

The malignant mechanisms that control the development of cutaneous T-cell lymphoma (CTCL) are starting to be identified. Recent evidence suggests that disturbances in specific intracellular signaling pathways, such as RAS-MAPK, TCR-PLCG1-NFAT and JAK-STAT, can play an essential role in the pathogenesis of CTCL.1,2 Our group previously reported a network of somatic mutations affecting genes with potential to affect critical T-cell signaling pathways in CTCL patients.1 As part of our findings we detected a number of mutations potentially affecting JAK/STAT signaling. These findings were recently confirmed by an independent group, suggesting that mutations in this pathway may contribute as disease mechanisms in CTCL.3 Deregulated JAK/STAT signaling is involved in many types of cancer. In fact, somatically acquired genetic alterations of JAK or STAT genes that induce aberrant activation of downstream signaling, via STAT phosphorylation, have been reported in some human hematologic malignancies including T-cell lymphomas.4,5 We decided to explore JAK/STAT signaling as part of an intricate network of malignant signaling that controls the pathogenesis of CTCL, on the basis of the following evidence: (i) we had detected mutations in the pseudokinase domain of JAK1 and JAK3 in two of 11 patients and one cell line; (ii) we had also found several mutations that can directly (i.e., IL6S/T) or indirectly (i.e., TRAF6, RELB and CARD11) activate JAK/STAT signaling; and (iii) activated STAT3 had been detected in a large proportion of patients with advanced CTCL.6,7
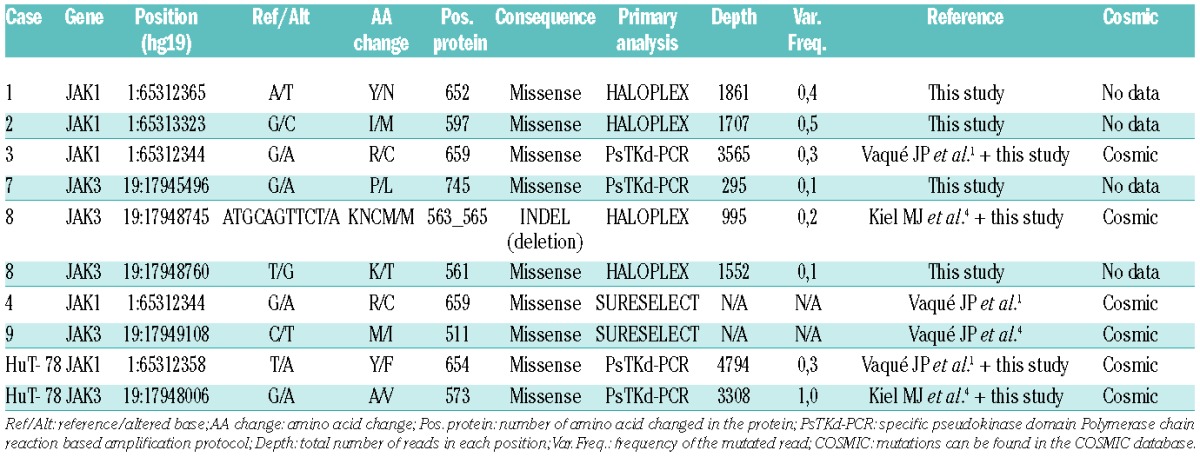
To explore the mutational status of JAK genes in a larger cohort of human CTCL patients’ samples and cell lines, two independent state-of-the-art ultrasequencing approaches were used: a targeted gene-enrichment kit (HaloPlex) coupled to Ion-PGM (Life Technologies) sequencing, and a specific polymerase chain reaction-based amplification protocol targeting the pseudokinase domains of JAK1, JAK2 and JAK3 genes (hereafter, referred to as PsTKd-PCR), followed by specific indexing and sequencing with MiSeq (Illumina; see the Online Supplementary Methods for details). These are two highly sensitive methods that can enable the detection of mutations even present at low frequencies in neoplastic cells or in minority clones which may be found in CTCL samples. Thus, taken together, the data from our series (including those already described by Vaqué et al.1) enabled us to detect and validate somatic mutations in either JAK1 or JAK3 genes in up to seven patients and one cell line (Table 1) from a total of 46 CTCL patients (clinical data described in Online Supplementary Table S1) and two cell lines. A recurrent mutation, JAK1-R659C, was found in two different samples, from patients 3 and 4 (Table 1). Most of the mutations were located within the pseudokinase domain of JAK proteins, a finding that is consistent with the results of other research groups that have found somatic mutations in the same domain of JAK1 and JAK3 kinases in prolymphocytic leukemia, other T-cell leukemias including CTCL and various human malignancies.3,4,8–10 Thus, it has been shown that JAK pseudokinase domains are auto-inhibitory and keep the kinase domain inactive until receptor dimerization stimulates transition to an active state.11
Table 1.
Somatic mutations of JAK in samples from CTCL patients and cell lines.
Molecular analysis of deregulated JAK/STAT signaling has provided a novel rationale for treating human cancers using targeted inhibition of JAK kinases. To explore this possibility in CTCL, we decided to study JAK/STAT activity in a panel of CTCL cell lines including HuT-78 cells carrying mutated JAK1 and JAK3 genes (Table 1 and Kiel et al.4). We first explored the biological effects of the specific inhibition of JAK/STAT signaling in human CTCL cells. MyLa, HuT-78 and HH cell lines were incubated with increasing doses of a specific JAK inhibitor (INCB018424, also known as ruxolitinib), which is currently used in the treatment of myeloproliferative disorders. This inhibitor caused a dose-dependent inhibition of cell proliferation (Figure 1A–C) that enabled us to calculate the IC50 concentration in each case. Molecularly, the cells showed activated basal STAT phosphorylation in the absence of serum, which could be due to multiple activating mechanisms including, but not restricted to, JAK mutations. In these conditions we incubated CTCL cells at different time points with IC50 concentrations of INCB018424 and observed a marked and rapid inhibition of STAT activation that was abolished after 3 h of treatment. Remarkably, this effect was greatly accentuated in HuT-78 cells that harbor mutations in JAK1 and JAK3 genes (Figure 1D–F). Thus, we found basal activation of JAK/STAT signaling in CTCL cells and also found that this is highly sensitive to the use of JAK inhibitors. We also studied the cytotoxic effects induced by JAK inhibition, using annexin V/7AAD binding and PARP cleavage, with flow activated cell sorting (FACS) and western blot analysis, respectively. We found a moderate effect on cell death (Figure 2A–F). However, we found that incubation with INCB018424 led to a marked inhibition of cell proliferation by a mechanism that impinges on the control of DNA synthesis, as shown by the FACS analyses illustrated in Figure 2G–I. Thus, blocking JAK/STAT signaling appears to target CTCL mechanisms of malignant cell growth more efficiently than occurs by simply inducing cytotoxic effects.
Figure 1.

Treatment with ruxolitinib inhibits CTCL cell proliferation and JAK/STAT activity in CTCL cell lines. Cell proliferation assay in (A) MyLa (B) HuT-78 and (C) HH cells incubated for 0, 24, or 48 h, using DMSO (control) or the indicated amount of INCB018424 (μM). N=3; error bars indicate SEM. (D, E) Analysis of basal STAT-1, 3 and 5 activity by western blot using total cell lysates from previously starved CTCL cell lines incubated for the indicated times with a specific JAK inhibitor (INCB018424) using the specific IC50 concentrations in each case.
Figure 2.

Treatment with ruxolitinib activates apoptosis and inhibits DNA synthesis in CTCL cells. Percentage of viable (A) Myla, (B) HuT-78 and (C) HH cells incubated for 24 h with vehicle (DMSO) or the indicated 1× or 2× IC50 concentration of INCB018424. Right plots are representative examples of annexin V (X-axis)/7AAD (Y-axis) staining data in each case. N=3; error bars indicate SEM. Representative western blot analysis using anti-PARP and anti-tubulin antibodies in whole-protein extracts from (D) Myla (E) HuT-78 and (F) HH cells treated with DMSO or the indicated concentrations of INCB018424. C+ indicates treatment of each cell line with 20 nM of okadaic acid for 48 h as a positive control. Percentage of total DNA synthesis in exponentially growing (G) Myla (H) HuT-78 and (I) HH cells incubated for 24 h with vehicle (DMSO) or the indicated concentration of INCB018424. Right plots are representative examples of FITC-A (X-axis) staining data and cell count (Y-axis) staining data in each case. N=3; error bars indicate SEM.
In summary, we show that JAK1 and JAK3 somatic mutations can contribute to deregulated JAK/STAT signaling in CTCL. Our study also provides new information that could help the development of new tools for molecular diagnosis (i.e., JAK mutations or STAT activation) as well as novel targets for therapy using specific JAK inhibitors.
Acknowledgments
We are indebted to the patients who contributed to this study. We especially thank José Revert and Carolina Santa Cruz from IDIVAL (Santander, Spain), and the staff members of the Biobank and the Pathology service at HUMV (Santander, Spain) for their exceptional work.
Footnotes
Funding: This work was supported by grants from the Instituto de Salud Carlos III of the Spanish Ministry of Economy and Competence (MINECO and RTIC) to MAP (SAF2013-47416-R and RD06/0020/0107-RD012/0036/0060) and from ISCIII-MINECO-AES-FEDER to JPV, MSB and POR (Plan Nacional I+D+I: 2012-2015-PI12/00357, 2011-2014-CP11/00018), and 2014-2017-FIS PI14/01784) and the Asociación Española Contra el Cáncer to MAP. CP is a recipient of a Sara Borrel contract from MINECO (CD13/00088). JG-R is a recipient of an iPFIS fellowship (IFI14/00003) from ISCIII-MINECO-AES-FEDER (P.E.I+D+I 2013-2016). Salary support to SG was provided by CP11/00018, from ISCIII-MINECO-AES-FEDER (P.N.I+D+I 2008-2011). MSB currently holds a Miguel Servet contract (CP11/00018) from the ISCIII- MINECO-AES-FEDER (P.N.I+D+I 2008-2011), Spain. JPV was partially supported by SODERCAN and is now supported by a Ramón y Cajal research program (RYC-2013-14097).
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Vaqué JP, Gómez-López G, Monsálvez V, et al. PLCG1 mutations in cutaneous T-cell lymphomas. Blood 2014;123(13):2034–2043. [DOI] [PubMed] [Google Scholar]
- 2.Kiessling MK, Oberholzer PA, Mondal C, et al. High-throughput mutation profiling of CTCL samples reveals KRAS and NRAS mutations sensitizing tumors toward inhibition of the RAS/RAF/MEK signaling cascade. Blood 2011;117(8):2433–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGirt LY, Jia P, Baerenwald DA, et al. Whole genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood 2015;126(4):508–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kiel MJ, Velusamy T, Rolland D, et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood 2014;124(9):1460–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crescenzo R, Abate F, Lasorsa E, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 2015;27(4):516–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Netchiporouk E, Litvinov IV, Moreau L, Gilbert M, Sasseville D, Duvic M. Deregulation in STAT signaling is important for cutaneous T-cell lymphoma (CTCL) pathogenesis and cancer progression. Cell Cycle 2014;13(21):3331–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sommer VH, Clemmensen OJ, Nielsen O, et al. In vivo activation of STAT3 in cutaneous T-cell lymphoma. Evidence for an anti-apoptotic function of STAT3. Leukemia 2004;18(7):1288–1295. [DOI] [PubMed] [Google Scholar]
- 8.Flex E, Petrangeli V, Stella L, et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med 2008;205(4):751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiang Z, Zhao Y, Mitaksov V, et al. Identification of somatic JAK1 mutations in patients with acute myeloid leukemia. Blood 2008; 111(9):4809–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergmann AK, Schneppenheim S, Seifert M, et al. Recurrent mutation of JAK3 in T-cell prolymphocytic leukemia. Genes Chromosomes Cancer 2014;53(4):309–316. [DOI] [PubMed] [Google Scholar]
- 11.Lupardus PJ, Ultsch M, Wallweber H, Bir Kohli P, Johnson AR, Eigenbrot C. Structure of the pseudokinase-kinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc Natl Acad Sci USA 2014;111(22):8025–8030. [DOI] [PMC free article] [PubMed] [Google Scholar]