

Abstract
The formation of disulfide bonds in the endoplasmic reticulum (ER) of eukaryotic cells is catalyzed by the sulfhydryl oxidase, ER oxidoreductin 1 (Ero1), and protein-disulfide isomerase (PDI). PDI is oxidized by Ero1 to continuously introduce disulfides into substrates, and feedback regulates Ero1 activity by manipulating the regulatory disulfides of Ero1. In this study we find that yeast Ero1p is enzymatically active even with its regulatory disulfides intact, and further activation of Ero1p by reduction of the regulatory disulfides requires the reduction of non-catalytic Cys90-Cys97 disulfide in Pdi1p. The principal client-binding site in the Pdi1p b′ domain is necessary not only for the functional Ero1p-Pdi1p disulfide relay but also for the activation of Ero1p. We also demonstrate by complementary activation assays that the regulatory disulfides in Ero1p are much more stable than those in human Ero1α. These new findings on yeast Ero1p-Pdi1p interplay reveal significant differences from our previously identified mode of human Ero1α-PDI interplay and provide insights into the evolution of the eukaryotic oxidative protein folding pathway.
Keywords: disulfide, endoplasmic reticulum (ER), protein-disulfide isomerase, protein-protein interaction, redox regulation, Ero1, oxidative protein folding, regulatory disulfide
Introduction
Correct disulfide bond formation is critical for the maturation and function of many secretory and membrane proteins. In the endoplasmic reticulum (ER)3 lumen of eukaryotic cells, the pivotal enzymatic pathway for catalyzing faithful disulfide formation is composed of sulfhydryl oxidase ER oxidoreductin 1 (Ero1) and protein-disulfide isomerase (PDI), which is conserved from yeast (Ero1p-Pdi1p) to human (Ero1α/β-PDI) (1–3). Both yeast Pdi1p and human PDI consist of four thioredoxin (Trx)-like domains, in the order of a, b, b′, and a′, with an x-linker between domains b′ and a′ and a carboxyl-terminal tail c (4, 5). The a and a′ domains each contain a -Cys-Gly-His-Cys- active site responsible for thiol-disulfide interchange reactions and the b′ domain possesses a hydrophobic pocket, which serves as the principal client-binding site (6–8). Yeast Pdi1p contains two additional non-catalytic cysteines within the a domain, which form a structural disulfide bridge (9), whereas human PDI has two non-essential cysteines in the b′ domain (10) (Fig. 1A, upper). Ero1 oxidase generates a disulfide de novo in its inner active site (Cys352-Cys355 in Ero1p) and a by-product H2O2 by transferring electron to molecular oxygen via its FAD cofactor (11, 12). The disulfide is then transferred to the active site of PDI for the subsequent oxidation of reducing substrates (13, 14) via the outer active site (Cys100-Cys105 in Ero1p) located on an intrinsically flexible loop (15, 16). In the human system, Ero1α binds to the b′ domain of PDI via a strong hydrophobic interaction (12, 17, 18), which results in the preferential oxidation of the a′ domain rather than the a domain of PDI (12, 19, 20). In contrast, the binding interaction between Ero1p and Pdi1p is weak (18), and Ero1p oxidizes domain a of Pdi1p faster than domain a′ (21).
FIGURE 1.

Activation of Ero1p depends on fully reduced Pdi1p. A, upper, cysteine connectivities of oxidized PDI and Pdi1p were shown schematically with the -CGHC- active sites in gray lines and structural disulfide in black line. Lower, cysteine connectivities of fully oxidized Ero1α and Ero1p were shown schematically with the disulfides of active sites in gray lines, regulatory disulfides in black curves, and structural disulfides in black lines. The starting and ending residue numbers of protein constructs were labeled on the upper portion of each diagram. B, upper, the glutathione redox buffers with different reduction potentials used in the oxygen consumption assay. The reduction potential was calculated according to the Nernst equation (Eh′ = E0′ − RT/nF × ln([GSH]2/[GSSG]), where E0′ = −258 mV, pH 7.3 (37), and n = 2). Lower, the oxygen consumption catalyzed by 2 μm Ero1p during oxidation of 20 μm Pdi1p in each glutathione redox buffer. C, enzymatically inactive Ero1p C100A/C105A-FLAG at 0.5 μm was incubated with or without 10 μm Pdi1p in corresponding glutathione redox buffer as numbered in B for 30 min. The oxidized (ox) and reduced (red) forms of Ero1p were monitored after NEM blocking by Western blotting of non-reducing SDS-9% PAGE using αFLAG. Fully reduced Ero1p was prepared with excess β-mercaptoethanol (β-ME). Note that the slow-migrating doublet of Ero1p is due to the heterogeneous reduction of Cys90-Cys349 and Cys150-Cys295 long-range disulfides. D, in the same reaction as in C, the redox states of Pdi1p were monitored by Coomassie staining after mPEG-5k modification; Pdi1p reduced by excess DTT was loaded as a marker for fully reduced Pdi1p. The number of free thiols (-SH) in each Pdi1p species was labeled on the right margin. Note that each of the 6 -SH, 4 -SH, and 2 -SH species was accompanied with a faster migrating band, probably due to incomplete alkylation. E, the fraction of reduced Ero1p doublet in C and fully reduced Pdi1p in D was quantified by densitometry and plotted against the redox conditions in B (mean ± S.D., n = 3 independent experiments), respectively.
The oxidase activity of Ero1 is important for oxidative protein folding, but unlimited production of H2O2 by Ero1 activity could be toxic to the cells. The idea that the activity of Ero1 can be regulated by its non-catalytic disulfides first came from observations in yeast. Disruption of several non-catalytic disulfides in Ero1p by mutagenesis increased its oxidase activity and inhibited yeast growth (22), suggesting that these non-catalytic disulfides might play inhibitory roles for Ero1p activity. Similar inhibitory disulfides were later discovered in both human Ero1 homologues, Ero1α (19, 23) and Ero1β (20). In human Ero1α, the formation of regulatory disulfides (Cys94-Cys131 and Cys99-Cys104) occupies the two catalytic cysteines in the outer active site (Cys94 and Cys99) (19, 20, 23), thus the reduction of these regulatory disulfides is necessary for the function of the outer active site. In the case of Ero1p, the formation of three non-catalytic disulfides, Cys90-Cys349, Cys143-Cys166, and Cys150-Cys295, decreases Ero1p activity not by occupying the catalytic cysteines, instead, by connecting the outer active site-containing “loop cap” to the inner active site-containing helical core and restricting the movement of the outer active site (15) (Fig. 1A, lower). Of these non-catalytic disulfides, Cys150-Cys295 was believed to play a key role in the regulation of Ero1p activity (22). Later elegant studies revealed that the activities of Ero1 in yeast and also in human are actually feedback regulated by PDI to maintain the redox balance in the ER. PDI predominantly in its reduced state can reduce the regulatory disulfides of Ero1 to generate more activated Ero1, and once sufficient PDI is oxidized by Ero1, it can re-oxidize the regulatory disulfides of Ero1 to inhibit its activity (18, 24–26). It has been found that human PDI reduces and also re-oxidizes the regulatory disulfides of Ero1α very fast (18), whereas yeast Pdi1p is more potent to inhibit Ero1p than to activate Ero1p (25). Coincidently, in cells endogenous Ero1α exists in mixed states of oxidized and semi-oxidized (24, 27), whereas Ero1p is almost fully oxidized (25).
Our previous studies on the human Ero1α-PDI interplay revealed that the working modes of human PDI as an efficient regulator and as a specific substrate of Ero1α are fairly different (18). However, the mechanism of the yeast Ero1p-Pdi1p interplay still poses many intriguing questions needing to be answered. Is the fully oxidized Ero1p with intact regulatory disulfides enzymatically inactive or active? Which redox conditions are favorable for Ero1p to be activated by Pdi1p? Why does Pdi1p act as a weak activator but a potent inhibitor of Ero1p? What roles do the non-catalytic elements of Pdi1p play for efficient Ero1p-Pdi1p interplay?
In this study, by using a reconstituted system we demonstrate that Ero1p is enzymatically active even with the regulatory disulfides intact, and that the activation of Ero1p by reduction of the regulatory disulfides occurs only under extremely reducing conditions when fully reduced Pdi1p accumulates. We discover for the first time that the Cys90-Cys97 pair of Pdi1p is a novel redox sensor that can initiate the activation of Ero1p. In addition, we identify critical residues in the client-binding b′ domain of Pdi1p for the activation of Ero1p as well as the functional Ero1p-Pdi1p disulfide relay. By integrating our new findings on Ero1p-Pdi1p interplay with previously identified human Ero1α-PDI interplay modes, we provide new insights into the evolution of the eukaryotic oxidative protein folding pathway.
Experimental Procedures
Plasmid Construction and Protein Preparation
pET28a-Ero1p (Phe56-Leu424) and pET23b-Pdi1p (Asp31-Leu522) plasmids were kind gifts, respectively, from Dr. Yi Yang (East China University of Science and Technology, China) and Dr. Lloyd W. Ruddock (University of Oulu, Finland). The coding sequences of truncated Pdi1p proteins and Ero1p C100A/C105A-FLAG, which contains a FLAG tag (DYKDDDDK) at the C terminus of Ero1p C100A/C105A were generated by PCR. Point mutations of Pdi1p and Ero1p were generated by using the Fast Mutagenesis System (TransGen Biotech). The sequences of all the constructs were verified by DNA sequencing (Life Technologies). Recombinant yeast Pdi1p and Ero1p, and human PDI and Ero1α proteins were expressed and purified as described (18). Escherichia coli Trx1 was expressed and purified as described (28).
Reduced Pdi1p proteins and human PDI were prepared by incubation with 20 mm DTT for 1 h in PBS at 25 °C unless otherwise specified. Reduced Trx1 was prepared by incubation with 100 mm DTT for 1 h in PBS at 25 °C. Oxidized Pdi1p proteins were prepared by incubation with 10 mm GSSG in PBS for 1 h at 25 °C. Purified Ero1p proteins were exclusively in the oxidized states. Fully oxidized Ero1α C99A/C104A/C166A was prepared by incubation with 50 mm K3[Fe(CN)6] at 4 °C for 12 h. Oxidized Ero1p C100A/C105A-FLAG and Ero1α C99A/C104A/C166A monomers were purified using a Superdex-200 10/300 GL column (GE Healthcare) pre-equilibrated with PBS. Reduced Ero1p C100A/C105A-FLAG was prepared by incubation with 100 mm DTT for 5 min in PBS at 25 °C. Excess oxidant or reductant was removed using a HiTrap desalting column (GE Healthcare) pre-equilibrated with PBS. The reduced proteins were kept on ice for use on the same day and the oxidized proteins were stored at −80 °C in aliquots.
Protein Redox State Determination
For the determination of the redox states of Ero1p and Pdi1p proteins in glutathione redox buffer, Ero1p C100A/C105A-FLAG was incubated with or without Pdi1p in buffer A (100 mm Tris-HAc, pH 8.0, 50 mm NaCl, 2 mm EDTA) containing various concentrations of GSH and GSSG for 30 min at 25 °C, and the samples were then divided into two parts. For Pdi1p, the samples were mixed with 1/3 volume of 100% (w/v) TCA pre-cooled at 4 °C for quenching reactions, and the precipitates were washed with 80% (v/v) acetone pre-cooled at −20 °C and then incubated with 1× non-reducing loading buffer containing 2 mm methoxy polyethyleneglycol 5000 maleimide (mPEG-5k, Sigma) for 1 h at 40 °C to block the free thiols. Excess mPEG-5k was quenched with 50 mm DTT and the samples were then subjected to SDS-10% PAGE and Coomassie staining. The parallel samples for Ero1p C100A/C105A-FLAG were treated with 100 mm N-ethylmaleimide (NEM, Sigma), and subjected to SDS-9% PAGE followed by Western blotting analysis using αFLAG (M2, F1804, Sigma).
To determine the redox states of reduced and oxidized Pdi1p proteins and reduced human PDI, Pdi1p proteins and PDI were mixed with 2× non-reducing loading buffer containing 4 mm mPEG-5k for 30 min at 25 °C for free thiol modifications. The samples were subjected to SDS-10% PAGE and Coomassie staining after consuming excess mPEG-5k by 50 mm DTT. To reduce the regulatory disulfides in Ero1p and Ero1α, 0.5 μm oxidized Ero1p C100A/C105A-FLAG or Ero1α C99A/C104A/C166A was incubated with 10 μm reduced Pdi1p or human PDI, respectively. To oxidize the regulatory disulfides in Ero1p, 0.5 μm reduced Ero1p C100A/C105A-FLAG was incubated with 10 μm oxidized Pdi1p proteins. Experiments were carried out in PBS at 25 °C, and aliquots were taken at different time points and immediately quenched with 20 mm NEM or 4 mm 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS, Life Technologies). Redox states of Ero1p and Ero1α were analyzed by non-reducing SDS-9% PAGE followed by Western blotting using αFLAG antibody and non-reducing SDS-8% PAGE followed by Western blotting using αEro1α rabbit serum, respectively. The αEro1α antiserum was generated by immunizing rabbits with purified recombinant Ero1α protein. For Western blotting, an equal volume of sample from each aliquot was loaded for analyses. The band intensities were quantified using ImageJ software (National Institutes of Health). The fraction of specific reduced or oxidized protein species in each lane was calculated as “band intensities of specific species/band intensities of all species.”
Oxygen Consumption Assay
Oxygen consumption was measured at 25 °C using an Oxygraph Clark-type oxygen electrode (Hansatech Instruments) as described (12). Briefly, reactions were initiated by adding Ero1p proteins to a final concentration of 2 μm into buffer A containing 20 μm Pdi1p proteins, 20 μm FAD, and various concentrations of GSH and GSSG. The oxygen consumption rate was calculated from the slope of the linear phase of oxygen decrease.
Chaperone Activity Assay
A variant of GFP “folding mutant” (C48S/F64L/S65T/Q80R/F99S/M153T/V163A/I167T) was subcloned into pQE30 at the BamHI/SacI sites. The purified GFP protein with an N-terminal MRGSH6GS tag was used as the substrate of chaperone assays performed as described (29) with minor alterations. 10 μm GFP in 50 mm Tris-HCl containing 0.3 mm EDTA, pH 7.5, was incubated with an equal volume of 125 mm HCl at room temperature for 1 h. The acid-denatured GFP was refolded by 100-fold dilution into the renaturing buffer (50 mm Tris-HCl, pH 7.5, 100 mm KCl, 25 mm MgCl2) containing 1 μm Pdi1p proteins at 25 °C, and the fluorescence emission at 538 nm with 485 nm excitation was measured after 10 min using the RF-5301PC spectrofluorophotometer (Shimazu).
RNase A Reactivation Assay
Bovine pancreatic RNase A (Sigma) was denatured and reduced as described (30). The reactivation was assayed quantitatively by monitoring the absorbance increase at 296 nm at 25 °C due to the hydrolysis of cCMP (Sigma), and the concentration of reactivated RNase A was calculated as described (12).
Reduction Potential Measurement
Pdi1p proteins at 10 μm were incubated with GSH and GSSG at various concentrations in 75 mm Hepes-NaOH, pH 7.0, 150 mm NaCl, and 1 mm EDTA at 25 °C for 1 h. The stock solutions of GSH and GSSG were adjusted to pH 7.0 using 12 m NaOH. For Pdi1p abb′x and Pdi1p bb′xa′c, an equal volume of 2× loading buffer containing 30 mm mPEG-5k was added to block free thiols. Pdi1p bb′xa′CGPCc was precipitated with 1/3 volume of pre-cooled 100% TCA, and washed with pre-cooled 80% acetone before incubation with 1× loading buffer containing 2 mm mPEG-5k at 40 °C for 1 h. After consuming excess mPEG-5k by 50 mm DTT, samples were resolved by SDS-10% PAGE and visualized by Coomassie staining.
The relative amount of reduced Pdi1p proteins in each reaction was determined by densitometry using ImageJ software. The equilibrium constant Keq for the redox reactions were calculated according to equation: R = ([GSH]2/[GSSG])/(Keq + [GSH]2/[GSSG]), where R is the fraction of reduced Pdi1p proteins. The reduction potential for the active sites of Pdi1p proteins was then derived from the Nernst equation [E0′(Pdi1p proteins) = E0′(GSH) − (RT/nF) × ln(Keq)] using the calculated Keq, T = 298 K, n = 2, and E0′(GSH) = −240 mV at pH 7.0 (31).
Results
Only Fully Reduced Pdi1p Can Reduce the Regulatory Disulfides of Ero1p
To study the relationship between the activity and redox states of Ero1p, we first measured oxygen consumption by Ero1p during oxidation of Pdi1p in the presence of different ratios of GSH/GSSG. As the reduction potential of the yeast ER is around −230 mV (32), reduction potentials in a range of −208 to −258 mV were used by mixing 20 mm GSH and different concentrations of GSSG (Fig. 1B, upper). As shown in the lower panel of Fig. 1B, the oxygen consumption rate increased with declining reduction potential of the glutathione redox buffer. Notably, in the reduction potential range of −208 to −228 mV the oxygen consumption rates were slow but with a stoichiometric excess of oxygen consumed, implying that Ero1p is functional even under more oxidizing conditions than the resting state of the ER. We further analyzed the redox states of Ero1p and Pdi1p under the corresponding redox conditions used for activity assays. Here the enzymatically inactive mutant Ero1p C100A/C105A was used instead of wild-type (wt) Ero1p to prevent the fast autonomous re-oxidation of the regulatory cysteines once reduced (25). Reduction of the long-range regulatory disulfides in Ero1p (Cys150-Cys295 and Cys90-Cys349, Fig. 1A, lower) can be readily discerned on a non-reducing gel by mobility retardation. As shown in Fig. 1C, even though the reduction potential decreased to −228 mV, the two regulatory disulfides of Ero1p were mainly kept intact and Ero1p showed considerable activity, suggesting that reduction of the regulatory disulfides is not a prerequisite for the activity of Ero1p. When the reduction potential was further decreased, the regulatory disulfides of Ero1p were reduced and the activity of Ero1p further increased, indicating the activation of Ero1p. Under highly reducing conditions with 20 mm GSH, the regulatory disulfides of Ero1p were mostly reduced. The redox states of Pdi1p were determined by mPEG-5k modification, as the reduced Pdi1p species will have more free thiols to be modified by mPEG-5k and migrate slower. As shown in Fig. 1D the emergence of 2 -SH and 4 -SH species, respectively, indicated the reduction of one active site and two active sites, and 6 -SH species (fully reduced Pdi1p with non-catalytic Cys90-Cys97 disulfide also reduced) appeared when the reduction potential decreased below −228 mV. It is worth noting that the appearance and accumulation of fully reduced Pdi1p positively correlated with the reduction of the long-range regulatory disulfides in Ero1p as shown in Fig. 1E.
We further prepared Pdi1p with only the active-site disulfides reduced by using an optimized redox buffer (7 mm GSH and 0.2 mm GSSG) and fully reduced Pdi1p by 20 mm DTT (Fig. 2A), and examined their abilities to reduce the regulatory disulfides of oxidized Ero1p C100A/C105A. GSH-reduced Pdi1p could hardly reduce the long-range disulfides in Ero1p, whereas DTT-reduced Pdi1p was nearly as potent as reduced Trx1, a commonly used activator of Ero1p (Fig. 2, B and C). Meanwhile, the reduction of the short-range regulatory disulfide Cys143-Cys166 in Ero1p was determined by using the AMS-alkylation method, which adds 0.5 kDa mass to each modified thiol. Again, DTT-reduced Pdi1p reduced the Cys143-Cys166 disulfide but GSH-reduced Pdi1p was incapable of doing so (Fig. 2D). Taken together, reduction of the regulatory disulfides in Ero1p can be achieved only by fully reduced Pdi1p.
FIGURE 2.

Fully reduced Pdi1p contributes to the reduction of all regulatory disulfides in Ero1p. A, the redox states of Pdi1p oxidized by 10 mm GSSG (oxidized), reduced by 7 mm GSH and 0.2 mm GSSG (GSH-reduced), or reduced by 20 mm DTT (DTT-reduced) were monitored by mPEG-5k modification and Coomassie staining. B, oxidized Ero1p C100A/C105A-FLAG at 0.5 μm was incubated with 10 μm reduced Pdi1p or Trx1 as indicated. Aliquots were taken at the indicated times, and the redox states of NEM-blocked Ero1p were analyzed under non-reducing conditions by Western blotting using αFLAG. C, the fraction of reduced Ero1p doublet in each lane in B was quantified by densitometry and plotted against time (mean ± S.D., n = 3 independent experiments). D, experiments were carried out as in B except that AMS was used for the alkylation of Ero1p. Ero1p C100A/C105A-FLAG, reduced by excess DTT, precipitated by TCA and blocked with AMS, was loaded as a marker for fully reduced Ero1p. The asterisk (*) indicated Ero1p C100A/C105A with only the Cys143-Cys166 regulatory disulfide reduced (34).
Pdi1p Cys90-Cys97 Pair Plays a More Significant Role in the Reduction Than in the Re-oxidation of Ero1p Regulatory Disulfides
Based on the above results that the non-catalytic Cys90-Cys97 pair of Pdi1p was required for the activation of Ero1p, we constructed three Pdi1p mutants: Pdi1p 2S (two non-catalytic cysteines mutated to serines), Pdi1p 4S (four cysteines in the two active sites mutated to serines), and Pdi1p 6S (all cysteines mutated to serines) to further dissect the role of the Cys90-Cys97 pair in the regulation of Ero1p. These cysteine mutants did not show gross structural alterations as determined by far UV circular dichroism and intrinsic fluorescence spectra (data not shown). We determined their abilities to reduce the regulatory disulfides of oxidized Ero1p C100A/C105A. Pdi1p proteins were reduced by 20 mm DTT to ensure all disulfides were fully reduced (Fig. 3A). As shown in Fig. 3, B and D, Ero1p was largely reduced by Pdi1p wt at 20 min, but only a small fraction was reduced by Pdi1p 2S, further proving the necessary role of the reduced Cys90-Cys97 pair in the efficient reduction of Ero1p regulatory disulfides. As expected, neither reduced Pdi1p 4S nor 6S mutant was able to reduce Ero1p. These results demonstrated that although the Cys90-Cys97 cysteine pair of Pdi1p in its dithiol form cannot directly reduce the regulatory disulfides of Ero1p, it is required for the efficient activation of Ero1p by Pdi1p active sites.
FIGURE 3.

The role of the Pdi1p Cys90-Cys97 cysteine pair in the regulation of Ero1p. A, the redox states of DTT-reduced Pdi1p (redPdi1p) and GSSG-oxidized Pdi1p (oxPdi1p) proteins were monitored as described in the legend to Fig. 2A. The number of free thiols (-SH) in each Pdi1p protein was labeled on the right margin. B, the reduction of the regulatory disulfides in Ero1p C100A/C105A-FLAG by reduced Pdi1p proteins was monitored as described in the legend to Fig. 2B. C, the re-oxidation of the regulatory disulfides in reduced Ero1p C100A/C105A-FLAG at 0.5 μm was monitored at indicated times, with or without 10 μm oxidized Pdi1p proteins. D and E, the fraction of reduced Ero1p doublet in B and oxidized Ero1p species in C was quantified by densitometry and plotted against time (mean ± S.D., n = 3 independent experiments), respectively.
Next we studied re-oxidation of the regulatory disulfides of reduced Ero1p C100A/C105A by the three Pdi1p mutants in oxidized states (Fig. 3A). As shown in Fig. 3, C and E, Pdi1p 4S and Pdi1p 6S only marginally promoted the re-oxidation of Ero1p, possibly through a mechanism independent of disulfide exchange. By contrast, Pdi1p 2S converted reduced Ero1p to the oxidized form to a similar extent as Pdi1p wt after 20 min, although the initial kinetics was slower. Therefore, the active sites of Pdi1p play critical roles in the re-oxidation of Ero1p with the non-catalytic disulfide playing a supporting role. Altogether, the Cys90-Cys97 pair of Pdi1p modulates the oxidoreduction of Ero1p regulatory disulfides, with a more significant role in the reduction than in the re-oxidation (compare Fig. 3, D with E).
The Non-catalytic Domains of Pdi1p Contribute More to the Reduction Than to the Re-oxidation of Ero1p Regulatory Disulfides
To analyze the contributions of the catalytic domains and the non-catalytic bb′x domains of Pdi1p to the regulation of Ero1p, we prepared Pdi1p domain truncations (see Fig. 5A, left) and tested their abilities to reduce and re-oxidize the regulatory disulfides of Ero1p C100A/C105A. Fully reduced Pdi1p a and a′c, containing four and two free thiols, respectively (Fig. 4A), were both unable to reduce Ero1p (Fig. 4B and D). By contrast, both oxidized Pdi1p a and a′c eventually promoted the re-oxidation of Ero1p similar to Pdi1p wt, albeit with a slower kinetics. Pdi1p a was somewhat less efficient than Pdi1p a′c probably due to a lower reduction potential (Fig. 4, C and E). These results indicated that the catalytic domains alone are able to re-oxidize but not to reduce the regulatory disulfides of Ero1p.
FIGURE 5.
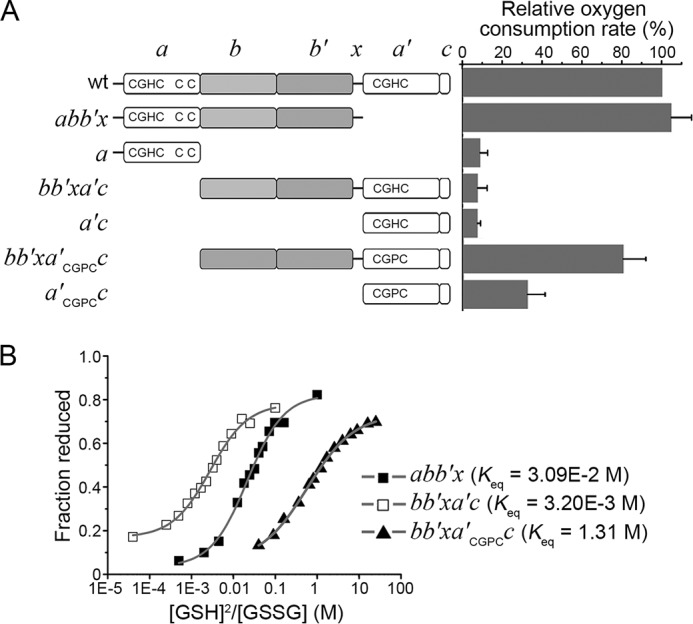
Catalytic oxidation of Pdi1p truncations by hyperactive Ero1p. A, left, schematic representation of Pdi1p mutants. The -CGHC- (-CGPC-) active sites and the non-catalytic cysteine pair were illustrated. Right, the oxygen consumption catalyzed by 2 μm hyperactive Ero1p C150A/C295A during the oxidation of 20 μm Pdi1p proteins in 10 mm GSH was monitored. The oxygen consumption rate was calculated from the slope of the linear phase of oxygen decrease. The relative oxygen consumption rate (%) was calculated as (R − R0)/(R1 − R0) × 100%, where R, rate in the presence of Pdi1p mutants; R1, rate in the presence of Pdi1p wt; R0, rate in the absence of Pdi1p. Data were expressed as mean ± S.D. (n = 3 independent experiments). B, the redox states of Pdi1p proteins at equilibrium with different concentrations of GSH and GSSG monitored by Coomassie staining after mPEG-5k modification. The intensities of the bands were quantified and the fractions of reduced Pdi1p proteins were plotted against to the ratio of [GSH]2/[GSSG]. The resulting Keq were shown and the reduction potentials calculated using Keq were −195 mV for Pdi1p abb′x, −166 mV for Pdi1p bb′xa′c and −244 mV for Pdi1p bb′xa′CGPCc.
FIGURE 4.

Regulation of Ero1p by Pdi1p truncations. A and F, the redox states of reduced and oxidized Pdi1p a and a′c (A), abb′x and bb′xa′c (F) were monitored as described in the legend to Fig. 2A. The number of free thiols (–SH) in each Pdi1p protein was labeled on the right margin. B and G, the reduction of the regulatory disulfides in Ero1p C100A/C105A-FLAG by reduced Pdi1p truncations was monitored as described in the legend to Fig. 3B. C and H, the re-oxidation of the regulatory disulfides in Ero1p C100A/C105A-FLAG by oxidized Pdi1p truncations was monitored as described in the legend to Fig. 3C. D, E, I, and J, the fraction of reduced Ero1p doublet in B and G, and oxidized Ero1p species in C and H was quantified by densitometry and plotted against time (mean ± S.D., n = 3 independent experiments), respectively.
Remarkably, the fully reduced Pdi1p abb′x (Fig. 4F) reduced Ero1p regulatory disulfides as efficiently as Pdi1p wt (Fig. 4, G and I), underscoring the critical role of the non-catalytic bb′x domains in the activation of Ero1p. However, reduced Pdi1p bb′xa′c barely reduced the regulatory disulfides of Ero1p (Fig. 4, G and I), implying that both the reduced Cys90-Cys97 pair and the bb′x domains of Pdi1p are required for the efficient activation of Ero1p. Comparison of Fig. 4, I and J with D and E, highlights the interactions between the non-catalytic bb′x domains and the redox-active a and a′c domains. It shows that the presence of the bb′x fragment had little effect on the ability of the a domain to promote the re-oxidation of Ero1p, while slightly enhancing the ability of the a′c domain. All of the above results indicated that the non-catalytic bb′x domains of Pdi1p are essential for the reduction of Ero1p regulatory disulfides, but contribute minorly to the opposite process.
The Non-catalytic Domains Are Indispensable for Catalytic Oxidation of Pdi1p by Ero1p
Previous studies showed that in the context of full-length Pdi1p both active sites can be efficiently oxidized by Ero1p, although the a′ domain is oxidized slower than the a domain (18, 21). To investigate the contribution of the non-catalytic bb′x domains to the catalytic oxidation of Pdi1p by Ero1p, we determined the oxygen consumption by Ero1p during the catalytic oxidation of various Pdi1p domain truncations (Fig. 5A, left). The hyperactive Ero1p variant C150A/C295A was used here to avoid inadequate activation by certain Pdi1p truncations (see Fig. 4, B and G), and to ensure Ero1p had maximal activity for all tested Pdi1p truncations. As shown in the right panel of Fig. 5A, Pdi1p bb′xa′c was poorly oxidized by hyperactive Ero1p at a rate of only about 10% of Pdi1p wt, whereas Pdi1p abb′x was oxidized as efficiently as Pdi1p wt. We then lowered the reduction potential of domain a′ from −166 to −244 mV by replacing the active site -CGHC- motif with -CGPC-, the active site of Trx (Fig. 5B). Indeed, the oxidation rate of Pdi1p bb′xa′CGPCc was greatly increased to be around 80% of that of Pdi1p wt (Fig. 5A), indicating that the relatively higher reduction potential of the Pdi1p a′ domain limits it as a good substrate of Ero1p. Importantly, the oxygen consumption rate during the oxidation of either a or a′CGPCc (with the bb′x domain deleted from either abb′x or bb′xa′CGPCc) catalyzed by hyperactive Ero1p was greatly decreased, strongly suggesting that the non-catalytic bb′x domains are indispensable for the oxidation of both active sites in Pdi1p during the catalytic cycles driven by Ero1p.
Critical Residues in Pdi1p b′ Domain for Ero1p-Pdi1p Interplay
The above results clearly indicated that the non-catalytic domains of Pdi1p are necessary for the interplay with Ero1p in terms of both the activation of Ero1p and catalytic oxidation of Pdi1p by Ero1p. Next, we deciphered the critical residues in the non-catalytic domains of Pdi1p for the interplay with Ero1p. The crystal structure of Pdi1p shows a highly hydrophobic pocket in domain b′, which was deduced to be the primary substrate binding site (4). We then prepared two Pdi1p client-binding mutants, Pdi1p F249D/F298D with two phenylalanines at the top of the hydrophobic pocket in domain b′ substituted by aspartic acids as well as Pdi1p L313P, a genetically screened binding mutant (33) with a leucine at the bottom of the hydrophobic pocket substituted by proline (Fig. 6A). Without gross conformational changes (data not shown), both mutants exhibited decreased chaperone activity as they did not promote the refolding yield of acid-denatured GFP (Fig. 6B).
FIGURE 6.

Effects of client-binding mutations in Pdi1p b′ domain on Ero1p-Pdi1p interplay. A, ribbon diagram of Pdi1p displaying the locations of Phe249, Phe298, and Leu313. B, the refolding yield of 50 nm acid-denatured green fluorescent protein was determined by fluorescence intensity in the absence or presence of 1 μm Pdi1p proteins as indicated. The fluorescence intensity of 50 nm native GFP was taken as 100% (mean ± S.D., n = 3 independent experiments). C, the redox states of reduced and oxidized Pdi1p client-binding mutants were monitored as described in the legend to Fig. 2A. D, the reduction of the regulatory disulfides in Ero1p C100A/C105A-FLAG by reduced Pdi1p substrate binding mutants was monitored as described in the legend to Fig. 3B. E, the re-oxidation of the regulatory disulfides in Ero1p C100A/C105A-FLAG by oxidized Pdi1p substrate-binding mutants was monitored as described in the legend to Fig. 3C. F and G, the fraction of reduced Ero1p doublet in D and oxidized Ero1p species in E was quantified by densitometry and plotted against time (mean ± S.D., n = 3 independent experiments), respectively. H and I, reactivation of 8 μm reduced and denatured RNase A by 3 μm Pdi1p proteins as indicated in the presence of 3 μm hyperactive Ero1p C150A/C295A (H) or 1 mm GSH and 0.2 mm GSSG (I) was determined (mean ± S.D., n = 3 independent experiments). J, the oxygen consumption catalyzed by 2 μm hyperactive Ero1p C150A/C295A during the oxidation of 20 μm Pdi1p in 10 mm GSH was determined as described in the legend to Fig. 5A (mean ± S.D., n = 3 independent experiments).
With their client-binding abilities impaired, the two fully reduced Pdi1p mutants (Fig. 6C) were much less effective to reduce Ero1p regulatory disulfides as compared with Pdi1p wt (Fig. 6, D and F). For the re-oxidation of Ero1p regulatory disulfides, the two mutants in oxidized states (Fig. 6C) were slightly inferior to Pdi1p wt (Fig. 6, E and G), in accordance with the above finding that the non-catalytic domains of Pdi1p contribute more to the reduction than to the re-oxidation of Ero1p regulatory disulfides. Moreover, the two mutants showed markedly impaired activities in cooperation with Ero1p to catalyze the oxidative folding of denatured and reduced RNase A (Fig. 6H), but were as active as Pdi1p wt in the GSSG-driven RNase A reactivation (Fig. 6I), suggesting that these residues are responsible for specific interaction with Ero1p rather than substrate proteins. Further oxygen consumption assays confirmed that the direct oxidation of Pdi1p mutants by hyperactive Ero1p was greatly compromised (Fig. 6J). All the above results strongly suggested that the client-binding ability of Pdi1p is indispensable for Pdi1p-Ero1p interplay, particularly for Pdi1p-mediated Ero1p activation and Ero1p-catalyzed Pdi1p oxidation.
The Regulatory Disulfides in Ero1p Are Much More Stable Than the Regulatory Disulfides in Ero1α
The above results that the non-catalytic elements of Pdi1p are necessary for Ero1p activation contrast with our previous finding that the two catalytic domains of human PDI can independently activate Ero1α (18). Because the disulfide bond organizations of Ero1 and PDI proteins in yeast and human are different (see Fig. 1A), we speculated that the regulatory disulfides in Ero1α have evolved to be regulated by human PDI active sites in a more prompt way. To further prove our speculation, we performed complementary activation assays by testing the abilities of human PDI and yeast Pdi1p to reduce the regulatory disulfides of yeast Ero1p and human Ero1α, respectively. Because Pdi1p wt in excess would interfere with the electrophoresis mobility of the Ero1α species (data not shown), a smaller fragment Pdi1p abb′x was used instead, which shows the same ability as full-length Pdi1p to regulate Ero1p activity (Fig. 4, G and I). DTT-reduced PDI, GSH-reduced Pdi1p abb′x, and DTT-reduced Pdi1p abb′x were prepared as shown in Fig. 7A. Human PDI with both active sites reduced, similar to GSH-reduced Pdi1p, was incapable of reducing the long-range and short-range regulatory disulfides of Ero1p (Figs. 2 and 7B). On the other hand, both reduced human PDI and GSH-reduced Pdi1p abb′x reduced Ero1α Cys94-Cys131 regulatory disulfide within 1 min, supporting that Ero1α can be activated very quickly by human PDI active sites (Fig. 7, C and D). Surprisingly, DTT-reduced Pdi1p abb′x not only reduced a large portion of the regulatory disulfide Cys94-Cys131 of Ero1α, but also reduced some of the structural disulfide Cys85-Cys391 (Fig. 7, C and D), which is much more stable than Cys94-Cys131 (18). Overall, the above results suggested that the regulatory disulfides in Ero1p are thermodynamically more stable than those in Ero1α. Ero1α can be promptly activated by reduced human PDI active sites, whereas Ero1p can be activated only when the non-catalytic Cys90-Cys97 disulfide of Pdi1p is reduced under very reducing conditions.
FIGURE 7.

Complementary activation of Ero1p and Ero1α by PDI and Pdi1p. A, reduced PDI and Pdi1p were prepared and monitored as described in the legend to Fig. 2A. The number of free thiols (-SH) in PDI and Pdi1p were labeled on the right margin. B, the reduction of the long-range (left) and short-range (right) regulatory disulfides in Ero1p C100A/C105A-FLAG by reduced PDI was monitored as described in the legend to Fig. 2, B and D, respectively. C, the activation of catalytically inactive Ero1α variant C99A/C104A/C166A retaining both long-range disulfides of Cys85-Cys391 and Cys94-Cys131 (18). Oxidized Ero1α C99A/C104A/C166A (ox2) at 0.5 μm was incubated in the absence or presence of 10 μm reduced PDI or Pdi1p as indicated. Aliquots were taken at the indicated times, and the redox states of NEM-blocked Ero1α were analyzed under non-reducing conditions by Western blotting using αEro1α rabbit serum. Ero1α C104A/C131A was loaded as an indicator for the reduction of Cys94-Cys131 disulfide (ox1). Fully reduced Ero1α C99A/C104A/C166A was prepared with excess β-mercaptoethanol (β-ME) (red). The asterisk indicated Ero1α species with both Cys85-Cys391 and Cys94-Cys131 disulfides reduced (18). D, the fraction of activated Ero1α (ox1 and red) in C was quantified by densitometry and plotted against time (mean ± S.D., n = 3 independent experiments).
Discussion
In eukaryotic cells, the Ero1-PDI system catalyzes oxidative protein folding and maintains the thiol-disulfide redox balance in the ER. If the ER becomes too reducing, the regulatory disulfides of Ero1 are reduced by PDI active sites in the dithiol form, increasing Ero1 activity and promoting disulfide formation. Conversely, if the ER becomes oxidizing enough, the regulatory disulfides of Ero1 are formed either by autonomous oxidation or by oxidized PDI, resulting in decreased Ero1 activity (18, 22, 24, 25). Although this feedback regulation model has been widely accepted, our present studies have unraveled unique features of the yeast Ero1p-Pdi1p interplay, which are very different from the human Ero1α-PDI system but were neglected in previous studies.
First, we have determined that under redox conditions approximate to or more oxidizing than −230 mV for the resting yeast ER, all the regulatory disulfides of Ero1p were intact, but Ero1p was enzymatically active. This result was surprising because fully oxidized Ero1p was presumed to be inactive in previous studies (22, 25). In this redox range the activity of Ero1p increased slightly with a decrease of reduction potential (Fig. 1, redox conditions 1 to 3), as more Pdi1p active sites were reduced to be the substrates for Ero1p (Fig. 1D). When the reduction potential fell below the steady state, the regulatory disulfides of Ero1p started to be reduced and Ero1p oxidase activity further increased (Fig. 1, redox conditions 4 to 7). Under these conditions almost all Pdi1p active sites are in reduced form, thus, the increase in Ero1p activity is likely due to allosteric activation by reduction of the regulatory disulfides. Accordingly, in yeast cells Ero1p is almost fully oxidized at steady state, and the reduction of Ero1p regulatory disulfides was observed when the cells were treated with DTT (25). Thus, the regulatory disulfides in Ero1p do not act as an “on/off switch” as in human Ero1α; instead, they function like a “derailleur” to control the transition between low-activity and high-activity states. Distinct from human Ero1α, the formation of the regulatory disulfides in Ero1p only restricts the movement of the outer active site (22), but does not break its integrity.
Again different from human PDI-mediated Ero1α activation, Pdi1p with active sites reduced alone is not able to reduce the regulatory disulfides of Ero1p, and only fully reduced Pdi1p with the non-catalytic Cys90-Cys97 disulfide further reduced is potent. This non-catalytic disulfide is much more stable than those in the two active sites, and is exclusively oxidized at steady state both in vitro (9) and in vivo (22). We determined that its reduction occurs under very reducing conditions (below −240 mV) and completes only in the presence of large excess of DTT. In line with our findings, Pdi1p reduced by 10 mm DTT can activate Ero1p (25), but Pdi1p reduced by 10 mm GSH is inefficient (34), which can now be explained by inadequate reduction of the Cys90-Cys97 disulfide. On the other hand, the Cys90-Cys97 cysteine pair alone cannot directly reduce the regulatory disulfides of Ero1p. It was reported that the presence of Cys90-Cys97 disulfide destabilizes the active site in domain a, making it a better oxidant by 18-fold (9). Therefore, reduction of the Cys90-Cys97 disulfide may lower the reduction potential of the active site in domain a, making it more potent to facilitate the reduction of the regulatory disulfides of Ero1p. We have found that the reduction of Cys90-Cys97 disulfide is also important for Pdi1p a′ domain to activate Ero1p, because the bb′xa′c fragment is unable to reduce Ero1p regulatory disulfides (Fig. 4, G and I), but DTT-reduced full-length Pdi1p with mutated active site in domain a is capable of doing so (25). Thus, we propose that the Cys90-Cys97 pair of Pdi1p is a novel redox sensor, which becomes reduced only under extremely reducing conditions and enables both Pdi1p active sites to activate Ero1p. In yeast cells, although the Cys90-Cys97 pair of Pdi1p is not essential for yeast growth and viability it is required for the efficient processing of a disulfide-containing vacuolar protein, carboxypeptidase Y (35). The non-catalytic cysteine pairs also exist in other three ER-located redox-active Pdi1p homologues, Mdp1p, Mpd2p, and Eps1p. Although it is currently unknown whether these non-catalytic disulfides have similar functions as Cys90-Cys97 of Pdi1p, it is rational to speculate that Pdi1p and its homologues could constitute an extensive network to regulate the activity of Ero1p according to the oxidative folding requirement of yeast ER.
An additional non-catalytic element important for Pdi1p-Ero1p interplay is the client-binding site located in the b′ domain of Pdi1p. Like in human PDI, the yeast Pdi1p b′ domain also has a highly hydrophobic pocket deduced as the primary client-binding site. We have demonstrated this client-binding site is important for Pdi1p-mediated Ero1p activation and Ero1p-catalyzed Pdi1p oxidation, although the binding interaction between Ero1p and Pdi1p is weaker than that between human Ero1α and PDI (18). In line with these results, a yeast strain carrying the Pdi1p L313P mutation shows a growth defect when exposed to DTT challenge (33), possibly due to the impaired Pdi1p-Ero1p interplay. Thus, the client-binding site together with the Cys90-Cys97 cysteine pair of Pdi1p are both required for efficient activation of Ero1p by Pdi1p active sites, underscoring the stringent control of Ero1p activity. On the contrary, the re-oxidation of Ero1p regulatory disulfide is readily promoted by Pdi1p active sites, less depending on the non-catalytic elements of Pdi1p. Our findings that the non-catalytic elements of Pdi1p contribute more to the activation of Ero1p than to the opposite process may also explain why Pdi1p behaves as a weak activator but a potent inhibitor of Ero1p (25).
By comparing our new findings in the yeast Ero1p-Pdi1p system with a previously elucidated mode of human Ero1α-PDI interplay (18), we summarize the differences between the two systems (Fig. 8). 1) The regulatory disulfides in Ero1p, like a derailleur, control the transition between low-activity and high-activity forms of Ero1p; the regulatory disulfides of Ero1α act as an on/off switch. 2) The activation of Ero1p by Pdi1p occurs under extremely reducing conditions and requires the non-catalytic elements of Pdi1p, whereas the inhibition of Ero1p is readily promoted by the oxidized Pdi1p active sites; the activation and inactivation of Ero1α by human PDI both occur very fast and are fulfilled by PDI active sites not requiring its non-catalytic element. 3) Client-binding b′ domain either in yeast Pdi1p or in human PDI plays a critical role in Ero1-driven oxidative substrate folding. However, the binding between Pdi1p and Ero1p is weak, and Ero1p can oxidize both active sites of Pdi1p; whereas Ero1α binds tightly to the human PDI b′ domain and preferentially oxidizes the PDI a′ domain, leaving a domain in the reduced state to catalyze efficient disulfide isomerization for the production of correctly folded substrates with complicated disulfides.
FIGURE 8.

Comparison of Ero1-PDI interplay in yeast and human systems. Left, in the resting state, Ero1p exists predominantly in the low-activity state with intact regulatory disulfides and catalyzes oxidative protein folding via oxidation of Pdi1p with slow turnover (thin red arrow). When cells encounter an extremely reductive challenge, the non-catalytic Cys90-Cys97 cysteine pair (magenta) of Pdi1p is reduced to make Pdi1p active sites more potent to reduce Ero1p regulatory disulfides (thin green arrow), with the assistance of the client-binding site located in domain b′ (orange oval). The activated Ero1p in the high activity form catalyzes the continuous oxidation of Pdi1p with fast turnover to quickly restore the redox balance in the ER (thick red arrow). Once the thiol-disulfide equilibrium is reestablished, the regulatory disulfides of Ero1p are re-oxidized to decrease its activity by oxidized Pdi1p active sites, a process less dependent on the non-catalytic elements of Pdi1p (only catalytic domain is shown). The re-oxidation of regulatory disulfides in Ero1p (thick dark-green arrow) by Pdi1p is more readily than the reduction of these regulatory disulfides (thin green arrow). Right, although Ero1α in the resting state tends to be in the oxidized and inactive state, the active sites of PDI and some of its homologues (PDIs, only catalytic domain is shown) can promptly respond to the redox fluctuation in the ER and rapidly reduce or re-oxidize the regulatory disulfides of Ero1α to modulate its oxidase activity (18). Note that the client-binding sites in both PDI and Pdi1p are important for their oxidation in Ero1-catalyzed reactions, and the binding interaction in Ero1α-PDI is strong, whereas that in Ero1p-Pdi1p is much weaker. The upper part (light blue) indicates the processes of regulation of Ero1 by PDI, and the lower part (pink) is for the oxidation of PDI by Ero1.
Yeast cells contain much less disulfide-containing proteins than human cells (18). At steady state, Pdi1p is largely in its oxidized state and Ero1p is maintained in the low-activity form, which may be adequate to provide basic oxidizing power for oxidative protein folding. When yeast cells encounter an extreme reductive challenge (e.g. anaerobic fermentation, DTT treatment), Ero1p would turn into the high-activity form to enhance the utilization of oxygen, which must be facilitated by the non-catalytic elements of Pdi1p, particularly the reduction of the non-catalytic disulfide. In highly evolved human cells with abundant disulfide-containing proteins, the requirement of oxidative folding capacity is very high especially in professional secretory cells, and the ER redox may fluctuate under different physiological and pathological conditions. Thus, the activity of Ero1α must be regulated very promptly and precisely to optimize the folding efficiency of large and complicated disulfide-containing secretory proteome and meanwhile to minimize the risks of over-oxidation in the ER. To this end, distinct regulatory disulfides in Ero1α (Cys94-Cys131 and Cys99-Cys104) have evolved to be rapidly modulated by the redox states of human PDI active sites. Recently, another disulfide (Cys208-Cys241) unique in Ero1α was reported to play an additional regulatory role (36), but its regulatory mechanism by human PDI active sites is not yet clear. On the other side, the tightly controlled regulatory disulfides (Cys143-Cys166 and Cys150-Cys295) of Ero1p disappeared in Ero1α, and the longest range disulfide (Cys90-Cys349) of Ero1p has remained in Ero1α (Cys85-Cys391) as an evolutionary trace, no longer having a regulatory function but instead playing a structural role.
Author Contributions
L. W. and C. C. W. conceived the project. Y. N., L. Z., and L. W. designed experiments. Y. N., L. Z., and J. Y. conducted experiments. Y. N., L. W., and C. C. W. interpreted the results and wrote the manuscript. All authors approved the final version of the manuscript.
Acknowledgments
We thank Yi Yang and Lloyd Ruddock for their generous gifts of constructs, Xi'e Wang for technical assistance, and Xi Wang and all other lab members for helpful discussions. We also thank Akash Mathew for careful language editing of the manuscript.
This work was supported by Chinese Ministry of Science and Technology Grants 2011CB910303 and 2012CB911002 (to C. C. W.) and National Natural Science Foundation of China Grants 31370775 and 31571163 (to L. W.). The authors declare that they have no conflict of interest with the contents of this article.
- ER
- endoplasmic reticulum
- AMS
- 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid
- Ero1
- endoplasmic reticulum oxidoreductin 1
- GSH
- reduced glutathione
- GSSG
- oxidized glutathione
- mPEG-5k
- methoxy polyethyleneglycol 5000 maleimide
- NEM
- N-ethylmaleimide
- PDI
- protein-disulfide isomerase
- Trx
- thioredoxin.
References
- 1.Oka O. B., and Bulleid N. J. (2013) Forming disulfides in the endoplasmic reticulum. Biochim. Biophys. Acta 1833, 2425–2429 [DOI] [PubMed] [Google Scholar]
- 2.Kojer K., and Riemer J. (2014) Balancing oxidative protein folding: the influences of reducing pathways on disulfide bond formation. Biochim. Biophys. Acta 1844, 1383–1390 [DOI] [PubMed] [Google Scholar]
- 3.Wang L., Wang X., and Wang C. C. (2015) Protein-disulfide isomerase, a folding catalyst and a redox-regulated chaperone. Free Radic. Biol. Med. 83, 305–313 [DOI] [PubMed] [Google Scholar]
- 4.Tian G., Xiang S., Noiva R., Lennarz W. J., and Schindelin H. (2006) The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell 124, 61–73 [DOI] [PubMed] [Google Scholar]
- 5.Wang C., Li W., Ren J., Fang J., Ke H., Gong W., Feng W., and Wang C. C. (2013) Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid. Redox Signal. 19, 36–45 [DOI] [PubMed] [Google Scholar]
- 6.Pirneskoski A., Klappa P., Lobell M., Williamson R. A., Byrne L., Alanen H. I., Salo K. E., Kivirikko K. I., Freedman R. B., and Ruddock L. W. (2004) Molecular characterization of the principal substrate binding site of the ubiquitous folding catalyst protein-disulfide isomerase. J. Biol. Chem. 279, 10374–10381 [DOI] [PubMed] [Google Scholar]
- 7.Byrne L. J., Sidhu A., Wallis A. K., Ruddock L. W., Freedman R. B., Howard M. J., and Williamson R. A. (2009) Mapping of the ligand-binding site on the b′ domain of human PDI: interaction with peptide ligands and the x-linker region. Biochem. J. 423, 209–217 [DOI] [PubMed] [Google Scholar]
- 8.Denisov A. Y., Määttänen P., Dabrowski C., Kozlov G., Thomas D. Y., and Gehring K. (2009) Solution structure of the bb′ domains of human protein disulfide isomerase. FEBS J. 276, 1440–1449 [DOI] [PubMed] [Google Scholar]
- 9.Wilkinson B., Xiao R., and Gilbert H. F. (2005) A structural disulfide of yeast protein-disulfide isomerase destabilizes the active site disulfide of the N-terminal thioredoxin domain. J. Biol. Chem. 280, 11483–11487 [DOI] [PubMed] [Google Scholar]
- 10.Schwaller M., Wilkinson B., and Gilbert H. F. (2003) Reduction-reoxidation cycles contribute to catalysis of disulfide isomerization by protein-disulfide isomerase. J. Biol. Chem. 278, 7154–7159 [DOI] [PubMed] [Google Scholar]
- 11.Gross E., Sevier C. S., Heldman N., Vitu E., Bentzur M., Kaiser C. A., Thorpe C., and Fass D. (2006) Generating disulfides enzymatically: reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. U.S.A. 103, 299–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L., Li S. J., Sidhu A., Zhu L., Liang Y., Freedman R. B., and Wang C. C. (2009) Reconstitution of human Ero1-Lα/protein-disulfide isomerase oxidative folding pathway in vitro: position-dependent differences in role between the a and a′ domains of protein-disulfide isomerase. J. Biol. Chem. 284, 199–206 [DOI] [PubMed] [Google Scholar]
- 13.Frand A. R., and Kaiser C. A. (2000) Two pairs of conserved cysteines are required for the oxidative activity of Ero1p in protein disulfide bond formation in the endoplasmic reticulum. Mol. Biol. Cell 11, 2833–2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertoli G., Simmen T., Anelli T., Molteni S. N., Fesce R., and Sitia R. (2004) Two conserved cysteine triads in human Ero1α cooperate for efficient disulfide bond formation in the endoplasmic reticulum. J. Biol. Chem. 279, 30047–30052 [DOI] [PubMed] [Google Scholar]
- 15.Gross E., Kastner D. B., Kaiser C. A., and Fass D. (2004) Structure of Ero1p, source of disulfide bonds for oxidative protein folding in the cell. Cell 117, 601–610 [DOI] [PubMed] [Google Scholar]
- 16.Sevier C. S., and Kaiser C. A. (2006) Disulfide transfer between two conserved cysteine pairs imparts selectivity to protein oxidation by Ero1. Mol. Biol. Cell 17, 2256–2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inaba K., Masui S., Iida H., Vavassori S., Sitia R., and Suzuki M. (2010) Crystal structures of human Ero1α reveal the mechanisms of regulated and targeted oxidation of PDI. EMBO J. 29, 3330–3343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L., Niu Y., Zhu L., Fang J., Wang X., Wang L., and Wang C. C. (2014) Different interaction modes for protein-disulfide isomerase (PDI) as an efficient regulator and a specific substrate of endoplasmic reticulum oxidoreductin-1α (Ero1α). J. Biol. Chem. 289, 31188–31199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker K. M., Chakravarthi S., Langton K. P., Sheppard A. M., Lu H., and Bulleid N. J. (2008) Low reduction potential of Ero1α regulatory disulphides ensures tight control of substrate oxidation. EMBO J. 27, 2988–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L., Zhu L., and Wang C. C. (2011) The endoplasmic reticulum sulfhydryl oxidase Ero1β drives efficient oxidative protein folding with loose regulation. Biochem. J. 434, 113–121 [DOI] [PubMed] [Google Scholar]
- 21.Vitu E., Kim S., Sevier C. S., Lutzky O., Heldman N., Bentzur M., Unger T., Yona M., Kaiser C. A., and Fass D. (2010) Oxidative activity of yeast Ero1p on protein-disulfide isomerase and related oxidoreductases of the endoplasmic reticulum. J. Biol. Chem. 285, 18155–18165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sevier C. S., Qu H., Heldman N., Gross E., Fass D., and Kaiser C. A. (2007) Modulation of cellular disulfide-bond formation and the ER redox environment by feedback regulation of Ero1. Cell 129, 333–344 [DOI] [PubMed] [Google Scholar]
- 23.Appenzeller-Herzog C., Riemer J., Christensen B., Sørensen E. S., and Ellgaard L. (2008) A novel disulphide switch mechanism in Ero1α balances ER oxidation in human cells. EMBO J. 27, 2977–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Appenzeller-Herzog C., Riemer J., Zito E., Chin K. T., Ron D., Spiess M., and Ellgaard L. (2010) Disulphide production by Ero1α-PDI relay is rapid and effectively regulated. EMBO J. 29, 3318–3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim S., Sideris D. P., Sevier C. S., and Kaiser C. A. (2012) Balanced Ero1 activation and inactivation establishes ER redox homeostasis. J. Cell Biol. 196, 713–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shepherd C., Oka O. B., and Bulleid N. J. (2014) Inactivation of mammalian Ero1α is catalysed by specific protein-disulfide isomerases. Biochem. J. 461, 107–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benham A. M., Cabibbo A., Fassio A., Bulleid N., Sitia R., and Braakman I. (2000) The CXXCXXC motif determines the folding, structure and stability of human Ero1-Lα. EMBO J. 19, 4493–4502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X., Wang L., Wang X., Sun F., and Wang C. C. (2012) Structural insights into the peroxidase activity and inactivation of human peroxiredoxin 4. Biochem. J. 441, 113–118 [DOI] [PubMed] [Google Scholar]
- 29.Mares R. E., Meléndez-López S. G., and Ramos M. A. (2011) Acid-denatured green fluorescent protein (GFP) as model substrate to study the chaperone activity of protein-disulfide isomerase. Int. J. Mol. Sci. 12, 4625–4636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lyles M. M., and Gilbert H. F. (1991) Catalysis of the oxidative folding of ribonuclease A by protein-disulfide isomerase: dependence of the rate on the composition of the redox buffer. Biochemistry 30, 613–619 [DOI] [PubMed] [Google Scholar]
- 31.Schafer F. Q., and Buettner G. R. (2001) Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 30, 1191–1212 [DOI] [PubMed] [Google Scholar]
- 32.Delic M., Mattanovich D., and Gasser B. (2010) Monitoring intracellular redox conditions in the endoplasmic reticulum of living yeasts. FEMS Microbiol. Lett. 306, 61–66 [DOI] [PubMed] [Google Scholar]
- 33.Gauss R., Kanehara K., Carvalho P., Ng D. T., and Aebi M. (2011) A complex of Pdi1p and the mannosidase Htm1p initiates clearance of unfolded glycoproteins from the endoplasmic reticulum. Mol. Cell 42, 782–793 [DOI] [PubMed] [Google Scholar]
- 34.Heldman N., Vonshak O., Sevier C. S., Vitu E., Mehlman T., and Fass D. (2010) Steps in reductive activation of the disulfide-generating enzyme Ero1p. Protein Sci. 19, 1863–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luz J. M., and Lennarz W. J. (1998) The nonactive site cysteine residues of yeast protein-disulfide isomerase are not required for cell viability. Biochem. Biophys. Res. Commun. 248, 621–627 [DOI] [PubMed] [Google Scholar]
- 36.Ramming T., Okumura M., Kanemura S., Baday S., Birk J., Moes S., Spiess M., Jenö P., Bernèche S., Inaba K., and Appenzeller-Herzog C. (2015) A PDI-catalyzed thiol-disulfide switch regulates the production of hydrogen peroxide by human Ero1. Free Radic. Biol. Med. 83, 361–372 [DOI] [PubMed] [Google Scholar]
- 37.Jones D. P. (2002) Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol. 348, 93–112 [DOI] [PubMed] [Google Scholar]