

Abstract
Our understanding of the biology of the normal hematopoietic stem cell niche has increased steadily due to improved murine models and sophisticated imaging tools. Less well understood, but of growing interest, is the interaction between cells in the bone marrow during the initiation, maintenance and treatment of hematologic neoplasms. This review summarizes the emerging concepts of the normal and leukemic hematopoietic bone marrow niche. Furthermore, it reviews current models of how the microenvironment of the bone marrow may contribute to or be modified by leukemogenesis. Finally, it provides the rationale for a “two-pronged” approach, directly targeting cancer cells themselves while also targeting the bone microenvironment to make it inhospitable to malignant cells and, ultimately, eradicating cancer stem-like cells.
Introduction
Interest in the leukemic stem-like cell (LSC) niche in the bone marrow (BM) developed due to the major advances made in the understanding of the normal hematopoietic stem/progenitor cell (HSPC) niche over the last 15 years. Given that leukemia does not propagate just anywhere in the body and is difficult to grow ex vivo, it was thought that leukemia cells depend on the bone marrow microenvironment (BMM). Furthermore, it was also thought that LSC interact with the BMM in ways that may affect both the LSC and the BMM, perhaps modulating the molecular pathways on which normal cells depend. Examining the BMM in leukemia may then provide opportunities for altering it in such a way that it becomes less hospitable to malignant cells.
The compostion of the BMM is complex and includes multiple different cell populations that have been reported to participate in normal hematopoietic stem and progenitor cell support. These include arterioloar and sinusoidal type endothelial cells, osteolineage cells, osteoclasts, osteocytes, adipocytes, sympathetic neurons, non-myelinating Schwann cells, mesenchymal stem cells, Cxcl12-abundant reticular cells, macrophages, megakaryoctyes and the extracellular matrix (Figure 1).1 These elements affect HSPC number, location, proliferation, self-renewal, and differentiation, and likely alter similar parameters of the LSC.2 Whether the BMM participates in the emergence of leukemia is unclear, though experimental support for this has emerged in animal studies reviewed below. Finally, there is the potential for the BMM to offer opportunities therapeutically targeting BMM-LSC interactions to disadvantage malignant cells. These aspects of leukemia biology will be discussed in the context of fundamental niche concepts, as first detailed by Schofield in 1978.3
Figure 1.
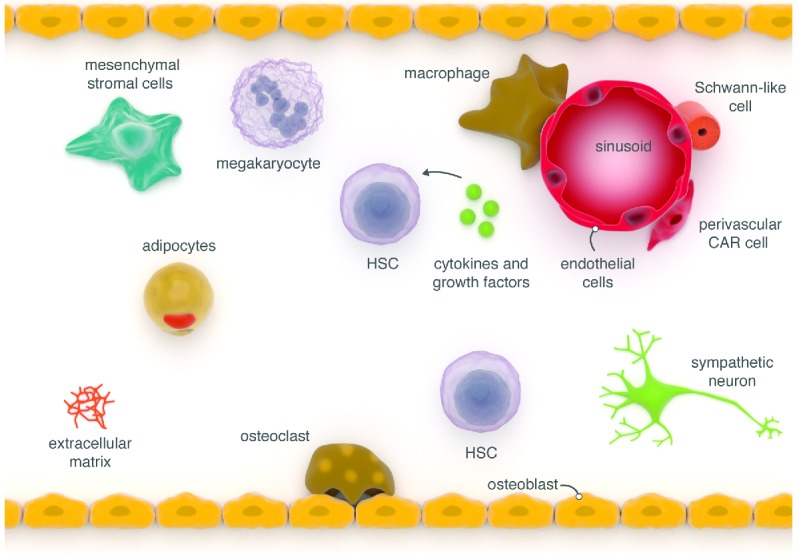
The normal bone marrow (BM) niche. The BM microenvironment is composed of multiple different cell populations that co-ordinately contribute to the regulation of hematopoiesis. The region near the endosteum is highly vascular and thought to be a site where transplanted hematopoietic stem/progenitor cells (HSPC) localize whereas sinusoids in the central portion of the marrow are thought to be the location of most HSPC under homeostasis. CAR cell: CXCL12-abundant reticular cell.
The normal bone marrow HSPC niche
The anatomy and physiology of the normal HSPC niche is still being elucidated, but it is likely that there is heterogeneity among niches as there is heterogeneity among stem cells (Figure 2). Indeed, probably there are niches that serve very different roles in different physiological contexts. For example, debate in the field still continues over the endosteal versus perivascular niche, yet it is likely that both exist. But different niches are important for different functions: the setting of transplantation stress (endosteal) compared with homeostasis (perivascular).1 This review does not aim to reconcile these debates but rather to outline concepts and pathways that are important for the maintenance of LSC in the BMM.
Figure 2.
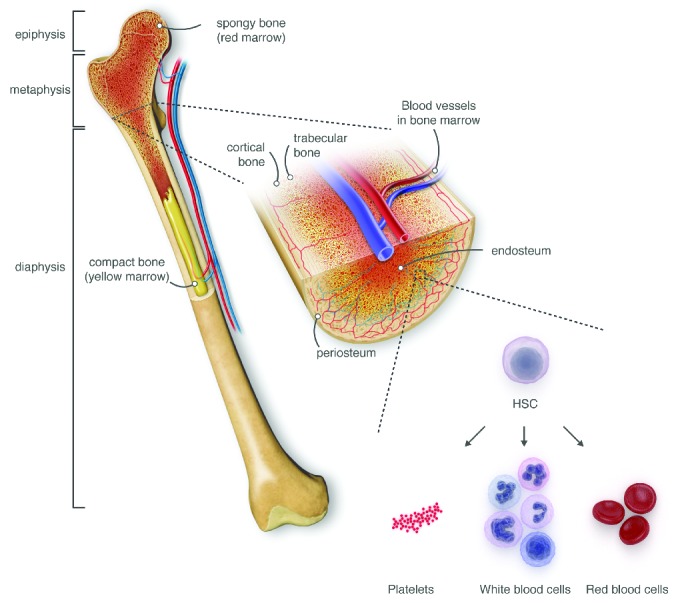
Bone marrow (BM) anatomy. The normal bone marrow anatomy (here using the example of the femur) is composed of different types of bone, blood vessels and red and yellow marrow. HSPC reside in the red marrow where they differentiate into red blood cells, white blood cells and platelets via different progenitor stages (not shown). Yellow marrow represents largely adipocyte-rich regions with minimal hematopoiesis.
The concept that vascular structures support HSPC has long been proposed and is in keeping with the growing idea that definitive hematopoiesis and establishment of a HSPC pool exists well before bone or bone marrow formation. Experimental evidence for vascular regulation of hematopoiesis was provided by the demonstration of hematopoietic regeneration occurring at sites of BM sinusoidal vascular regeneration.4 Several in vitro5,6 and in vivo7,8 models support the notion that sinusoidal endothelial cells regulate hematopoiesis, in part by soluble factors9,10 and by anatomic proximity of CD150+ CD244− CD48− lineage-(SLAM marker+) HSC to BM sinusoidal vessels.11 Osteoblastic cells were demonstrated to support hematopoiesis in ex vivo culture systems.12 Evidence in vivo was provided by two independent studies using transgenic mice with osteoblast-specific, constitutively activated receptors for parathyroid hormone (PTH) and PTH-related peptide and mice with conditional inactivation of bone morphogenetic protein (BMP) receptor type IA (BMPRIA). In these studies, it was respectively demonstrated that a PTH-induced increased number of osteoblastic cells13 and an increase in the number of spindle-shaped N-cadherin+ CD45− osteoblastic (SNO) cells14 was associated with an increase in HSPC number. Conversely, the ablation of developing osteoblastic cells by conditional expression of thymidine kinase and cell killing using ganciclovir, led to a loss of progenitors of the lymphoid, erythroid and myeloid lineages.15 These were the first in vivo demonstrations of specific niche cell participants in a mammalian tissue.
These discoveries were followed by evidence that more immature perivascular mesenchymal stromal cells (MSC) maintained HSC under homeostasis. Nestin-GFP marked MSC were found in close proximity to HSC and adrenergic nerve fibers, and their depletion led to reduction of HSC.16 The majority of HSC were found in the vicinity of cells expressing high amounts of CXC chemokine ligand (CXCL) 12 (CXCL12), called CXCL12-abundant reticular (CAR) cells, which are distributed throughout the BM. Deletion of CXCR4, a receptor for CXCL12, led to a reduction in HSC frequency and increased sensitivity to myelotoxic drugs.17 Cell-restricted deletion of CXCL12 from endothelium or Prx1+ or leptin receptor (leptinR)+ cells resulted in decreased HSC. It should be noted, however, that both studies used models in which the Cre was not inducibly activated. Therefore, Cre was active throughout development and therefore all descendents of Prx1+ and leptinR+ cells including all bone cells could be implicated. This is balanced against the absence of an effect on HSC when osteblastic cell-specific promoter-driven Cre activation was induced.18,19 In complementary studies, it was shown that stem cell factor (SCF) is highly expressed by perivascular cells and that HSC were lost from the BMM if SCF was deleted from endothelial cells or leptin receptor (LEPR)-expressing perivascular stromal cells.20 The same was not true if SCF was deleted from osteolineage or nestin+ cells. However, the recombination efficiency in the different cell types was not reported. Other work demonstrated that quiescent HSC were located close to small arterioles, frequently found in the endosteal area of the BMM and enveloped by NG2+ pericytes. Activation of the cell cycle in HSC led to a redistribution from NG2+ periarteriolar niches to LEPR+ perisinusoidal niches, suggesting that periarteriolar niches are important for HSC quiescence.21 Nestin+ MSC in vivo are located in association with adrenergic neural fibres and HSC, which they support via the secretion of HSC-maintaining factors. The mobilization of HSPC is dependent on circadian oscillations of noradrenaline secretion and fluctuating expression of the chemokine CXCL12, suggesting that the sympathetic nervous system is heavily involved with BMM regulation.16
Blood cells themselves have been shown to be important in the BMM. Depletion of macrophages in Macrophage Fas-induced apoptosis (Mafia) transgenic mice or via administration of liposomes containing clodronate to normal mice led to loss of macrophages, reduction of HSC-supportive cytokines at the endosteum, and mobilization of HSC into the peripheral blood.22 In addition, loss of bone marrow macrophages enhanced mobilization induced by a CXCR4 antagonist or granulocyte colony-stimulating factor.23
Another important component of the BMM is the extracellular matrix (ECM). An early work showed that heparin sulfate, a protein produced by marrow-derived stromal cells of undefined phenotype and found in the ECM of the bone marrow, is important for the adhesion of hematopoietic blast colony-forming cells to the ECM.24 Consistent with this, inhibition of heparan sulfate led to mobilization of more and more functionally competent HSC.25,26 Mice deficient for tenascin-C, another ECM protein normally expressed by stromal and endothelial cells, were unable to reconstitute hematopoiesis after myeloablation or support hematopoiesis after transplantation.26 Furthermore, osteopontin, secreted by osteoblastic cells and other cell types, was found to negatively regulate HSC number and function through influencing the expression of Jagged1 and Angiopoietin-1.27,28
Oxygen tension within a given environment was thought to regulate stem cell behavior, but methods to directly study the distribution of oxygen in the BMM have only recently become available. The bioprobe pimonidazole measures reducing intermediates and in conjunction with in vivo microscopy suggested that the endosteal region is hypoxic.29 HSPC with pimonidazole retention and increased expression of hypoxia-inducible transcription factor-1α (HIF-1α) consistent with a hypoxic state preferentially localized in endosteal zones.30 Using a Hoechst 33342 perfusion gradient to assess perfusion, HSC were found in areas of the BMM with the lowest Hoechst level. Furthermore, the hypoxic cytotoxic agent tirapazamine was selectively toxic to HSC.31 Direct measurement of the local oxygen tension (pO2) using two-photon phosphorescence lifetime microscopy showed that the lowest (pO2 ~9.9 mm Hg) measurements were in perisinusoidal regions with the arteriole-rich endosteal region being less hypoxic. Cytotoxins increased pO2, suggesting that oxygen consumption accounts for the relatively low ambient oxygen level in the central bone marrow.
Alterations of the BMM can lead to hematologic abnormalities
Based on findings in the normal bone marrow, alteration of the BMM was tested and found to lead to hematologic abnormalities. Mice with deficiency of retinoic acid receptor (RAR) γ are characterized by an increase in granulocytic/macrophagic progenitors and increased granulocytes in peripheral blood, bone marrow and spleen. However, when wild-type bone marrow was transplanted into these mice, the recipient mice, also developed this myeloproliferation. These data suggest that the myeloproliferation was not intrinsic to the hematopoietic cells, but arose due to alterations in the microenvironment. Increased levels of tumor necrosis factor α (TNFα) in the RARγ-deficient BMM conributed to the myeloproliferation.32 When retinoblastoma protein (Rb) was deleted in the hematopoietic system, a similar myeloproliferation was found and HSCs were lost from the BMM due to mobilization; but these findings were dependent on Rb deletion in both hematopoietic cells and their BMM, as deletion in either one alone was not sufficient to create the phenotype.33 Similarly, fatal microenvironment-dependent myeloproliferation also developed in mice with inactivation of mind bomb 1 (Mib1), a molecule necessary for the processing and presentation of Notch ligands.34 In mice with osteocyte-specific deletion of the Gsα subunit of the G-protein signaling cascade, a signaling molecule downstream of the receptors for parathyroid hormone and prostaglandin, and certain β-adrenergic receptors myeloproliferation also developed. In the latter example, the myeloproliferation was due to increased GCSF secretion from mutated osteocytes.35 The hyperproliferation in these examples was not accompanied by hematopoietic cell transformation or evidence of abnormal differentiation.
In constrast, osteoprogenitor cell-specific disruption of Dicer1, required for microRNA processing, led to a myelodysplasia-like syndrome. Furthermore, few mice developed acute myeloid leukemia (AML) with new complex genetic abnormalities that did not involve Dicer1.36 These data are the first evidence that a specific distinct stromal cell type, in this case an osteolineage cell, can be the inciting abnormality that eventually leads to malignancy in a distinct, parenchymal cell. Subsequently, it has been reported that mutations activating β-catenin in osteoblasts in mice resulted in AML with high penetrance in association with increased Notch activation. These investigators also found increased β-catenin signaling in osteoblastic cells and increased Notch signaling in hematopoietic cells in 38% of patients with myelodysplastic syndromes (MDS) or AML.37 Taken together, these findings suggest that an altered microenvironment can serve as the inciting event in hematologic neoplasia.
The phenomenon of donor-derived leukemia in patients receiving allogeneic transplantation is uncommon but may represent a dysfunctional microenvironment sufficient to rapidly induce neoplasia.38 Along these lines, studies have reported genetic abnormalities in bone marrow stromal cells, isolated via adherence to plastic, from patients with MDS or AML, although whether these can cause disease has still not been proved.39,40
The LSC niche
Adhesion to the LSC niche
In 2006, the important role of the cell-surface glycoprotein CD44, which binds the extracellular matrix proteins hyaluronan, osteopontin,41 and possibly E-selectin,42,43 was investigated for the interaction between AML44 and CML45 cells with their BMM. Repopulation by human AML cells in non-obese diabetic-severe combined immune-deficient (NOD/SCID) mice treated with an activating antibody to CD44 was decreased, likely due to defective transport of LSC to supportive niches.44 Others showed that high levels of CD44 were important for AML induction or relapse in mouse models of AML.46
In CML, CD44 expression was shown to be increased and to contribute functional E-selectin ligands on CML-initiating cells. Lack of CD44 ed to reduced homing and engraftment of CML-initating cells in the well described retroviral model of CML-like myeloproliferative neoplasia.47,48 It also led to decreased efficiency of induction of CML-like myeloproliferative neoplasia in wild-type recipients. CD44, in contrast, was shown to be dispensable for the induction of B-cell acute lymphoblastic leukemia.45 In support of these data, lower levels of soluble CD44 in autologous hematopoietic stem cell grafts from patients with AML, plasma cell myeloma and non-Hodgkin lymphoma, shed from the surface of malignant cells, correlated with improved outcome after autologous hematopoietic stem cell transplantation.49 Furthermore, treatment of humanized RAG2-/-gc-/-mice transplanted with human CD34+ blast crisis CML cells with a CD44 monoclonal antibody alone or in combination with the tyrosine kinase inhibitor dasatinib led to a reduction of human progenitor cells in niches and a significant reduction of LSC self-renewal capacity upon serial transplantation.50 Polymeric nanoparticle-mediated silencing of CD44 decreased surface levels of CD44 in AML cells, induced apoptosis and reduced adhesion of AML cells to mesenchymal stem cells from reamed human bone marrow.51 In addition, treatment with a combination of anti-CD44 (and particularly anti-CD44v1052) and anti-CD49d antibodies dislodged CD44-expressing lymphoid cell lines, EL4 and Jurkat, in mouse bone marrow and spleen, leading to increased sensitivity to chemotherapy.53 These data strongly support targeting CD44 as a means of compromising myeloid and possibly acute lymphoid malignancies, though current clincial trials are only targeting CD44 in terms of a cancer stem cell marker in solid tumors.
The importance of specialized niches for the engraftment of tumor cells in the BMM was shown by dynamic in vivo confocal imaging. The adhesion molecule E-selectin and stromal-derived-factor 1 (SDF-1) are expressed in certain domains in the endothelium of the BMM and represent the sites at which circulating leukemic cells, a B-ALL cell line, as well as normal HSPC and lymphocytes can engraft.54 Functionally, a critical role of selectins and their ligands was shown for the engraftment of CML-initiating cells.55 Also, inhibition of E-selectin with a small molecule antagonist, GMI1271, reduced the number of CML LSC.56 Finally, E-selectin was reported to be over-expressed on bone marrow endothelium in AML and the absence of E-selectin markedly increased sensitivity of AML to cytarabine.57
Other adhesion molecules, such as CD82 on CD34+ CD38− AML cells, were shown to inhibit adhesion to fibronectin and impaired engraftment of AML cells in immunodeficient mice.58
In the 1990s, significant in vitro work demonstrated a role for β1-integrins in the adhesion of CML cells to fibronectin. An activating antibody to α5β1, for example, led to increased adhesion of leukemia cells to fibronectin and decreased their proliferation.59 Interestingly, interferon α restored β1 integrin-mediated adhesion of CML to stromal cells of undefined phenotype inibiting proliferation of CML cells.60,61 Similarly, β1 integrins may play a role in AML, as increased binding of soluble vascular cell adhesion molecule (VCAM)-1 via the β1 integrin VLA-4 was associated with longer overall survival in patients.62
Understanding of the pathways by which leukemia cells interact with their BMM (Figure 3) will continue to be of great interest, as these pathways may be targetable and thereby make the LSC more sensitive to chemotherapy and other agents.
Figure 3.

Leukemia stem cells. Leukemia stem cells (LSC) in acute myeloid leukemia (AML; red), chronic myelogenous leukemia (CML; dark blue), B-cell acute lymphoblastic leukemia (B-ALL; green), chronic lymphocytic leukemia (CLL; pale blue), myelodysplastic syndrome (MDS; orange) and JAK2 V617F positive myeloproliferative neoplasia (MPN; yellow) interact with their bone marrow microenvironment via specific pathways. Specific details are provided in the text. SDF-1α: stromal-derived factor 1α; BMP: bone morphogenetic protein; BMPR: bone morphogenetic protein receptor; Gas6: growth-arrest-specific-gene 6; VEGF (A): vascular endothelial growth factor (A); SCF: stem cell factor; IL-8: interleukin-8; PlGF: placental growth factor; TPO: thrombopoietin; LIF: leukemia-inhibitory factor; PDGF: platelet-derived growth factor; Ang2: angiopoietin2; TGFβ: transforming growth factor β.
Bi-directional interactions between leukemia cells and their niche
There are several examples of the BMM influencing leukemia. For example, depletion of osteoblastic cells in the BMM of mice with AML worsened the disease with increased circulating blasts, higher tumor burden in bone marrow and spleen, and shorter survival. On the other hand, preserving osteoblastic cells by an inhibitor of serotonin production restored normal bone marrow function, reduced tumor burden and prolonged survival.63 Furthermore, mixed lineage leukemias arising due to the MLL-AF9 fusion adopt differing lineage fates, depending on the cytokine milieu in which the cells were treated in vitro prior to injection or by altering the strain of mice.64 Preliminary data generated by in vivo imaging suggest that BCR-ABL1+ LSC may be found further away from the endosteum than normal HSC, yet this was reversed if the BCR-ABL1+ LSC had previously been treated with imatinib. This may be due to upregulation of CXCR4,65 or possible harboring of the T315I mutation conferring imatinib resistance.66 And, finally, as described below, modulation of the BMM with parathyroid hormone led to opposite changes of the phenotype in CML versus AML.67
Just as the BMM may influence leukemia, a leukemia can shape the BMM. In a murine model of AML, osteoblastic cells were reduced and inhibited by the leukemia, as measured by decreased levels of the bone formation marker, osteocalcin. Mineralized bone was also lost. The cytokine CCL3, known to be involved in bone loss, was increased in AML cells in mice and humans suggesting that CCL3 (macrophage inflammatory protein 1α; MIP 1α) may be involved in the observed phenotype.68 It is tempting to speculate that inhibition of this “molding” of the niche by leukemia cells, which likely shapes the BMM in a way most beneficial to surivival of the leukemia, but most detrimental to the survival of normal HSPC, would have a negative impact on leukemia progression. Future research endeavors may be aimed at this.
In a transgenic model of BCR-ABL1+ CML, the endosteal BMM was reported to be remodeled into a leukemic niche. Multipotent murine stromal cells were stimulated by leukemia cells in vitro to produce functionally abnormal osteoblastic cells impaired in their capacity to generate HSC-retaining and –maintaining factors. Thrombopoietin, CCL3, and direct cell-cell interactions were found to be involved in promoting the expansion of osteoblastic cells, whereas TGF-β, Notch, and inflammatory signaling were found to play a role in the remodeling of the osteoblastic niche.69 Long-term LSC in CML exhibited reduced homing and retention in the bone marrow, associated with increased granulocyte colony-stimulating factor (G-CSF) produced by leukemia cells and decreased levels of CXCL12 in the BMM,70 as was observed in pediatric B-ALL.71 Altered expression of cytokines in the murine CML BMM favored the growth of LSC versus normal HSC, which was partially restored by treatment with imatinib.70 Ablation of osteoblastic cells in the same model of CML led to an acceleration of the leukemia phenotype and reduced survival compared to the control mice. The CML BMM is also thought to over-express the Notch ligand Jagged-1, affecting hematopoietic cell cycling.72
In the perivascular niche, autocrine and paracrine secretion of vascular endothelial growth factor (VEGF)73 and other factors by leukemia cells led to proliferation of leukemia and endothelial cells in ALL74 and AML.75 Neo-angiogenesis is a well-established phenomenon observed in AML76 and ALL,77 but, mechanistically, it has not received much attention, though it may provide another Achilles’ heel in the treatment of leukemia.
Mesenchymal stromal cells derived from bone marrow stroma have also been shown to play a role in the LSC niche. In a xenotransplantation model of transplanted MDS-initiating Lin− CD34+ CD38− stem cells, only orthotopic co-transplantation of patient-derived MSC, isolated via adherence to plastic, which are characterized by an altered differentiation pattern, gives rise to MDS-like disease in murine recipients. Factors such as N-Cadherin, insulin-like growth factor binding protein 2, VEGFA, and leukemia inhibitory factor increase the ability of MSC in MDS to promote MDS and may be involved in altering molecular expression patterns in healthy MSC when co-cultured with hematopoietic MDS cells.78
In another report, levels of placental growth factor (PlGF), secreted by bone marrow stromal cells positive for the osteogenic/fibroblastoid markers Runx2, Col1a1 and alkaline phosphatase upon stimulation by the leukemia cells themselves, were elevated in CML. PlGF led to stimulation of BM angiogenesis, promotion of CML proliferation and metabolism, thereby contributing to disease aggressiveness.79
In a murine model of JAK2 V617F+ myeloproliferative neoplasia abrogation of the regulatory innervation of the BMM by sympathetic nerve fibres was shown to be an essential component of the pathogenesis of MPN. In patients and mice with MPN sympathetic nerve fibres, supporting Schwann cells ensheathing the neural fibres and nestin+ MSC were reduced in the BMM. The loss of MSC and neural damage were due to the production of interleukin-1β by malignant HSC, and depletion of nestin+ cells led to acceleration of MPN. However, treatment of mice with β3-adrenergic agonists as sympathomimetic agents restored the loss of nestin+MSCs and blocked MPN progression via an indirect reduction of MPN cells.80 These findings have led to initiation of a clinical trial, as described below. In AML, leukemia-induced impairment of the sympathetic nervous system in the BMM led to an increased infiltration of the BMM with leukemic cells in a murine model of MLL-AF9+ AML. AML development altered the quiescence of Nestin+ niche cells, favoring the differentiation of mesenchymal stem and progenitor cells towards osteoblastic cells, while not favoring the periarteriolar niche cells known to support HSC. Leukemogenesis was promoted by the β2 adrenergic receptor expressed on stromal cells in the BMM, suggesting that the sympathetic nerve system may be “hijacked” by the malignant cells in order to promote and advance the neoplasm.81
Just as in the normal HSC niche, hypoxia may play a role in the leukemic BMM. Expression of HIF-1 α, up-regulated by hypoxia and other factors, though this correlation may be controversial, was associated with a negative prognostic impact on survival of patients with pre-B-cell ALL. Stroma-mediated AKT/mTOR signaling further induced HIF-1 α, while blockade of mTOR restored the chemosensitivity of B-ALL cells.82 In a murine model of CML, HIF-1 α is involved in regulating cell cycle propagation and apoptosis and, therefore, promotes LSC survival via increased expression of p16Ink4a and p19Arf in LSCs.83 Hypoxia also led to decreased apoptotic rates and increased clonogenicity and repopulating efficiency of BCR-ABL1+ cells, while knockdown of HIF1-α reversed the enhanced clonogenicity during hypoxic conditions.84 High expression of HIF-1α is found in AML1-ETO+ AML cells and is associated with inferior outcomes while co-expression of AML1-ETO and HIF1α leads to an increased cellular proliferation rate in vitro and more aggressive leukemia in mice.85 Knockdown of HIF-1 α and the use of the HIF inhibitor echinomycin abrogated the colony-forming unit activity and eradicated LSC in human AML, as tested by serial transplantation assays in xenotransplants.86 Deregulation of HIF-2 α, which impaired the long-term repopulating ability of human CD34+ cells, impaired the engraftment of human AML, suggesting that HIF-2 α also affects AML cell interactions with the BMM.87 These studies suggest that hypoxia with deregulation of HIF1-α, and possibly HIF2-α, alters LSC function, including chemoresistance. However, the hypoxia field in the LSC (and HSC) niche, the role of HIF-1 in hypoxia, as well as the location of LSC (and HSC) in relation to hypoxic areas in the BMM all remain a subject of scientific debate and warrant further research.
Exosomes
Exosomes are lipid vesicles or microparticles (<1 μm) secreted by cancer cells, MSC, macrophages, dendritic cells, B and T cells, mast cells and endothelial cells.88 They are associated with the bidirectional transfer of mRNAs, microRNAs, drug efflux transporters and other proteins between cancer and stroma cells leading to alteration of gene expression in neighboring cells.89 They may be a means by which a cancer modulates its environment. It was demonstrated that primary leukemia cells and leukemia cell lines release microvesicles containing coding and non-coding RNAs that enter cells of the microenvironment, altering their secretion of growth factors and reprogramming the LSC niche.90,91 Co-culture of a CML cell line with endothelial cells led to exosomes containing the microRNA miR-126 being shuttled into the endothelial cells, led to reduced expression of VCAM-1 and CXCL12 in endothelial cells, and decreased motility and adhesion of LAMA84 cells. Transfection of a miR-126 inhibitor into the endothelial cells reversed these effects.92 Treatment of LAMA84 cells or mice xenografted with human CML cells promoted the colony-forming ability of LAMA84 cells in methylcellulose and tumor size, respectively, due to an increase in the anti-apoptotic molecules BCL-w, BCL-xl, and survivin, and a reduction of the pro-apoptotic molecules BAD, BAX and PUMA in the leukemia cells.93 Exosomes appear to be a means by which cancer cells communicate with each other and their environment, though their role has not yet been fully elucidated. Exactly how exosomes are formed, how they incorporate their “message”, and how this may be functionally blocked represent important issues for further research.
The LSC niche in B-cell acute lymphoblastic leukemia
The niche in B-ALL appears to be distinct from the LSC niches in AML and CML, possibly reflecting differences in myeloid and lymphoid niches of normal cells.32,35,94
The metabolomic95 and functional abnormalities of niche cells from patients with B-ALL have long been known.96,97 For example, some Philadelphia chromosome-positive (Ph+) ALL cells coexpress markers of endothelial cells (VE-cadherin and others) and B-lineage progenitors (Ph+ VE-cadherin+).98 E2A/PBX1-positive B-cell precursor ALL interacts with the BMM through Gas6/Mer with Gas6 promoting survival of ALL cells and preventing their apoptosis after chemotherapy.99
Acute lymphoblastic leukemia blasts adhere to osteopontin secreted by osteoblastic and ALL cells. Inhibition of osteopontin led to better eradication of B-ALL cells in synergism with Ara-C.100 The plasma levels of the chemokines CCL2 and IL-8 were increased in children with B-ALL due to stimulation of bone marrow stromal cells by leukemia cells. CCL2 and IL-8 promoted the adhesion of CD105+ CD29+ CD44+ CD14− CD34− and CD45− MSC generated via adherence to plastic, to B-ALL cells, and enhanced the survival and proliferation of MSC.101 Similar to the role of the SDF-1α/CXCR4 axis in AML, CXCR4 mediates homing of B-ALL cells to the BMM, as evidenced by inhibition with a small molecule antagonist of CXCR4. Furthermore, the combination of nilotinib, a second-line tyrosine kinase inhibitor, or vincristine with a CXCR4 antagonist prolonged survival or reduced leukemia burden in murine models.102
An interesting pathway employed by B-ALL cells is the β1-integrin/focal adhesion kinase (FAK) pathway. FAK is a non-receptor tyrosine kinase constitutively active in BCRABL1+ B-ALL cells. Integrin α5β1 (VLA-5) is more highly expressed on B-ALL cells after serum starvation, and its inhibition leads to decreased adhesion of Ph+ leukemia cells to fibronectin and synergistically induces apoptosis in conjuntion with imatinib. Various strategies to block the activity of integrin led to impaired engraftment of leukemia cells in xenotransplantation experiments.103 BCR-ABL1+ B-ALL, positive for the Ikaros (IKZF) mutation, are associated with a worse prognosis in patients, led to a more aggressive phenotype in mice, and are characterized by chemoresistance to tyrosine kinase inhibitors in association with a stromal adhesion phenotype. Treatment with the FAK inhibitors VS-4718 and VS-6063 abolished stromal adhesion of Ikzf1-mutant B-ALL cells and induced apoptosis, but had virtually no effect on Ikzf1 WT B-ALL cells. In combination with dasatinib, VS-4718 and VS-6063 decreased the viability of Ikzf1-mutant BCRABL1+ B-ALL cells from mice and human patients cultured on stroma.104 Inhibiting FAK may be a means of impairing B-ALL interaction with the BMM and has been introduced into clinical trials in solid tumors and B-ALL.
Chemoresistance mediated by the BMM in leukemia
Several mechanisms leading to chemoresistance in leukemia cells mediated by the BMM have been described. LSC in AML home to and engraft within the endosteal, osteoblast-rich area of the bone marrow in newborn NSG mice. AML cells appeared to be protected from apoptosis induced by chemotherapy at that site.105 In a murine model of ALL, and in human patients, small foci of leukemia-propagating cells remained after chemotherapy. These persisting leukemia cells secreted cytokines leading to the recruitment of Nestin+, mostly Vimentin+ and partially α-smooth muscle actin+ stromal cells and the formation of a protective niche. The niche itself provided the protease Furin, in order to process the growth differntiation factor 15 (GDF15), a member of the transforming growth factor β superfamily, which conferred chemoresistance on the leukemia-propagating cells.106 The tumor microenvironment also inhibits the attack of tumor cells, which have been targeted by antibodies, by macrophages and macrophage-mediated killing. Cyclophosphamide, however, a frequently used chemotherapeutic agent, induced a secretory phenotype in ALL cells leading to the release of CCL4, IL8, VEGF, and TNFα from treated tumor cells, inducing infiltration and phagocytosis by macrophages in the BMM.107 These data suggest that the choice of the chemotherapeutic regimen may influence the immunological homeostasis in the BMM. The interaction between vascular cell adhesion molecule 1 (VCAM-1) expressed in the bone marrow micorenvironment and the β1 integrin very late antigen-4 (VLA-4) expressed on leukemia cells was shown to play an important role in the activation of NF-κB and chemoresistance in leukemia cells in the BMM.108 Another protein known to mediate chemoresistance in CML109 and ALL110 is Galectin-3 (Gal-3), a lectin with the ability to bind β-galactosides. Co-culture of 5 CML cell lines with HS-5 stromal cells induced the expression of Gal-3 in the leukemic cell lines. The signaling pathways activated by enforced expression of Gal-3 induced resistance to BCRABL1-specific tyrosine kinase inhibitors, as well as chemotherapeutic agents due to increased leukemia cell proliferation and decreased induction of apoptosis. In vivo overexpression of Gal-3 led to enhanced lodgement of leukemia cells in the BMM.109 Treatment of CML cells with a tyrosine kinase inhibitor (TKI) promotes migration towards stromal cells, characterized by their adhesion to plastic, in vitro111 and towards osteoblastic cells in vivo66 where it promotes resistance to imatinib via redistribution of CXCR4 into the lipid raft fraction in the cell membrane.111
Altered expression of c-Myc112 or activation of Stat3113 by the BMM have been reported to affect chemosensitivity of AML and CML, respectively. Co-culture experiments with human mesenchymal stromal cells and CML led to the identification of N-cadherin receptor as a mediator of stroma-induced resistance to TKIs. N-cadherin-mediated adhesion to MSCs, previously adhered to plastic, increased the nuclear translocation and transcriptional activity of β-catenin, thereby linking Wnt-mediated β-catenin signaling with protection of leukemia cells from TKI therapy by the BMM.114 In myeloproliferative neoplasia positive for the JAK2(V617F) mutation, treatment with the potent JAK2 inhibitor atiprimod only inhibited the growth of the malignant cells in in vitro experiments in the absence of stroma, which was shown to be due to the secretion of various cytokines by the stromal cells in co-culture experiments.115
Modeling the LSC niche in 3D
Much effort has been made to model the LSC niche in 3 dimensions. In 2009 a “biomimetic osteoblast niche” was constructed using bio-derived bone as a scaffold. On this scaffold, MSC from CML patients were seeded and differentiation into osteoblastic cells was induced. Compared to cultures in a 2-dimensional system, the 2-dimensional system yielded a higher number of CD34+ and CD34+ CD38− cells, higher numbers of colony-forming units and a higher number of long-term culture-initiating cells.116 Another strategy was the creation of ectopic niches using polyurethane scaffolds that had been coated with human MSC. Upon subcutaneous implantation of these scaffolds, which, eventually, became vascularized and showed the presence of osteoclasts and adipocytes, into NOD/SCID mice, engraftment of primary human AML cells was supported.117 Furthermore, the leukemia cell lines K562, HL60 and Kasumi-6 were shown to grow best on polyurethane scaffolds that had been precoated with collagen type I compared to scaffolds made of other materials.118 Decellularized matrices have been employed for the expansion of hematopoietic progenitor cells from umbilical cord blood119 and a mixture of human MSC, generated via prior adhesion to plastic, endothelial colony-forming cells and matrigel has been implanted subcutaneously into immunodeficient mice, leading to engraftment of normal hematopoietic cells and leukemic cells in one study,120 and in hypoxic conditions in another study.121 3D models used to test drug resistance have demonstrated their superiority over 2D models, as leukemic cells cultured in 3D exhibited greater resistance to drug-induced apoptosis compared to cells cultured 2-dimensionally or in suspension, and differences were observed in the response of leukemia cell lines to chemotherapy when cultured in a 3D system.122 Improved modeling of the LSC (and HSC) niche may help improve pre-clinical assessment of drug response. However, controversy still remains as to the most suitable composition of the scaffold. Furthermore, most 3D scaffolds are inefficient at modeling the physico-chemical properties of the niche, such as pH, shear forces, oxygen and cytokine gradients.
Targeting the LSC niche
Most of the current strategies to target the BMM in leukemia involve impairment or inhibition of the CXCR4/SDF1α axis. The CXCR4 inhibitor AMD3100 arrested the proliferation of AML cell lines and initiated changes suggesting differentiation123 and in a murine model of acute promyelocytic leukemia, co-treatment with the CXCR4 inhibitor AMD3100 and chemotherapy led to a decreased tumor burden and prolonged survival.124 Similarly, the effects of the FLT3 inhibitor lestaurtinib, the BCR-ABL1-targeting tyrosine kinase inhibitor nilotinib or vincristine were enhanced in various models of ALL when combined with an antagonist of CXCR4.102,125 These results may be due to altered localization in the bone marrow. A clinical trial employing an antibody to CXCR4 in AML (clinicaltrials.gov identifier:01120457) or a peptidic inhibitor to CXCR4 in CML (clinicaltrials.gov identifier: 02115672) are in progress or recruiting, respectively.
An RNA oligonucleotide in L-configuration, the Spiegelmer NOX-A12, neutralized CXCL12, leading to decreased chemotaxis of CLL cells and increased sensitivity to chemotherapy.126 But in the retroviral model of BCRABL1-induced CML, combination of AMD3100 with dasatinib led to improved control of disease; however, this was at the price of an increased rate of symptoms due to involvement of the central nervous system by leukemia cells.127 This suggests that the combination of CXCR4 antagonism with tyrosine kinase inhibitors or chemotherapy may be beneficial in the setting of minimal residual disease.
Other strategies to inhibit the LSC niche are encouraging (Figure 4). Using a murine transduction/transplantation model of CML and recipient mice with osteoblastic-cell specific activation of the receptor for PTH- and PTH-related peptide, it was demonstrated that modulation of the BMM can lead to an alteration of the leukemia phenotype, as these transgenic recipient mice died significantly later than the wild-type controls, and the incidence of CML was reduced. Even treatment of wild-type mice with PTH led to a 15-fold reduction of LSC, as measured by limiting dilution analysis in secondary recipients of saline- versus PTH-treated BCR-ABL1+ donor bone marrow. Upon extension of these studies to the xenotransplant setting, it was shown that treatment of NOD SCID IL2Rγ−/− (NSG) mice with PTH led to significantly decreased engraftment of human BCR-ABL1+ cells. It was shown that PTHinduced bone remodeling led to a release of su-praphysiological levels of TGFβ1 from the bone matrix suppressing the growth of CML cells, which express the receptor for TGFβ1. However, the same was not applicable to AML, suggesting that the niches in these two myeloid leukemias differ significantly.67 A clinical trial is being initiated in eight centers in Germany in which patients who have been receiving a tyrosine kinase inhibitor for at least six months will also receive PTH, in order to test if any remaining transcript levels of BCR-ABL1 can be rendered undetectable.
Figure 4.

Leukemia stem cell targeting. Depending on the disease entity certain therapies (red) may specifically target the interaction of leukemic stem cells with their bone marrow niche. As detailed in the text, some of these novel therapies have already entered clinical trials. FAK: focal adhesion kinase. Other abbreviations as in Figure 3.
In the vascular niche in AML57 and CML56 preliminary data show that treatment with a small molecule inhibitor of E-selectin, which leads to an increase of HSC quiescence and self-renewal in the normal HSC niche,128 may lead to reduction of LSC. In AML, a clinical trial using the pan-E-selectin inhibitor GMI-1271 has been initiated (clinicaltrials.gov identifier 02306291). Based on findings described above, the sympathomimetic agent mirabegron is being used in a clinical trial testing its efficacy in Jak2+ myeloproliferative neoplasia (clinicaltrials.gov identifier 02311569) and patients with B-ALL are currently being recruited for a clinical trial testing the efficacy of the FAK inhibitor VS-4718 (clinicaltrials.gov identifier 02215629). The inhibition of Axl and its interaction with Gas6, secreted by stromal cells with fibroblastic/mesenchymal morphology in the BMM, and abolishment of the chemoprotective niche established by the Gas6-Axl paracrine axis in AML129 or inhibition of CCL3, as mentioned above, in conjunction with chemotherapy may prove a feasible strategy for targeting the niche. The Bruton tyrosine kinase (BTK) inhibitor PCI-32765 inhibits CXCL12-mediated migration and α4β1 integrin-mediated adhesion to fibronectin and VCAM-1 of lymphoma cell lines and primary CLL cells.130 Other modalities of treatment resulting from mechanisms involved in the interaction of leukemia cells with their niche stated above, are inhibition of receptors for stromal-derived cytokines, such as inhibition of placental growth factor (PlGF)/VEGFR1 signaling or inhibitors of adhesive interactions of CML stem cells with stroma, including inhibition of N-cadherin–mediated activation of Wnt-β-catenin114 or β1 integrin-mediated adhesion.104,108 An exciting and novel idea would be to inhibit leukemia-mediated “molding” of the niche, for example in the form of inhibition of exosome-mediated changes such as neovascularization in the BMM. In this respect, carboxyamidotriazole-orotate may have properties targeting both the cancer cells directly, as well as the tumor microenvironment, though this needs to be tested in further studies.131 Furthermore, it was recently shown that cytokines such as IL-6, secreted by CML cells, modulate normal HSPC leading to their increased proliferation, altered differentiation ability, and reduced potential for repopulation and self-renewal. This effect, which also led to a similar gene expression pattern in normal and malignant cells, could be efficiently blocked by anti-IL6 therapy and, thereby, treated the disease.132 In MPN, similarly, it has recently been established that malignant and non-malignant cells equally produce inflammatory cytokines, which signal via the Jak-Stat3 pathway and lead to splenomegaly and systemic symptoms; effects which are efficiently inhibited by inhibitors of the Jak kinase.133 This suggests that targeting of the cytokines secreted by hematologic malignancies, which influence their microenvironment to promote the survival of malignant cells, may be another feasible strategy to improve current therapies.
Finally, progress has been made in modeling the interaction of the BMM with LSC in vitro to test for new inhibitory compounds. Using an in vitro stromal co-culture with either LSC or their normal counterparts, a sufficiently robust screening platform was generated with which over 14,000 compounds could be tested; some that would not have been found using leukemia cell lines alone.134 By defining compounds capable of inhibiting LSC but not normal cells or stroma, compounds that were validated in human samples, and some that were active strictly in the presence of stroma, were defined. A clinical trial to test for at least one result is being planned. Therefore, the interplay between LSC and the BMM might be used for direct targeting or it may be used for compound discovery, with the view that cells will respond differently in the context of supportive stromal cells.
An outlook on LSC niche biology
Targeting the LSC niche with an effort to augment existing therapies in the form of chemotherapy or therapy with tyrosine kinase inhibitors, other small molecule inibitors or antibodies in order to, ultimately, eradicate LSC is a powerful strategy, which is still not being fully exploited. However, even our knowledge of the normal HSC niche is only recent and is in no way complete, so efforts must first be directed at understanding the pathophysiology of the LSC niche. Only then can treatment modalities modulating the LSC niche be rationally designed. While exciting progress is being made with regards to 3D and mathematical modeling, in vivo imaging of the LSC (and HSC) niche and in vivo models for hypo-thesis testing, understanding the niches is a complex process. Multiple cell types, extracellular matrices, pH, oxygenation status, cytokine networks, cell-cell interactions and mechanical forces all come into play. Understanding niche biology with human leukemia samples is difficult, as diagnoses are primarily made on bone marrow aspirates that disrupt bone marrow architecture. Therefore, xenogeneic models may ultimately prove to be extremely useful.
Importantly, however, the microenvironment is increasingly recognized to be an important component of cancer and its response to therapy. Clinicians and researchers recognize that, even with targeted leukemia therapy, we are frequently not eradicating LSC, a prerequisite for cure of our patients, and that adjuvant therapies are needed. Targeting the tumor microenvironment in leukemia may prove a useful approach to complement existing therapies, increasing our armamentarium against hematologic cancers.
Acknowledgments
DSK receives research funding and salary support from Glycomimetics Inc. and has acted as a consultant for Novartis Oncology. We apologize to our colleagues whose work could not be cited due to space constraints.
Footnotes
Funding
This work was supported in part by the LOEWE Center for Cell and Gene Therapy Frankfurt (CGT) and institutional funds of the Georg-Speyer-Haus to D.S.K. The Georg-Speyer-Haus is funded jointly by the German Federal Ministry of Health (BMG) and the Ministry of Higher Education, Research and the Arts of the State of Hessen (HMWK). The LOEWE Center for Cell and Gene Therapy Frankfurt is funded by HMWK, reference number: III L 4-518/17.004 (2010). Grants U54 CA163191 suppported D.T.S.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature 2014;505(7483):327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krause DS, Scadden DT, Preffer FI. The hematopoietic stem cell niche - home for friend and foe? Cytometry B Clin Cytom. 2013;84(1):7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schofield R. The relationship between the spleen colony-forming cell and the haematopoietic stem cell. Blood Cells 1978;4(1–2):7–25. [PubMed] [Google Scholar]
- 4.Knospe WH, Gregory SA, Husseini SG, Fried W, Trobaugh FE., Jr Origin and recovery of colony-forming units in locally curetted bone marrow of mice. Blood 1972; 39(3):331–340. [PubMed] [Google Scholar]
- 5.Rafii S, Shapiro F, Pettengell R, et al. Human bone marrow microvascular endothelial cells support long-term proliferation and differentiation of myeloid and megakaryocytic progenitors. Blood 1995;86(9):3353–3363. [PubMed] [Google Scholar]
- 6.Davis TA, Robinson DH, Lee KP, Kessler SW. Porcine brain microvascular endothelial cells support the in vitro expansion of human primitive hematopoietic bone marrow progenitor cells with a high replating potential: requirement for cell-to-cell interactions and colony-stimulating factors. Blood 1995;85(7):1751–1761. [PubMed] [Google Scholar]
- 7.Salter AB, Meadows SK, Muramoto GG, et al. Endothelial progenitor cell infusion induces hematopoietic stem cell reconstitution in vivo. Blood 2009;113(9):2104–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brandt J, Bartholemew A, Fortman J, et al. Ex vivo expansion of autologous bone marrow CD34+ cells with porcine microvascular endothelial cells results in a graft capable of rescuing lethally irradiated baboons. Blood 1999;94(1):106–113. [PubMed] [Google Scholar]
- 9.Chute JP, Muramoto GG, Fung J, Oxford C. Soluble factors elaborated by human brain endothelial cells induce the concomitant expansion of purified human BM CD34+CD38- cells and SCID-repopulating cells. Blood 2005;105(2):576–583. [DOI] [PubMed] [Google Scholar]
- 10.Himburg HA, Muramoto GG, Daher P, et al. Pleiotrophin regulates the expansion and regeneration of hematopoietic stem cells. Nat Med 2010;16(4):475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kiel MJ, Yilmaz OH, Iwashita T, et al. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005; 121(7):1109–1121. [DOI] [PubMed] [Google Scholar]
- 12.Taichman RS, Reilly MJ, Emerson SG. Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood 1996; 87(2):518–524. [PubMed] [Google Scholar]
- 13.Calvi LM, Adams GB, Weibrecht KW, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003; 425(6960):841–846. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Niu C, Ye L, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003; 425(6960):836–841. [DOI] [PubMed] [Google Scholar]
- 15.Visnjic D, Kalajzic Z, Rowe DW, et al. Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood 2004;103(9):3258–3264. [DOI] [PubMed] [Google Scholar]
- 16.Mendez-Ferrer S, Michurina TV, Ferraro F, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010;466(7308):829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006;25(6):977–988. [DOI] [PubMed] [Google Scholar]
- 18.Greenbaum A, Hsu YM, Day RB, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. 2013;495(7440):227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013;495(7440):231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012;481(7382):457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kunisaki Y, Bruns I, Scheiermann C, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013;502(7473):637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winkler IG, Sims NA, Pettit AR, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 2010;116(23):4815–4828. [DOI] [PubMed] [Google Scholar]
- 23.Chow A, Lucas D, Hidalgo A, et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med 2011;208(2):261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gordon MY, Riley GP, Clarke D. Heparan sulfate is necessary for adhesive interactions between human early hemopoietic progenitor cells and the extracellular matrix of the marrow microenvironment. Leukemia 1988;2(12):804–809. [PubMed] [Google Scholar]
- 25.Saez B, Ferraro F, Yusuf RZ, et al. Inhibiting stromal cell heparan sulfate synthesis improves stem cell mobilization and enables engraftment without cytotoxic conditioning. Blood 2014;124(19):2937–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura-Ishizu A, Okuno Y, Omatsu Y, et al. Extracellular matrix protein tenascin-C is required in the bone marrow microenvironment primed for hematopoietic regeneration. Blood 2012;119(23):5429–5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stier S, Ko Y, Forkert R, et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med 2005;201(11):1781–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nilsson SK, Johnston HM, Whitty GA, et al. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 2005;106(4):1232–1239. [DOI] [PubMed] [Google Scholar]
- 29.Lévesque JP, Winkler IG, Hendy J, et al. Hematopoietic progenitor cell mobilization results in hypoxia with increased hypoxia-inducible transcription factor-1 alpha and vascular endothelial growth factor A in bone marrow. Stem Cells 2007;25(8):1954–1965. [DOI] [PubMed] [Google Scholar]
- 30.Nombela-Arrieta C, Pivarnik G, Winkel B, et al. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol 2013; 15(5):533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parmar K, Mauch P, Vergilio JA, et al. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci USA 2007; 104(13):5431–5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walkley CR, Olsen GH, Dworkin S, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell 2007; 129(6): 1097–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walkley CR, Shea JM, Sims NA, et al. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell 2007;129(6):1081–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim YW, Koo BK, Jeong HW, et al. Defective Notch activation in microenvironment leads to myeloproliferative disease. Blood 2008;112(12):4628–4638. [DOI] [PubMed] [Google Scholar]
- 35.Fulzele K, Krause DS, Barry K, et al. Myelopoiesis is regulated by osteocytes through Gsalpha-dependent signaling. Blood 2013;121(6):930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raajimakers MH, Mukherjee S, Guo S, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010;464(7290):852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kode A, Manavalan JS, Mosialou I, et al. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature 2014;506(7487):240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sala-Torra O, Hanna C, Loken MR, et al. Evidence of donor-derived hematologic malignancies after hematopoietic stem cell transplantation. Biol Blood Marrow Transpl 2006;12(5):511–517. [DOI] [PubMed] [Google Scholar]
- 39.Blau O, Hofmann WK, Baldus CD, et al. Chromosomal aberrations in bone marrow mesenchymal stroma cells from patients with myelodysplastic syndrome and acute myeloblastic leukemia. Exp Hematol 2007;35(2):221–229. [DOI] [PubMed] [Google Scholar]
- 40.Blau O, Baldus CD, Hofmann WK, et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood 2011;118(20):5583–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lesley J, Hyman R, Kincade PW. CD44 and its interaction with extracellular matrix. Adv Immunol 1993;54:271–335. [DOI] [PubMed] [Google Scholar]
- 42.Miyake K, Underhill CB, Lesley J, Kincade PW. Hyaluronate can function as a cell adhesion molecule and CD44 participates in hyaluronate recognition. J Exp Med 1990; 172(1):69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katayama Y, Hidalgo A, Chang J, et al. CD44 is a physiological E-selectin ligand on neutrophils. J Exp Med 2005;201(8):1183–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin L, Hope KJ, Zhai Q, et al. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med 2006; 12(10): 1167–1174. [DOI] [PubMed] [Google Scholar]
- 45.Krause DS, Lazarides K, von Andrian UH, Van Etten RA. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med 2006;12(10):1175–1180. [DOI] [PubMed] [Google Scholar]
- 46.Quéré R, Andradottir S, Brun AC, et al. High levels of the adhesion molecule CD44 on leukemic cells generate acute myeloid leukemia relapse after withdrawal of the initial transforming event. Leukemia 2011;25 (3):515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990;247(4944):824–830. [DOI] [PubMed] [Google Scholar]
- 48.Li S, Ilaria RL, Million RP, et al. The P190, P210, and P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia-like syndrome in mice but have different lymphoid leukemogenic activity. J Exp Med 1999;189(9):1399–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krause DS, Spitzer TR, Stowell CP. The concentration of CD44 is increased in hematopoietic stem cell grafts of patients with acute myeloid leukemia, plasma cell myeloma, and non-Hodgkin lymphoma. Arch Pathol Lab Med 2010;134(7):1033–1038. [DOI] [PubMed] [Google Scholar]
- 50.Holm FL, Hellqvist E, Mason CN, et al. Malignant Reprogramming of Progenitors into Leukemia Stem Cells Is Enhanced By Upregulation of CD44 Transcript Variant 3 in Malignant Microenvironments. Blood (ASH Annual Meeting Abstracts). 2014; Abstract 511. [Google Scholar]
- 51.Gul-Uluda H, Valencia-Serna J, Kucharski C, et al. Polymeric nanoparticle-mediated silencing of CD44 receptor in CD34(+) acute myeloid leukemia cells. Leuk Res 2014;38(11):1299–1308. [DOI] [PubMed] [Google Scholar]
- 52.Erb U, Megaptche AP, Gu X, et al. CD44 standard and CD44v10 isoform expression on leukemia cells distinctly influences niche embedding of hematopoietic stem cells. J Hematol Oncol 2014;7:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singh V, Erb U, Zöller M. Cooperativity of CD44 and CD49d in leukemia cell homing, migration, and survival offers a means for therapeutic attack. J Immunol 2013;191(10): 5304–5316. [DOI] [PubMed] [Google Scholar]
- 54.Sipkins DA, Wei X, Wu JW, et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 2005;435(7044):969–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krause DS, Lazarides K, Lewis JB, et al. Selectins and their ligands are required for homing and engraftment of BCR-ABL1+ leukemic stem cells in the bone marrow niche. Blood 2014;123(9):1361–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aggoune D, Weissenberger E, Magnani JL, Van Etten RA, Krause DS. The vascular niche is involved in regulating leukemic stem cells in murine chronic myelogenous leukemia. ASH Abstract. 2014; #516. [Google Scholar]
- 57.Winkler IG, Barbier V, Pattabiraman DR, Gonda TJ, Magnani JL, Levesque J-P. Vascular Niche E-Selectin Protects Acute Myeloid Leukaemia Stem Cells from Chemotherapy. ASH Abstract. 2014; #620. [Google Scholar]
- 58.Nishioka C, Ikezoe T, Furihata M, et al. CD34/CD38 acute myelogenous leukemia cells aberrantly express CD82 which regulates adhesion and survival of leukemia stem cells. Int J Cancer 2013;132(9):2006–2019. [DOI] [PubMed] [Google Scholar]
- 59.Lundell BI, Mc Carthy JB, Kovach NL, Verfaillie CM. Activation of beta1 integrins on CML progenitors reveals cooperation between beta1 integrins and CD44 in the regulation of adhesion and proliferation. Leukemia 1997;11(6):822–829. [DOI] [PubMed] [Google Scholar]
- 60.Bhatia R, McCarthy JB, Verfaillie CM. Interferon-alpha restores normal beta 1 integrin-mediated inhibition of hematopoietic progenitor proliferation by the marrow microenvironment in chronic myelogenous leukemia. Blood 1996;87(9):3883–3891. [PubMed] [Google Scholar]
- 61.Bhatia R, Wayner EA, McGlave PB, Verfaillie CM. Interferon-alpha restores normal adhesion of chronic myelogenous leukemia hematopoietic progenitors to bone marrow stroma by correcting impaired beta 1 integrin receptor function. J Clin Invest 1994;94(1):384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Becker PS, Kopecky KJ, Wilks AN, et al. Very late antigen-4 function of myeloblasts correlates with improved overall survival for patients with acute myeloid leukemia. Blood 2009;113(4):866–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krevvata M, Silva BC, Manavalan JS, et al. Inhibition of leukemia cell engraftment and disease progression in mice by osteoblasts. Blood 2014;124(18):2834–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wei J, Wunderlich M, Fox C, et al. Microenvironment determines lineage fate in a human model of MLL-AF9 Leukemia. Cancer Cell 2008;13(6):483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jin L, Tabe Y, Konoplev S, et al. CXCR4 upregulation by imatinib induces chronic myelogenous leukemia (CML) cell migration to bone marrow stroma and promotes survival of quiescent CML cells. Mol Cancer Ther 2008;7(1):48–58. [DOI] [PubMed] [Google Scholar]
- 66.Meister M, Spencer JA, Wu J, et al. The Microanatomy of the Leukemic Stem Cell Niche in Murine Chronic Myelogenous Leukemia. ASH Abstract 2014;351. [Google Scholar]
- 67.Krause DS, Fulzele K, Catic A, et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat Med 2013;19(11):1513–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frisch BJ, Ashton JM, Xing L, Becker MW, Jordan CT, Calvi LM. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood 2012;119(2):540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schepers K, Pietras EM, Reynaud D, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013;13(3):285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang B, Ho YW, Huang Q, et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell 2012;21(4):577–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van den Berk LC, van der Veer A, Willemse ME, Theeuwes MJ, Luijendijk MW, Tong WH, et al. Disturbed CXCR4/CXCL12 axis in paediatric precursor B-cell acute lymphoblastic leukaemia. Br J Haematol 2014;166(2):240–249. [DOI] [PubMed] [Google Scholar]
- 72.Bowers M, Zhang B, Ho Y, et al. Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood 2015;125(17):2678–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dias S, Hattori K, Zhu Z, et al. Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J Clin Invest 2000;106(4):511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Veiga JP, Costa LF, Sallan SE, et al. Leukemia-stimulated bone marrow endothelium promotes leukemia cell survival. Exp Hematol 2006;34(5):610–621. [DOI] [PubMed] [Google Scholar]
- 75.Hatfield K, Oyan AM, Ersvaer E, et al. Primary human acute myeloid leukaemia cells increase the proliferation of microvascular endothelial cells through the release of soluble mediators. Br J Haematol 2009; 144(1):53–68. [DOI] [PubMed] [Google Scholar]
- 76.Aguayo A, Kantarjian H, Manshouri T, et al. Angiogenesis in acute and chronic leukemias and myelodysplastic syndromes. Blood 2000;96(6):2240–2245. [PubMed] [Google Scholar]
- 77.Perez-Atayde AR, Sallan SE, Tedrow U, et al. Spectrum of tumor angiogenesis in the bone marrow of children with acute lymphoblastic leukemia. Am J Path 1997; 150(3):815–821. [PMC free article] [PubMed] [Google Scholar]
- 78.Medyouf H, Mossner M, Jann JC, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014;14(6):824–837. [DOI] [PubMed] [Google Scholar]
- 79.Schmidt T, Kharabi Masouleh B, Loges S, et al. Loss or inhibition of stromal-derived PIGF prolongs survival of mice with imatinib-resistant Bcr-Abl1(+) leukemia. Cancer Cell 2011;19(6):740–753. [DOI] [PubMed] [Google Scholar]
- 80.Arranz L, Sanchez-Aguilera A, Martin-Perez D, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014; 512(7512):78–81. [DOI] [PubMed] [Google Scholar]
- 81.Hanoun M, Zhang D, Mizoguchi T, et al. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 2014;15(3):365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Frolova O, Samudio I, Benito JM, et al. Regulation of HIF-1alpha signaling and chemoresistance in acute lymphocytic leukemia under hypoxic conditions of the bone marrow microenvironment. Cancer Biol Ther 2012;13(10):858–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang H, Li H, Xi HS, Li S. HIF1 is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012; 119(11): 2595–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ng KP, Manjeri A, Lee KL, et al. Physiologic hypoxia promotes maintenance of CML stem cells despite effective BCR-ABL1 inhibition. Blood 2014;123(21):3316–3326. [DOI] [PubMed] [Google Scholar]
- 85.Gao XN, Yan F, Lin J, et al. AML1/ETO cooperates with HIF1 to promote leukemogenesis through DNMT3a transactivation. Leukemia. 2015;March 2 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 86.Wang Y, Liu Y, Malek SN, et al. Targeting HIF1 eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 2011;8(4):399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rouault-Pierre K, Lopez-Onieva L, Foster K, et al. HIF-2alpha protects human hematopoietic stem/progenitors and acute myeloid leukemic cells from apoptosis induced by endoplasmic reticulum stress. Cell Stem Cell 2013;13(5):549–563. [DOI] [PubMed] [Google Scholar]
- 88.Jaiswal R, Luk F, Gong J, Mathys JM, Grau GE, Bebawy M. Microparticle conferred microRNA profiles--implications in the transfer and dominance of cancer traits. Mol Cancer 2012;11:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Muralidharan-Chari V, Clancy JW, Sedgwick A, D’Souza-Schorey C. Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci. 123(Pt 10):1603–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huan J, Hornick NI, Shurtleff MJ, et al. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res 2013;73(2):918–929. [DOI] [PubMed] [Google Scholar]
- 91.Corrado C, Raimondo S, Saieva L, et al. Exosome-mediated crosstalk between chronic myelogenous leukemia cells and human bone marrow stromal cells triggers an interleukin 8-dependent survival of leukemia cells. Cancer Lett 2014;348(1–2):71–76. [DOI] [PubMed] [Google Scholar]
- 92.Taverna S, Amodeo V, Saieva L, et al. Exosomal shuttling of miR-126 in endothelial cells modulates adhesive and migratory abilities of chronic myelogenous leukemia cells. Mol Cancer 2014;13:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Raimondo S, Saieva L, Corrado C, et al. Chronic myeloid leukemia-derived exosomes promote tumor growth through an autocrine mechanism. Cell Commun Signal 2015;13:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu JY, Purton LE, Rodda SJ, et al. Osteoblastic regulation of B lymphopoiesis is mediated by Gs{alpha}-dependent signaling pathways. Proc Natl Acad Sci USA 2008;105(44):16976–16981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tiziani S, Kang Y, Harjanto R, et al. Metabolomics of the tumor microenvironment in pediatric acute lymphoblastic leukemia. PLoS One 2013;8(12):e82859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Conforti A, Biagini S, Del Bufalo F, et al. Biological, functional and genetic characterization of bone marrow-derived mesenchymal stromal cells from pediatric patients affected by acute lymphoblastic leukemia. PLoS One 2013;8(11):e76989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vicente Lopez A, Vazquez Garcia MN, Melen GJ, et al. Mesenchymal stromal cells derived from the bone marrow of acute lymphoblastic leukemia patients show altered BMP4 production: correlations with the course of disease. PLoS One 2014;9(1): e84496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang L, O’Leary H, Fortney J, Gibson LF. Ph+/VE-cadherin+ identifies a stem cell like population of acute lymphoblastic leukemia sustained by bone marrow niche cells. Blood 2007;110(9):3334–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shiozawa Y, Pedersen EA, Taichman RS. GAS6/Mer axis regulates the homing and survival of the E2A/PBX1-positive B-cell precursor acute lymphoblastic leukemia in the bone marrow niche. Exp Hematol 2010;38(2):132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Boyerinas B, Zafrir M, Yesilkanal AE, et al. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood 2013; 121(24):4821–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.de Vasconcellos JF, Laranjeira AB, Zanchin NI, et al. Increased CCL2 and IL-8 in the bone marrow microenvironment in acute lymphoblastic leukemia. Pediatr Blood Cancer 2011;56(4):568–577. [DOI] [PubMed] [Google Scholar]
- 102.Parameswaran R, Yu M, Lim M, et al. Combination of drug therapy in acute lymphoblastic leukemia with a CXCR4 antagonist. Leukemia 2011;25(8):1314–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hu Z, Slayton WB. Integrin VLA-5 and FAK are Good Targets to Improve Treatment Response in the Philadelphia Chromosome Positive Acute Lymphoblastic Leukemia. Front Oncol 2014;4:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Joshi I, Jena N, Yoshida T, et al. Focal Adhesion Kinase Inhibitors Reverse the Stromal Adhesion Phenotype of Ikaros-Mutant B-ALL, Induce Apopotosis, and Synergize with ABL1 Tyrosine Kinase Inhibitors: A New Paradigm for Pathogenesis and Therapy of High-Risk BALL. ASH Abstract. 2014;285. [Google Scholar]
- 105.Ishikawa F, Yoshida S, Saito Y, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol 2007;25(11):1315–1321. [DOI] [PubMed] [Google Scholar]
- 106.Duan CW, Shi J, Chen J, et al. Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell 2014;25(6):778–793. [DOI] [PubMed] [Google Scholar]
- 107.Pallasch CP, Leskov I, Braun CJ, et al. Sensitizing protective tumor microenvironments to antibody-mediated therapy. Cell 2014;156(3):590–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jacamo R, Chen Y, Wang Z, et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-kappaB mediates chemoresistance. Blood 2014;123(17):2691–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yamamoto-Sugitani M, Kuroda J, Ashihara E, et al. Galectin-3 (Gal-3) induced by leukemia microenvironment promotes drug resistance and bone marrow lodgement in chronic myelogenous leukemia. Proc Natl Acad Sci USA 2011; 108(42):17468–17473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fei F, Abdel-Azim H, Lim M, et al. Galectin-3 in pre-B acute lymphoblastic leukemia. Leukemia 2013;27(12):2385–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tabe Y, Jin L, Iwabuchi K, et al. Role of stromal microenvironment in nonpharmacological resistance of CML to imatinib through Lyn/CXCR4 interactions in lipid rafts. Leukemia 2012;26(5):883–892. [DOI] [PubMed] [Google Scholar]
- 112.Xia B, Tian C, Guo S, et al. c-Myc plays part in drug resistance mediated by bone marrow stromal cells in acute myeloid leukemia. Leuk Res 2015;39(1):92–99. [DOI] [PubMed] [Google Scholar]
- 113.Bewry NN, Nair RR, Emmons MF, Boulware D, Pinilla-Ibarz J, Hazlehurst LA. Stat3 contributes to resistance toward BCR-ABL inhibitors in a bone marrow microenvironment model of drug resistance. Mol Cancer Ther 2008;7(10):3169–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang B, Li M, McDonald T, et al. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood 2013;121 (10):1824–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Manshouri T, Estrov Z, Quintas-Cardama A, et al. Bone marrow stroma-secreted cytokines protect Jak2(V617F)-mutated cells from the effects of a JAK2 inhibitor. Cancer Res 2011;71(11):3831–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hou L, Liu T, Tan J, et al. Long-term culture of leukemic bone marrow primary cells in biomimetic osteoblast niche. Int J Hematol 2009;90(3):281–291. [DOI] [PubMed] [Google Scholar]
- 117.Vaiselbuh SR, Edelman M, Lipton JM, Liu JM. Ectopic human mesenchymal stem cellcoated scaffolds in NOD/SCID mice: an in vivo model of the leukemia niche. Tissue Eng Part C Methods 2010;16(6):1523–1531. [DOI] [PubMed] [Google Scholar]
- 118.Blanco TM, Mantalaris A, Bismarck A, Panoskaltsis N. The development of a three-dimensional scaffold for ex vivo biomimicry of human acute myeloid leukaemia. Biomaterials 2010;31(8):2243–2251. [DOI] [PubMed] [Google Scholar]
- 119.Tiwari A, Tursky ML, Kirkland MA, Pande G. Expansion of human hematopoietic stem/progenitor cells on decellularized matrix scaffolds. Curr Protoc Stem Cell Biol. 2014;28:Unit 1C.15. [DOI] [PubMed] [Google Scholar]
- 120.Chen Y, Jacamo R, Shi YX, et al. Human extramedullary bone marrow in mice: a novel in vivo model of genetically controlled hematopoietic microenvironment. Blood 2012;119(21):4971–4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Griessinger E, Anjos-Afonso F, Pizzitola I, et al. A niche-like culture system allowing the maintenance of primary human acute myeloid leukemia-initiating cells: a new tool to decipher their chemoresistance and self-renewal mechanisms. Stem Cells Transl Med 2014;3(4):520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Aljitawi OS, Li D, Xiao Y, et al. A novel three-dimensional stromal-based model for in vitro chemotherapy sensitivity testing of leukemia cells. Leuk Lymphoma 2014;55(2):378–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tavor S, Eisenbach M, Jacob-Hirsch J, et al. The CXCR4 antagonist AMD3100 impairs survival of human AML cells and induces their differentiation. Leukemia 2008;22(12): 2151–5158. [DOI] [PubMed] [Google Scholar]
- 124.Nervi B, Ramirez P, Rettig MP, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood 2009;113(24):6206–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sison EA, Rau RE, McIntyre E, et al. MLL-rearranged acute lymphoblastic leukaemia stem cell interactions with bone marrow stroma promote survival and therapeutic resistance that can be overcome with CXCR4 antagonism. Br J Haematol 2013; 160(6):785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hoellenriegel J, Zboralski D, Maasch C, et al. The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood 2014;123(7): 1032–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Agarwal A, Fleischman AG, Petersen CL, et al. Effects of plerixafor in combination with BCR-ABL kinase inhibition in a murine model of CML. Blood 2012;120(13):2658–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Winkler IG, Barbier V, Nowlan B, et al. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat Med 2012;18(11):1651–1657. [DOI] [PubMed] [Google Scholar]
- 129.Ben-Batalla I, Schultze A, Wroblewski M, et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood 2013; 122(14):2443–2452. [DOI] [PubMed] [Google Scholar]
- 130.de Rooij MF, Kuil A, Geest CR, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 2012; 119(11):2590–2594. [DOI] [PubMed] [Google Scholar]
- 131.Corrado C, Flugy AM, Taverna S, et al. Carboxyamidotriazole-orotate inhibits the growth of imatinib-resistant chronic myeloid leukaemia cells and modulates exosomes- stimulated angiogenesis. PLoS One 2012;7(8):e42310. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 132.Welner RS, Amabile G, Bararia D, et al. Treatment of chronic myelogenous leukemia by blocking cytokine alterations found in normal stem and progenitor cells. Cancer Cell 2015;27(5):671–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kleppe M, Kwak M, Koppikar P, et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov 2015;5(3):316–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hartwell KA, Miller PG, Mukherjee S, et al. Niche-based screening identifies small-molecule inhibitors of leukemia stem cells. Nat Chem Biol 2013;9(12):840–848. [DOI] [PMC free article] [PubMed] [Google Scholar]