Pani B., Banerjee S., Chalissery J., Muralimohan A., Abishek M., Loganathan R. M., Suganthan R. B., and Sen R. (2006) Mechanism of inhibition of Rho-dependent transcription termination by bacteriophage P4 protein Psu. J. Biol. Chem. 281, 26491–26500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ranjan A., Sharma S., Banerjee R., Sen U., and Sen R. (2013) Structural and mechanistic basis of anti-termination of Rho-dependent transcription termination by bacteriophage P4 capsid protein Psu. Nucleic Acids Res. 41, 6839–6856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rabhi M., Espéli O., Schwartz A., Cayrol B., Rahmouni A. R., Arluison V., and Boudvillain M. (2011) The Sm-like RNA chaperone Hfq mediates transcription antitermination at Rho-dependent terminators. EMBO J. 30, 2805–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pichoff S., Alibaud L., Guédant A., Castanié M. P., and Bouché J. P. (1998) An Escherichia coli gene (yaeO) suppresses temperature-sensitive mutations in essential genes by modulating Rho-dependent transcription termination. Mol. Microbiol. 29, 859–869 [DOI] [PubMed] [Google Scholar]
- 48.Gutiérrez P., Kozlov G., Gabrielli L., Elias D., Osborne M. J., Gallouzi I. E., and Gehring K. (2007) Solution structure of YaeO, a Rho-specific inhibitor of transcription termination. J. Biol. Chem. 282, 23348–23353 [DOI] [PubMed] [Google Scholar]
- 49.Harinarayanan R., and Gowrishankar J. (2003) Host factor titration by chromosomal R-loops as a mechanism for runaway plasmid replication in transcription termination-defective mutants of Escherichia coli. J. Mol. Biol. 332, 31–46 [DOI] [PubMed] [Google Scholar]
- 50.Cheeran A., Babu Suganthan R., Swapna G., Bandey I., Achary M. S., Nagarajaram H. A., and Sen R. (2005) Escherichia coli RNA polymerase mutations located near the upstream edge of an RNA:DNA hybrid and the beginning of the RNA-exit channel are defective for transcription antitermination by the N protein from lambdoid phage H-19B. J. Mol. Biol. 352, 28–43 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.