

Abstract
mTOR – the mammalian/mechanistic target of rapamycin – has been implicated as a key signaling node for promoting survival of cancer cells. However, clinical trials that have targeted mTOR with rapamycin or rapamycin analogs have had minimal impact. In spite of the high specificity of rapamycin for mTOR, the doses needed to suppress key mTOR substrates have proved toxic. We report here that rapamycin when combined with AICAR – a compound that activates AMP-activated protein kinase makes rapamycin cytotoxic rather than cytostatic at doses that are tolerated clinically. AICAR by itself is able to suppress mTOR complex 1 (mTORC1), but also stimulates a feedback activation of mTORC2, which activates the survival kinase Akt. However, AICAR also suppresses production of phosphatidic acid (PA), which interacts with mTOR in a manner that is competitive with rapamycin. The reduced level of PA sensitizes mTORC2 to rapamycin at tolerable nano-molar doses leading reduced Akt phosphorylation and apoptosis. This study reveals how the use of AICAR enhances the efficacy of rapamycin such that rapamycin at low nano-molar doses can suppress mTORC2 and induce apoptosis in human cancer cells at doses that are clinically tolerable.
Keywords: AMP-activated protein kinase, AICAR, Akt, mTOR, phospholipase D, phosphatidic acid, rapamycin
Abbreviations
- 4E-BP1
eIF4E-binding protein-1
- AICAR
5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside
- AMPK
AMP-activated protein kinase
- DMEM
Dulbecco's modified Eagle medium
- FK-BP12
FK506-binding protein 12
- mTOR
mammalian/mechanistic target of rapamycin
- PA
phosphatidic acid
- PBS
phosphate buffered saline
- PCNA
proliferating cell nuclear antigen
- PLD
phospholipase D
- PARP
poly-ADP-ribose polymerase
- S6K
S6 kinase
Introduction
In the progression of a normal cell to a cancer cell, it is critical that there be a means to suppress default apoptotic programs that arguably are the first line of defense of cancer.1 A critical signaling node that promotes the survival of cancer cells is mTOR – the mammalian/mechanistic target of rapamycin. There are 2 mTOR complexes mTORC1 and mTORC2 that have both been implicated in cancer cell survival signals. It has been suggested that the signals that regulate mTOR are the most dysregulated signals in human cancer cells.2 The activation of mTOR in cancer cells leads to a critical metabolic transformation whereby cells shift from catabolic to anabolic metabolism.2, 3 As a consequence, there has been substantial interest in mTOR and metabolism as therapeutic targets for many human cancers.4 Compounds that target mTOR have been employed in many clinical trials5,6 – albeit without much success. There are distinct classes of compounds that target mTOR: rapamycin and rapamycin analogs (rapalogs) and ATP-competitive inhibitors. Rapamycin is a natural product that acts as an allosteric inhibitor that preferentially inhibits mTORC1.7 Both classes of inhibitors have inherent problems. The ATP-competitive inhibitors are good in that they target both mTORC1 and mTORC2, which both contribute to survival; however, as with most ATP-competitive inhibitors, there is concern as to specificity for mTOR. In contrast, rapamycin is highly specific for mTOR, but there are peculiar dosage issues associated with rapamycin.7
Rapamycin inhibits different cells with different dose responses. For example, phosphorylation of the mTORC1 substrate ribosomal subunit S6 kinase (S6K) in MCF7 breast cancer cells is suppressed at 0.5 nM, but in MDA-MB-231 cells, you need 20 nM to suppress S6K.8 This was due at least in part to the levels of phospholipase D (PLD) activity in the 2 cell lines. PLD generates the metabolite phosphatidic acid (PA), which interacts with mTOR in a manner that is competitive with rapamycin.8-10 Elevating PLD activity in MCF7 cells increased the dose of rapamycin to suppress phosphorylation of S6K, and similarly, reducing PLD activity in MDA-MB-231 cells reduced the dose needed to suppress S6K phosphorylation.8 There is also a problem in that different doses of rapamycin are needed to inhibit the phosphorylation of different mTORC1 substrates. The phosphorylation of S6K can be suppressed by low nano-molar levels of rapamycin; whereas phosphorylation of eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) requires micro-molar doses.11 This is an important issue because the apoptotic effects of rapamycin are due to suppressing phosphorylation of 4E-BP1.11 The doses that can be achieved in the clinic do not approach the levels needed to inhibit 4E-BP1 phosphorylation.12 This is likely why rapalogs have been largely disappointing in clinical trials in that you cannot deliver doses of rapamycin that overcome the survival effect of mTORC1, which involves primarily the phosphorylation of 4E-BP1.11
Another problem with rapamycin is that by suppressing S6K phosphorylation, it suppresses a negative feedback loop that keeps mTORC2 from phosphorylating and activating the survival kinase Akt, and as a consequence, rapamycin activates Akt.13,14 Whereas, the catalytic ATP-competitive inhibitors suppress both mTORC1 and mTORC2,5 under most conditions, rapamycin suppresses only mTORC1.11 Thus, activating mTORC2 by rapamycin treatment can lead to elevated Akt activity and suppress the apoptotic effects of rapamycin, which has been observed in pancreatic cancer cells.15 Therefore, in order to take advantage of the high specificity of rapamycin for mTOR, there needs to be a means for making rapamycin effective at lower doses. We reported previously that partial suppression of PLD activity in breast cancer cells resulted in the suppression of Akt at the mTORC2 site at Ser473 with 200 nM rapamycin.10 Thus, suppression of PLD activity can improve the efficacy of rapamycin for both mTORC1 and mTORC2. We recently reported that PLD activity is suppressed by stimulating AMP-activated protein kinase (AMPK) activity with the AMP-mimetic compound AICAR (5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside).16 We therefore investigated the effect of treating cancer cells with the combination of rapamycin and AICAR. In this report we provide evidence that tolerable doses of rapamycin in combination with AICAR suppresses both 4E-BP1 and Akt phosphorylation and induces apoptosis in cancer cells.
Results
AICAR treatment causes S-phase cell cycle arrest
We previously reported that cells arrested in S-phase of the cell cycle could be killed with rapamycin.17 In this regard, it was of interest that activation of AMPK has been shown to cause S-phase cell cycle arrest.18 AMPK can be activated by AICAR – a cell-permeable nucleoside that is metabolically converted by adenosine kinase to 5-aminoimidazole-4-carboxamide ribonucleoside monophosphate or ZMP, which mimics the allosteric effects of AMP and activates AMPK.19 As shown in Figure 1A, there was dramatic increase in cells with S-phase DNA content in both MDA-MB-231 and MCF7 breast cancer cells treated with AICAR as determined by flow cytometry. AICAR suppressed proliferation of the MDA-MB-231 and MCF7 cells – indicating that the cells were actually arresting in S-phase and not just changing the duration of the cell cycle phases (Fig. 1B). As shown in Figure 1C, there were decreased levels of cyclin D1, phosphorylated Rb, and proliferating cell nuclear antigen (PCNA); and increased levels of cyclin A2 in both cell lines – consistent with the apparent S-phase cell cycle arrest. Dose responses to AICAR were determined for cell cycle arrest (Fig. 1D) and for the phosphorylation of AMPK and the AMPK substrate acetyl-CoA carboxylase (Fig. 1E). These doses are consistent with those used by others.20,21 These data demonstrate that AICAR induces S-phase cell cycle arrest in these 2 breast cancer cell lines at doses that activate AMPK.
Figure 1.
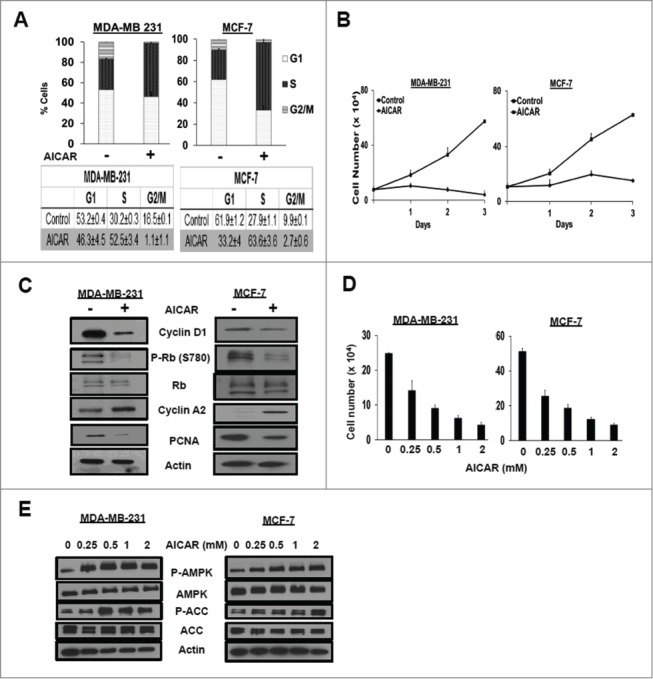
AICAR treatment causes S-phase cell cycle arrest. (A) MDA-MB-231 and MCF-7 cells were plated at 30% confluence in 10-cm plates in DMEM containing 10% serum. After 24 hr, the cells were treated with AICAR (0.5 mM) for 48 hr. After 48 h, the cells were harvested, fixed, stained with propidium iodide, and analyzed for cell cycle distribution by measuring DNA content/cell as described in Methods. The error bars represent the standard error of the mean for experiments repeated 3 times. (B) Cells were plated at 20% confluence in 6-well plates in complete media containing 10% serum. After 24 hr, AICAR (0.5mM) was added. Cells were harvested at indicated time points, stained using crystal violet, and quantified by light microscopy as described in Methods. Error bars represent the standard error for an experiment repeated 3 times. (C) Cells were plated at 30% confluence in 10-cm plates in complete media containing 10% serum for 24 h at which time they were treated with AICAR (0.5 mM) for 48 hr. The cells were subsequently harvested and cell lysates were collected. The indicated protein levels were determined by Western blot analysis. The data shown are representative of experiments repeated at least 2 times. (D) Cells were seeded as in B and treated with various concentrations of AICAR (0.25–2mM) for 48 hr, at which time the cells were harvested, stained using crystal violet, and quantified by light microscopy as described in Experimental Procedures. Error bars represent the standard error for an experiment repeated 3 times. (E) Cells were seeded as in (C)and treated with various concentrations of AICAR (0.25–2mM) for 24 hr. Cells were harvested and the levels of phospho-AMPK, AMPK, phospho-acetyl-CoA carboxylase (P-ACC), ACC, and actin were determined by Western blot analysis. The data shown are representative of experiments repeated at least 2 times.
AICAR treatment reduces the concentration of rapamycin to induce apoptosis
We next treated the MDA-MB-231 cells with rapamycin in combination with AICAR and looked for cleavage of the caspase 3 substrate poly-ADP-ribose polymerase (PARP) as an indicator of apoptosis. As expected based on our previous study,17 arresting cells in S-phase with AICAR resulted in a sharp increase in the level of cleaved PARP when rapamycin was included (Fig. 2A). What was not expected was that the dose required for induction of PARP cleavage was 1000-fold lower than that observed previously.8,11, 17 PARP cleavage was induced at 20 nM rapamycin in the presence of AICAR; whereas previously, rapamycin, by itself, induced PARP cleavage at 20 μM in MDA-MB-231 cells (Fig. 2B). As shown in Figure 2C, the combination of AICAR and 200 nM rapamycin led to increased levels of sub-G1 DNA content in the MDA-MB-231 and MCF7 cells – further supporting an apoptotic cell death. This was also observed in Calu1 lung cancer cells (Fig. 2C) – indicating that the effect is relevant for a variety of cancer cells. Importantly the apoptotic effect was not observed in the non-cancerous BJ-hTERT human fibroblast cell line. We also performed a dose response curve for induction of PARP cleavage by rapamycin on MCF7 cells in the presence of AICAR and as shown in Figure 2D, PARP cleavage could be detected at 0.5 nM. We previously reported that MCF7 cells are much more sensitive to rapamycin than MDA-MB-231 cells and demonstrated that loss of viability in MCF7 cells was observed at 100 nM.8 Thus, like the MDA-MB-231 cells, the presence of AICAR reduced the effective dose of rapamycin needed to induce apoptosis. Consistent with the lack of BJ-hTERT cells containing sub-genomic DNA (Fig. 2E), the combination of AICAR and 200 nM rapamycin also failed to induce PARP cleavage in these cells. The data in Figure 2 reveal that AICAR reduces the concentration of rapamycin needed to induce apoptosis in cancer cells, while not inducing apoptosis in in the non-cancer BJ-hTERT human fibroblast cell line.
Figure 2.
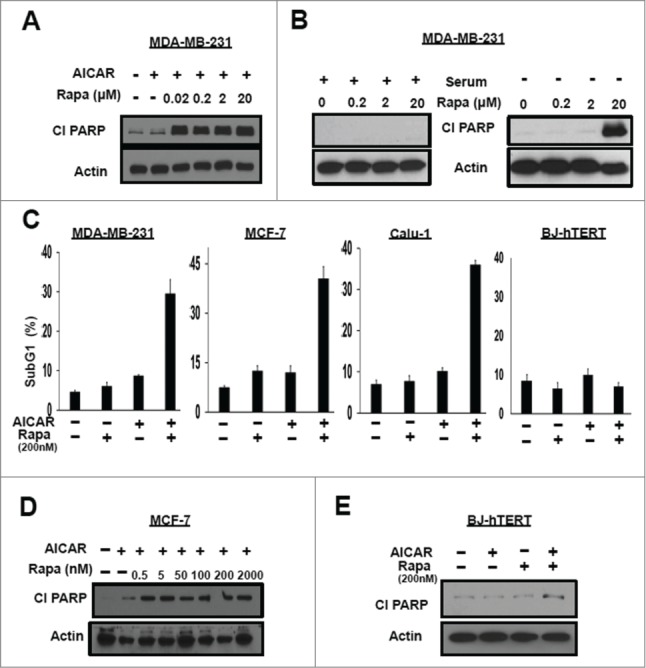
AICAR treatment reduces the concentration of rapamycin to induce apoptosis. (A) MDA-MB-231 cells were plated at 60% confluence in 60mm plates in DMEM containing 10% serum. Twenty-four hr later the cells were treated with AICAR (2mM) and/or different doses of rapamycin as indicated for 24 hr. The cells were then harvested and levels of cleaved PARP (Cl PARP) and actin were determined by Western blot analysis. (B) MDA-MB-231 cells were plated as in A. Twenty-four hr later of plating, the cells were shifted to complete medium or medium lacking serum and treated with rapamycin at different doses for 24 hr. The cells were then harvested and indicated protein levels were determined as in A. (C) MDA-MB-231, MCF-7, Calu-1 and BJ-hTERT cells were plated at 40% confluence and treated with AICAR (0.5 mM) and/or rapamycin (200 nM) for 48 hr, after which these were collected and subjected to flow cytometric analysis. Total subgenomic DNA is plotted as indicated. Error bars represent SD values for at least 2 independent experiments. (D) MCF-7 cells were plated in A. The cells were treated with AICAR (2 mM) and/or varying doses of rapamycin as indicated for 24 hr. The cells were then harvested and indicated protein levels were determined as in A. (E) BJ-hTERT cells were plated in A. The cells were treated with AICAR (2 mM) and/or rapamycin (200 nM) for 24 hr. The cells were then harvested and indicated protein levels were determined as in A. The data shown are representative of experiments repeated at least 2 times.
Apoptotic effects of AICAR and rapamycin is dependent on the suppression of mTORC2 by low dose rapamycin
We next examined the efficacy of AICAR and rapamycin on mTORC1 and mTORC2 substrates in MDA-MB-231 cells. As shown in Figure 3A, AICAR treatment suppressed the phosphorylation of the mTORC1 substrates S6K and 4E-BP1. However, AICAR stimulated phosphorylation of Akt at the mTORC2 site at Ser473 (Fig. 3A). No PARP cleavage was detected with AICAR treatment alone (Fig. 3A). Rapamycin (200 nM) suppressed phosphorylation of S6K and weakly suppressed phosphorylation of 4E-BP1 (Fig. 3A), which is consistent with our previous report that micro-molar concentrations of rapamycin were needed to suppress phosphorylation of 4E-BP1.11 Rapamycin, by itself, had no effect on the level of Akt phosphorylation or PARP cleavage (Fig. 3A). The most significant difference between the use of either rapamycin or AICAR by themselves vs rapamycin and AICAR in combination was that rapamycin strongly suppressed Akt phosphorylation in the presence of AICAR and induced PARP cleavage (Fig. 3A). Thus, AICAR also reduced the concentration of rapamycin needed to inhibit mTORC2 and suppress phosphorylation of Akt at Ser473. The ability of rapamycin to suppress Akt phosphorylation is sufficient to induce PARP cleavage and apoptotic cell death in combination with AICAR, which efficiently suppressed S6K and 4E-BP1 phosphorylation in the absence of rapamycin.
Figure 3.
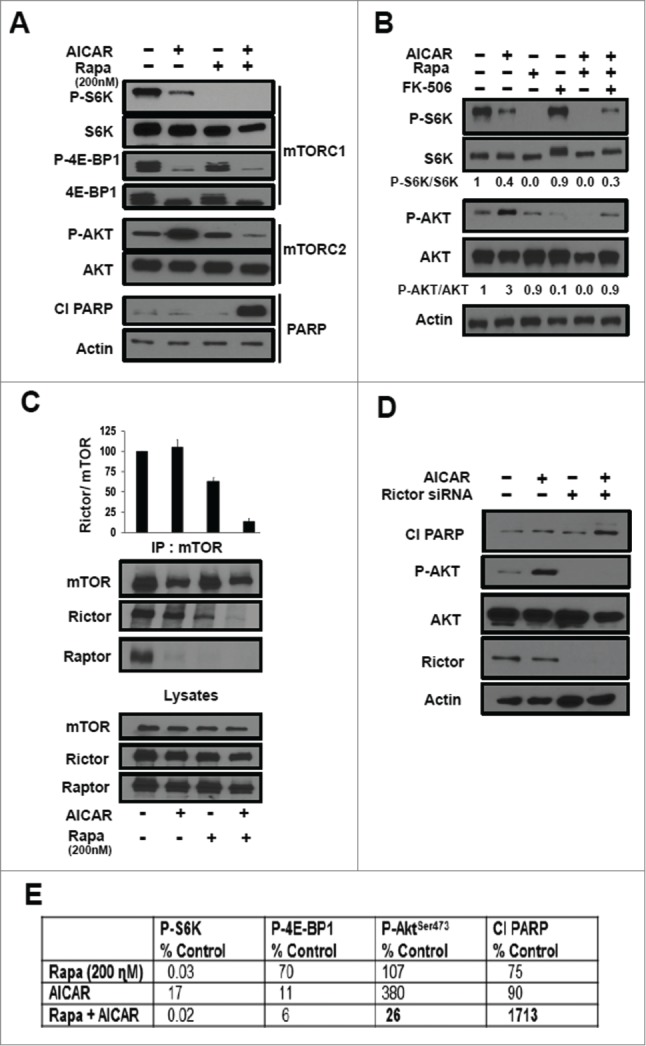
Apoptotic effects of AICAR and rapamycin is dependent on the suppression of mTORC2 by low dose rapamycin. (A) MDA-MB-231 cells were plated as in Figure 2A. The cells were treated with AICAR (2 mM) and/or rapamycin (200 nM) for 24 hr. The cells were then harvested and levels of the indicated proteins or phospho-proteins were determined by Western blot analysis. (B) MDA-MB-231 cells were plated as in A and were treated with AICAR (2 mM), rapamycin (200 nM), FK-506 (10μM) for 24 h. The cells were then harvested and indicated protein levels were determined by Western blot analysis. The relative levels of S6K and Akt phosphorylation were normalized to total S6K and total Akt respectively, and quantified using LI-COR image studio software. (C) MDA-MB-231 cells were plated and treated with AICAR (2 mM) and/or rapamycin (200 nM) for 24 hr as in A. At this time, lysates were prepared and subjected to immunoprecipitation with anti-mTOR antibody overnight, and then the mTOR immunoprecipitate (IP:mTOR) was subjected, along with the lysates to Western blot analysis for Rictor or Raptor. Because AICAR reduced the levels of mTOR in the immunoprecipitates, we determined the relative protein levels of Rictor normalized to mTOR and quantified using LI-COR image studio software. The data shown are representative of experiments repeated 3 times. Error bars for the graph represent the standard error for an experiment repeated 3 times. (D) MDA-MB-231 cells were plated in 6 well plates at 30% confluence overnight. The cells were then transfected with siRNAs for either scrambled control siRNA, or Rictor as indicated. Six hr later, the cells were treated with fresh medium containing 10% serum for 48hr. AICAR (2 mM) was then added for an additional 24 hr where indicated. The cells were then harvested and the levels of indicated proteins were determined by Western blot analysis. The data shown are representative of experiments repeated at least 2 times. (E) The key data for Figure 3 are summarized in table form where the most critical numbers are in bold highlighting the key effects of the combination of AICAR and rapamycin. Relative levels of phosphorylated proteins were normalized to respective total protein and quantified using LI-COR image studio software. Relative levels of Cl PARP were normalized to actin and quantified. The values were then normalized to controls, which were given a value of 100%.
To establish that the suppression of Akt phosphorylation was due to rapamycin, we examined the effect of FK506 on S6K and Akt phosphorylation. Rapamycin inhibits mTOR by combining with FK506-binding protein 12 (FK-BP12) and then binding to mTOR. FK506 also binds FK-BP12 and competes with rapamycin, and thusly has been used to reverse the effects of rapamycin.10 As shown in Figure 3B, FK506 reversed the suppression of S6K phosphorylation by 75% (compare lanes 2 and 6). FK506 reversed the rapamycin-induced suppression Akt phosphorylation by 30% (compare lanes 2 and 6). The suppression of Akt phosphorylation caused by treatment with both AICAR and rapamycin was achieved at 200 nM rapamycin. These data demonstrate that rapamycin is responsible for the suppression of Akt phosphorylation at the mTORC2 site at Ser473.
It was previously reported that under some conditions, rapamycin induces dissociation of mTORC2 components mTOR and Rictor.10,22 We therefore examined whether the combination of AICAR and lower dose rapamycin could dissociate mTOR from Rictor. mTOR was immunoprecipitated from MDA-MB-231 cell lysates and then subjected to Western blot analysis for both Raptor (mTORC1) and Rictor (mTORC2). As shown in Figure 3C, both AICAR and rapamycin, by themselves, cause dissociation of mTOR and Raptor. However, significant dissociation of mTOR from Rictor occurred only when rapamycin treatment was combined with AICAR (Fig. 3C). Since AICAR treatment suppresses the levels of mTOR that could be immunoprecipitated, we quantified the levels of Rictor detected relative to the level of mTOR that was immunoprecipitated. This is shown graphically in the upper panel where it can be seen that rapamycin by itself suppresses the level of Rictor co-immunoprecipitated with mTOR by roughly 30%. However the combination of AICAR and rapamycin reduces the level of Rictor to about 10%.
We also examined the effect of AICAR treatment in MDA-MB-231 cells with knockdown of Rictor, and as shown in Figure 3D, the knockdown of Rictor mimicked the effect of low dose rapamycin with regard to stimulating PARP cleavage and inhibiting Akt phosphorylation. The key results of Figure 3 are summarized in Figure 3E. These data further indicate that the key apoptotic effect of rapamycin in combination with AICAR is suppression of the mTORC2-catalyzed phosphorylation of Akt. Collectively, the data in Figure 3 indicate that the ability of AICAR and rapamycin to kill MDA-MB-231 cells is the result of AICAR suppressing mTORC1 and rapamycin suppressing the feedback activation mTORC213,14 in response to the AICAR suppression of mTORC1. This is a critical point in that it is the phosphorylation of Akt at S473 by mTORC2 that prevents apoptosis – and in the presence of AICAR, rapamycin can inhibit Akt phosphorylation at nano-molar concentrations of rapamycin.
Effect of AICAR on Rapamycin Efficacy is Due to Suppression of PLD Activity
mTOR requires PA for stabilizing both mTORC1 and mTORC2 complexes10 and for mTOR kinase activity.9 Although there are several sources of PA, the most significant is likely PLD, which catalyzes the hydrolysis of phosphatidylcholine to PA and free choline.23 Importantly, the highly conserved PA-binding domain on mTOR24 is at the same site where rapamycin binds;9 and rapamycin binds mTOR in a manner that is competitive with PA.8-10 We reported previously that suppressing PA production by PLD reduced the level of rapamycin needed to inhibit both mTORC1 and mTORC2.8,10 We also reported very recently that activating AMPK with AICAR suppressed PLD activity16 – suggesting the possibility that AICAR was reducing the dose of rapamycin needed to inhibit mTORC2 by suppressing PLD activity. As reported previously,16 AICAR treatment of MDA-MB-231 cells reduced PLD activity (Fig. 4A). To determine whether the reduction in PLD activity was responsible for the increased sensitivity of rapamycin, we added PA to determine whether it would reverse the effect of AICAR on the dose of rapamycin needed to suppress Akt phosphorylation. As shown in Figure 4B, the ability of rapamycin to suppress the AICAR-induced phosphorylation of Akt at Ser473 was reversed by PA. As shown in Figure 4C, the dissociation of mTOR from Rictor observed with AICAR and rapamycin in Figure 3C, was also reversed with PA. These data are consistent with the effect of AICAR on the rapamycin dose needed to suppress mTORC2 being due to suppressing PLD activity and the production of PA – leading to dissociation of mTOR and Rictor making free mTOR accessible to rapamycin (Fig. 5).
Figure 4.
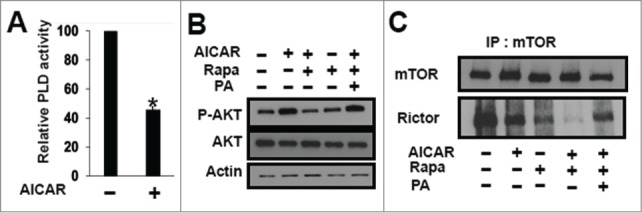
Effect of AICAR on rapamycin efficacy is due to suppression of PLD activity. (A) MDA-MB-231 cells were plated at 70% confluence in 60mm plates. Twenty-four hr later the cells were treated with AICAR (2 mM) for 45 min. [3H]-myristic acid was also added for 4 hr to label lipids. One-BtOH was added for 20 minutes, and the amount of the PLD-catalyzed transphosphatidylation product, phosphatidyl-butanol, was determined as described in the Methods section. Values were normalized to the levels of PLD activity in controls, which were given a value of 100%. Error bars for PLD assays represent SD values for at least 2 independent experiments. The statistical significance (P value) was determined by a 2-tailed, paired Student's t-test. *P ≤ 0.01 compared with control. (B) MDA-MB-231 cells were plated as in A and treated with AICAR (2 mM) and/or rapamycin (200 nM) for 8 hr. PA (300μM) was added where indicated. After 8h, the cells were harvested and the levels of the indicated proteins and phosphor-proteins were analyzed by Western blot analysis. The data shown are representative of experiments repeated at least 2 times. (C) MDA-MB-231 cells were plated and treated with AICAR (2 mM) and/or rapamycin (200 nM) for 24 hr as in Figure 3C. PA (300μM) was added where indicated 45 min prior lysate preparation. At this time, lysates were prepared and subjected to immunoprecipitation with anti-mTOR antibody overnight. The immunoprecipitates were then subjected to Western blot analysis for mTOR and Rictor. The data shown are representative of experiments repeated at least 2 times.
Figure 5.
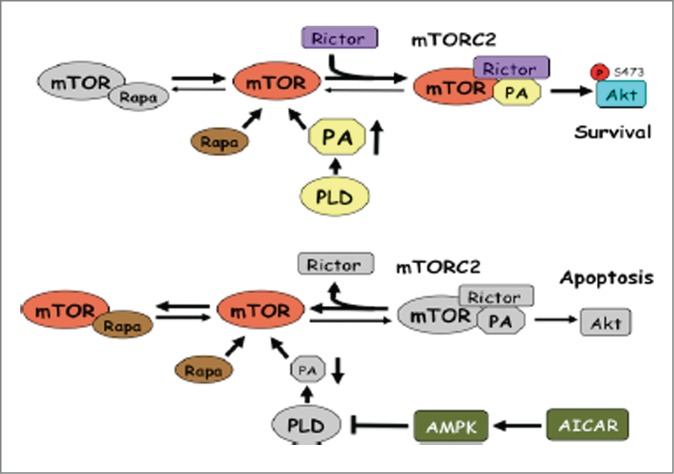
Model for differential doses of rapamycin needed to suppress mTORC2 in the presence and absence of AICAR. In the upper model, PA levels are high, which strongly favors formation of the highly stable mTORC2 complex. It was proposed that rapamycin inhibits mTORC2 only by binding newly synthesized mTOR before the complex forms.22 Since mTORC2 is so stable, mTOR complexed with Rictor (mTORC2) effectively never becomes available for binding to rapamycin. However, as indicated in the lower model, when AICAR is present, PLD activity is suppressed and PA levels are reduced. In this case, mTORC2 is destabilized and the equilibrium shifts toward free mTOR, which can bind rapamycin at low doses – and prevent re-assembly.
Discussion
We previously reported that arresting cells in S-phase renders most cancer cells sensitive to the apoptotic effects of rapamycin.17 The ability of rapamycin to induce apoptosis was dependent on the ability to suppress the phosphorylation of the mTORC1 substrate 4E-BP1, which required micro-molar doses of rapamycin.11 A serious problem with rapamycin-based therapeutic strategies is that the micro-molar doses that are required for the apoptotic effect are toxic.12 Another problem is that rapamycin suppresses a negative feedback suppression of Akt phosphorylation by mTORC2 leading to elevated levels of phosphorylated Akt13,14 that can overcome the apoptotic effect of rapamycin.15 In this report, we describe a surprising finding that activating AMPK with AICAR not only promotes S-phase arrest, which sensitizes cells to the apoptotic effect of suppressing mTORC1, it also makes mTORC2 sensitive to nano-molar doses of rapamycin that are tolerated in the clinic.
The activation of AMPK by AICAR leads to the phosphorylation of tuberous sclerosis complex (TSC1/2), which acts as a GTPase activating protein for Rheb, and thusly turns off Rheb.25 Rheb is required for the activity of both mTORC1 and PLD1.25 Of interest, AICAR can suppress phosphorylation of both S6K and 4E-BP1 with equal efficiency. This is not the case with rapamycin, which inhibits S6K at 1000-fold lower doses than it inhibits 4E-BP1.11 Thus, AICAR can accomplish more than rapamycin accomplishes at conventional nano-molar doses. However, there was an unanticipated benefit of combining AICAR with rapamycin – that being the suppression of PLD activity by AICAR,16 which sensitizes mTORC2 to rapamycin due to the reduced levels of PA generated.10 This turned out to be critical because, like rapamycin, AICAR suppressed the negative feedback suppression of Akt,13,14 which led to the phosphorylation and activation of Akt. Activated Akt suppresses the apoptotic effect of suppressing mTORC1.15 Thus mechanistically, AICAR stimulates S-phase arrest, which sensitizes cells to suppression of mTORC1. However, AICAR also suppresses PLD activity – making mTORC2 sensitive to rapamycin at clinically tolerated doses that can prevent the Akt phosphorylation stimulated by AICAR. The ability of AICAR to reduce the level of rapamycin needed to suppress mTORC2 by suppressing PA levels is shown schematically and described in Figure 5. Thus, while the combination of AICAR and rapamycin might seem redundant – they both suppress mTORC1 – the ability of AICAR to suppress PLD activity, and as a consequence, make mTORC2 responsive to tolerated doses of rapamycin leads to suppression of mTORC2 as well as mTORC1. Moreover, because AICAR suppresses 4E-BP1 phosphorylation more efficiently than rapamycin, the combination of AICAR and rapamycin leads to better suppression of this mTORC1 substrate that is the most critical for the survival effects of mTORC1.11
A key motivation for investigating AICAR in combination with rapamycin was the observation that AICAR arrests cells in S-phase of the cell cycle18 and that rapamycin kills cells arrested in S-phase.17 That is effectively what was observed with the combination of AICAR and rapamycin. As it turned out AICAR was able to efficiently suppress the phosphorylation of 4E-BP1, effectively negating the need for rapamycin. However, rapamycin, unlike AICAR arrests cells in G1,17 indicating that activating AMPK does more than suppress mTORC1 in causing S-phase rather than G1 arrest. A critical factor for the apoptotic effect of mTORC1 suppression is arresting the cells in S-phase.17,26 Ironically, the critical contribution of rapamycin to the apoptotic effect of rapamycin in combination with AICAR is the suppression of mTORC2-mediated activation of Akt.
With regard to the importance of targeting mTORC2 and the phosphorylation of Akt, ATP-competitive catalytic mTOR kinase inhibitors have been developed that target both mTORC1 and mTORC2 and can suppress the phosphorylation of S6K, 4E-BP1 and Akt at Ser 473.27 Thus, in principle, the catalytic inhibitors are ideal therapeutic agents for treating cancers where the activation of Akt by suppression of mTORC1 is preventing apoptosis. Consistent with this idea, we have found that torin1 kills BxPC3 pancreatic and that AZD8055 kills MCF7 breast cancer cells where Akt phosphorylation is elevated in response to rapamycin treatment.11,28 However, we have also observed that Torin1 arrests MDA-MB-231 cells under conditions where rapamycin induces apoptosis (our unpublished observations). Thus, conditions exist where rapamycin is more toxic than a catalytic inhibitor. In addition, several adverse effects have been noted for the catalytic inhibitors due to off-target effects of ATP analogs as well as potential additional effects of suppressing mTORC2.29 Therefore, the high specificity of rapamycin for mTOR – even at the high micro-molar doses11 – still has some advantages over the catalytic inhibitors. The impact of AICAR on the dose of rapamycin needed reported here further enhances the relevance of rapamycin as a therapeutic anti-cancer agent.
The findings reported here have potential clinically important implications. AMPK activators have been in use for many years to treat type II diabetes.30 Rapamycin and rapalogs have been widely employed in clinical trials.6 Thus, targeting both AMPK and rapamycin has been widely employed in the clinic suggesting the feasibility using AMPK activators in combination with rapalogs. This combination has previously been reported to be effective in suppressing the proliferation of acute lymphoblastic leukemia cells.31 In addition, a phase I study with the rapalog temsirolimus in combination with metformin in advanced solid tumors has been performed with some positive responses.32 Although no dramatic responses were reported, the study was restricted to limited set of solid tumors. But the study did reveal that the combination of AMPK activation and rapalog is well tolerated. It will be important to establish whether there is a subset of cancers with specific genetic alterations that are especially sensitive to this 2-pronged therapeutic approach.
Methods
Cells, cell culture conditions
The MDA-MB-231, Calu-1, MCF-7 and BJ-hTERT cells used in this study were obtained from the American Tissue Type Culture Collection. All cells were maintained in Dulbecco's modified Eagle medium (DMEM) (D6429) supplemented with 10% fetal bovine serum (Sigma) and penicillin/streptomycin (Sigma).
Materials
Reagents were obtained from the following sources. Antibodies against S6 kinase, P-S6 kinase (Thr389), 4E-BP1, P-4E-BP1 (Thr37/46), AKT, P-AKT (Ser473), mTOR, Raptor, P-Rb (Ser780), Rb, Cyclin D1, Cyclin A2, Rictor, phospho-AMPK (Thr-172), AMPK, Acetyl-CoA Carboxylase (ACC), phospho-ACC (Ser-79), cleaved PARP and actin were obtained from Cell Signaling. Antibody against PCNA and siRNA targeting Rictor (sc-61478), control siRNA were obtained from Santa Cruz Biotechnology. AICAR, FK506 was obtained from Tocris Bioscience. Rapamycin was obtained from LC Laboratories. PA (1-palmitoyl 2-oleoyl) in chloroform was purchased from Avanti-Polaris Lipids.
Transient transfections
Cells were plated on 6-well plates at 30% confluence in medium containing 10% serum. After 24 h, cells were transfected with siRNA at 100nM concentration using Lipofectamine RNAiMAX (Invitrogen). After 6 h, the medium was changed to fresh medium containing 10% serum and 72 h later, cells were lysed and analyzed by Western blot.
Western blot analysis
Cell lysates were collected using M-PER (Thermo Scientific, 78501), and proteins were separated on denaturing SDS-PAGE gels. Electrophoresed proteins were transferred to nitrocellulose membrane. After transfer, membranes were blocked in an isotonic solution containing 5% non-fat dry milk in phosphate buffered saline (PBS). Membranes were incubated with primary antibodies as described in text, and depending on the origin of the primary antibody, either anti-mouse or anti-rabbit HRP conjugated IgG was used for detection using ECL system (Pierce).
Preparation of PA
Immediately before use, the appropriate amount of PA was dried under nitrogen and resuspended by vortexing briefly in Dulbecco's phosphate buffered saline (SAFC Biosciences, 59321C). The lipid suspension was then sonicated in a water bath for 5 min. The resulting PA suspension was immediately added to cell culture to a final concentration of 300μM.
PLD activity
PLD activity was determined by accumulation of the transphosphatidylation product [3H]-phosphatidylbutanol as described previously.10 Lipid membranes were labeled with [3H]-myristic acid (60 Ci/mmol; 1.5 μCi/ml; Perkin-Elmer) for 4 hours. One-BtOH was added for 20 min before lipids where collected. Lipids were extracted and separated by thin layer chromatography along with phosphatidylbutanol standard (Enzo Life Sciences, BML-ST401-0050). The phosphatidylbutanol fraction was identified through co-migration with standards and the levels of the PLD product [3H]-phosphatidylbutanol was determined by scintillation counting after scraping the phosphatidylbutanol band from thin layer chromatography plates.
Flow cytometric analysis
Cells were washed twice in PBS and harvested. Cell suspensions were resuspended in the following fixing solution: 7ml PBS, 2% bovine serum albumin, 5mM EDTA, 0.1% NaN3; and 3ml of 100% ethanol was added dropwise. Fixed cells were centrifuged, washed using PBS, and then resuspended in 500µl sorting buffer: PBS, 0.1% Triton-X 100, 2% BSA, 5 mM EDTA, 40µg/ml propidium iodide, 100 µg/ml RNAse A, and incubated at 37C for 30 min. The cells were filtered through 70-µm mesh to remove cell aggregates. The DNA content was analyzed by flow cytometry (FACSCalibur; Becton Dickinson), and percentages of cells within each phase of the cell cycle were determined using WinCycle software (Phoenix Flow Systems).
Immunoprecipitation
Cells were grown in 10-cm-diameter plates. Immediately before lysing, culture plates were rinsed once with cold PBS and lysed on ice for 30 min in 500 µl of ice-cold 3-[(3-cholamidopropyl) - dimethylammonio]-1-propanesulfonate (CHAPS) immunoprecipitation buffer (40 mM HEPES [pH 7.5], 120 mM NaCl, 1 mM EDTA, 10 mM pyrophosphate, 10 mM glycerophosphate, 50 mM NaF, 0.5 mM orthovanadate, protease inhibitors [Millipore]) containing 0.3% CHAPS. A 500 µg sample of protein was then incubated with appropriate antibodies, and the immunoprecipitates were recovered 16 h later with protein G-Sepharose. The immunoprecipitates were then subjected to Western blot analysis along with 40µg of total cell lysate.
Cell proliferation assay
At indicated times, cells in 6-well plates were washed once with PBS, trypsinized with 500 µl trypsin, resuspended in 500 µl complete medium. The cells were stained using 0.1% crystal violet solution (Sigma), and then counted twice using a hemocytometer.
Funding
This work was supported by grants from the National Cancer Institute (R01-CA046677; R01-CA179542). Research Centers in Minority Institutions award RR-03039 from the National Center for Research Resources of the National Institutes of Health, which supports infrastructure and instrumentation in the Biological Sciences Department at Hunter College, is also acknowledged.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Zhivotovsky B, Kroemer G. Apoptosis and genomic instability. Nat Rev Mol Cell Biol 2004; 5:752-62; PMID:15340382; http://dx.doi.org/ 10.1038/nrm1443. [DOI] [PubMed] [Google Scholar]
- 2.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012; 21:297-308; PMID:22439925; http://dx.doi.org/ 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 2008; 7:11-20; PMID:18177721; http://dx.doi.org/ 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Liu Q, Thoreen C, Wang J, Sabatini D, Gray NS. mTOR Mediated Anti-Cancer Drug Discovery. Drug Discov Today Ther Strateg 2009; 6:47-55; PMID:20622997; http://dx.doi.org/ 10.1016/j.ddstr.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang YJ, Duan Y, Zheng XF. Targeting the mTOR kinase domain: the second generation of mTOR inhibitors. Drug Discov Today 2011; 16:325-31; PMID:21333749; http://dx.doi.org/ 10.1016/j.drudis.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Don AS, Zheng XF. Recent clinical trials of mTOR-targeted cancer therapies. Rev Recent Clin Trials 2011; 6:24-35; PMID:20868343; http://dx.doi.org/ 10.2174/157488711793980147. [DOI] [PubMed] [Google Scholar]
- 7.Foster DA, Toschi A. Targeting mTOR with rapamycin: one dose does not fit all. Cell Cycle 2009; 8:1026-9; PMID:19270529; http://dx.doi.org/ 10.4161/cc.8.7.8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Zheng Y, Foster DA. Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene 2003; 22:3937-42; PMID:12813467; http://dx.doi.org/ 10.1038/sj.onc.1206565. [DOI] [PubMed] [Google Scholar]
- 9.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 2001; 294:1942-5; PMID:11729323; http://dx.doi.org/ 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 10.Toschi A, Lee E, Xu L, Garcia A, Gadir N, Foster DA. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol Cell Biol 2009; 29:1411-20; PMID:19114562; http://dx.doi.org/ 10.1128/MCB.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yellen P, Saqcena M, Salloum D, Feng J, Preda A, Xu L, Rodrik-Outmezguine V, Foster DA. High-dose rapamycin induces apoptosis in human cancer cells by dissociating mTOR complex 1 and suppressing phosphorylation of 4E-BP1. Cell Cycle 2011; 10:3948-56; PMID:22071574; http://dx.doi.org/ 10.4161/cc.10.22.18124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, Hsueh T, Chen Y, Wang W, Youngkin D, et al.. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med 2008; 5:e8; PMID:18215105; http://dx.doi.org/ 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res 2005; 65:7052-8; PMID:16103051; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 14.O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al.. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006; 66:1500-8; PMID:16452206; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le Gendre O, Sookdeo A, Duliepre SA, Utter M, Frias M, Foster DA. Suppression of AKT Phosphorylation Restores Rapamycin-Based Synthetic Lethality in SMAD4-Defective Pancreatic Cancer Cells. Mol Cancer Res 2013; 11:474-81; PMID:23443316; http://dx.doi.org/ 10.1158/1541-7786.MCR-12-0679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukhopadhyay S, Saqcens M, Chatterjee A, Garcia A, Frias MA, Foster DA. Reciprocal regulation of AMP-activated protein kinase and phospholipase D. J Biol Chem 2015; 290, 6986-93; PMID:25632961; http://dx.doi.org/ 10.1074/jbc.M114.622571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gadir N, Jackson DN, Lee E, Foster DA. Defective TGF-β signaling sensitizes human cancer cells to rapamycin. Oncogene 2008; 27:1055-62; PMID:17700525; http://dx.doi.org/ 10.1038/sj.onc.1210721. [DOI] [PubMed] [Google Scholar]
- 18.Guan TJ, Qin FJ, Du JH, Geng L, Zhang YY, Li M. AICAR inhibits proliferation and induced S-phase arrest, and promotes apoptosis in CaSki cells. Acta Pharmacol Sin 2007; 28:1984-90; PMID:18031613; http://dx.doi.org/ 10.1111/j.1745-7254.2007.00675.x. [DOI] [PubMed] [Google Scholar]
- 19.Corton JM, Gillespie JG, Hawley SA, Hardie DG. Five-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem 1995; 229:558-65; PMID:7744080; http://dx.doi.org/ 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- 20.Sui X, Xu Y, Yang J, Fang Y, Lou H, Han W, Zhang M, Chen W, Wang K, Li D, et al.. Use of metformin alone is not associated with survival outcomes of colorectal cancer cell but AMPK activator AICAR sensitizes anticancer effect of 5-fluorouracil through AMPK activation. PLoS One 2014; 9:e97781; PMID:24849329; http://dx.doi.org/ 10.1371/journal.pone.0097781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cuthbertson DJ, Babraj JA, Mustard KJ, Towler MC, Green KA, Wackerhage H, Leese GP, Baar K, Thomason-Hughes M, Sutherland C, et al.. Five-aminoimidazole-4-carboxamide 1-β-D-ribofuranoside acutely stimulates skeletal muscle 2-deoxyglucose uptake in healthy men. Diabetes 2007; 56:2078-84; PMID:17513706; http://dx.doi.org/ 10.2337/db06-1716. [DOI] [PubMed] [Google Scholar]
- 22.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 2006; 22:159-68; PMID:16603397; http://dx.doi.org/ 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 23.Foster DA, Salloum D, Menon D, Frias MA. Phospholipase D and the Maintenance of Phosphatidic Acid Levels for Regulation of Mammalian Target of Rapamycin (mTOR). J Biol Chem 2014; 289:22583-8; PMID:24990952; http://dx.doi.org/ 10.1074/jbc.R114.566091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foster DA. Phosphatidic acid and lipid-sensing by mTOR. Trends Endocrinol Metab 2013; 24:272-8; PMID:23507202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun Y, Chen J. mTOR signaling: PLD takes center stage. Cell Cycle 2008; 7:3118-23; PMID:18927511; http://dx.doi.org/ 10.4161/cc.7.20.6881. [DOI] [PubMed] [Google Scholar]
- 26.Saqcena M, Patel D, Menon D, Mukhopadhyay S, Foster DA. Apoptotic effects of high-dose rapamycin occur in S-phase of the cell cycle. Cell Cycle 2015; 14, 2285-92; PMID:25945415; http://dx.doi.org/ 10.1080/15384101.2015.1046653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang YJ, Duan Y, Zheng XF. Targeting the mTOR kinase domain: the second generation of mTOR inhibitors. Drug Discov Today 2011; 16:325-31; PMID:21333749; http://dx.doi.org/ 10.1016/j.drudis.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le Gendre O, Sookdeo A, Duliepre SA, Utter M, Frias M, Foster DA. Suppression of AKT phosphorylation restores rapamycin-based synthetic lethality in SMAD4-defective pancreatic cancer cells. Mol Cancer Res 2013; 11:474-81; PMID:23443316; http://dx.doi.org/ 10.1158/1541-7786.MCR-12-0679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pallet N, Legendre C. Adverse events associated with mTOR inhibitors. Expert opinion on drug safety 2013; 12:177-86; PMID:23252795; http://dx.doi.org/ 10.1517/14740338.2013.752814. [DOI] [PubMed] [Google Scholar]
- 30.Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circulation Res 2007; 100:328-41; PMID:17307971; http://dx.doi.org/ 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- 31.Sengupta TK, Leclerc GM, Hsieh-Kinser TT, Leclerc GJ, Singh I, Barredo JC. Cytotoxic effect of 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) on childhood acute lymphoblastic leukemia (ALL) cells: implication for targeted therapy. Mol Cancer 2007; 6:46; PMID:17623090; http://dx.doi.org/ 10.1186/1476-4598-6-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacKenzie MJ, Ernst S, Johnson C, Winquist E. A phase I study of temsirolimus and metformin in advanced solid tumours. Invest New Drugs 2012; 30:647-52; PMID:20978924; http://dx.doi.org/ 10.1007/s10637-010-9570-8. [DOI] [PubMed] [Google Scholar]