

Abstract
DNA damage response is required for male fertility. DNA damage repair mediates recombination between homologous chromosomes in meiotic prophase, which is essential for proper chromosome segregation during meiotic division. Interestingly, some DNA damage response proteins are also required for the survival of premeiotic germ cells, but their roles in these cells are still unclear. CHFR was recently shown to participate in DNA damage response, but it remains to be established if CHFR is required for male fertility. In this study, we characterized Chfr knockout male mice and found that around 30% of them were infertile. The onset of spermatogenesis was delayed and there was significant increase in apoptosis in premeiotic germ cells. This resulted in complete loss of germ cells in testes in 3 months and azoospermia in these mice. We further demonstrated that ATM activation was compromised in the testes of these mice. Therefore, CHFR is important for the survival of male premeiotic germ cells, which is likely through maintaining genomic stability in spermatogonial stem cells.
Keywords: ATM activation, DNA damage repair, premeiotic germ cells
Introduction
Many factors contribute to male infertility. Non-obstructive azoospermia, the most severe form of male infertility, is often caused by impaired spermatogenesis in testes.1 In particular, genetic defects that disrupt proper DNA damage response often cause non-obstructive azoospermia and male infertility, but the roles of many DNA damage response proteins in this process are not well understood.
Male knockout mice of key DNA damage response proteins, which are often infertile, have greatly advanced our understanding of the functions of these proteins in male fertility. Most DNA damage response proteins function during meiosis.2 In meiotic prophase, programmed DNA double-stranded breaks are generated globally and are repaired to promote meiotic recombination between homologous chromosomes.3,4 DNA damage response proteins are required to ensure accurate repair of the breaks. In addition, these proteins are assembled through a unique mechanism to repair the breaks on male sex chromosomes, which share little homology.5 In agreement with these important functions, meiosis in knockout mice for H2AX, MDC1, and ATM arrests before the onset of meiotic division.6-9 Another group of DNA damage response proteins functions in spermiogenesis, during which haploid round spermatids elongate and compact their nucleus. It is believed that DNA damage occurs during this process.10 Accordingly, developing haploid germ cells in knockout mice for RNF8 and RAD6B arrest at this stage.11,12 Although it remains elusive how DNA breaks are generated at this stage, it is likely that DNA repair play a critical role during sperm nucleus compaction.
Interestingly, DNA damage response is also important for the survival of premeiotic germ cells. Spermatogonial stem cells are the progenitors of all germ cells in testes.13 The availability of these cells is required to maintain sustained production of germ cells in adults. Some DNA damage response proteins, such as ATM, RAD18 and RNF168, are required for genomic stability and viability of spermatogonial stem cells in adult mice. Absence of these proteins leads to depletion of spermatogonial stem cells pool and complete loss of germ cells in testes and causes azoospermia in adult mice.14-17
CHFR is an E3 ubiquitin ligase and participates in DNA damage response.18,19 It is recruited to DNA damage sites by poly(ADP-ribose) and is important for the first wave of protein ubiquitination there.19 It functions synergistically with RNF8 to activate ATM in response to DNA damage.18 Chfr knockout mice have been generated and are tumor-prone.20 When combined with RNF8 deficiency, these mice have high incidences of thymic lymphoma.18 The tumor phenotypes in Chfr knockout mice are in accordance with CHFR's function in DNA damage response. However, it is not clear if CHFR plays any roles in male fertility. Here we show that CHFR also regulates sustained germ cell production. Around 30% of Chfr knockout male mice are infertile. The onset of spermatogenesis is delayed and all germ cells are lost in the adult mice. Therefore CHFR is important for the survival of premeiotic germ cells.
Results
We have previously generated Chfr knockout mice to study the function of CHFR in tumor prevention. During these studies, we noticed that some Chfr knockout male mice are infertile. To reveal the role of CHFR in spermatogenesis, we compared the fertility of wild type and Chfr knockout adult male mice of different ages by mating them with wild type female mice. Interestingly, while all wild type male mice were fertile, around 30% Chfr knockout male mice were infertile (Fig. 1a). We harvested the epididymides of all the male mice and little sperm were recovered from infertile Chfr knockout male mice (Fig. 1b). Histological analysis also revealed that there were little sperm inside the epididymides of these mice (Fig. 1c). These observations revealed that a large portion of Chfr knockout male mice were defective in spermatogenesis. On the contrary, CHFR deficiency had no impact on the fertility of female mice (data not shown), suggesting that CHFR had a unique role in male fertility.
Figure 1.
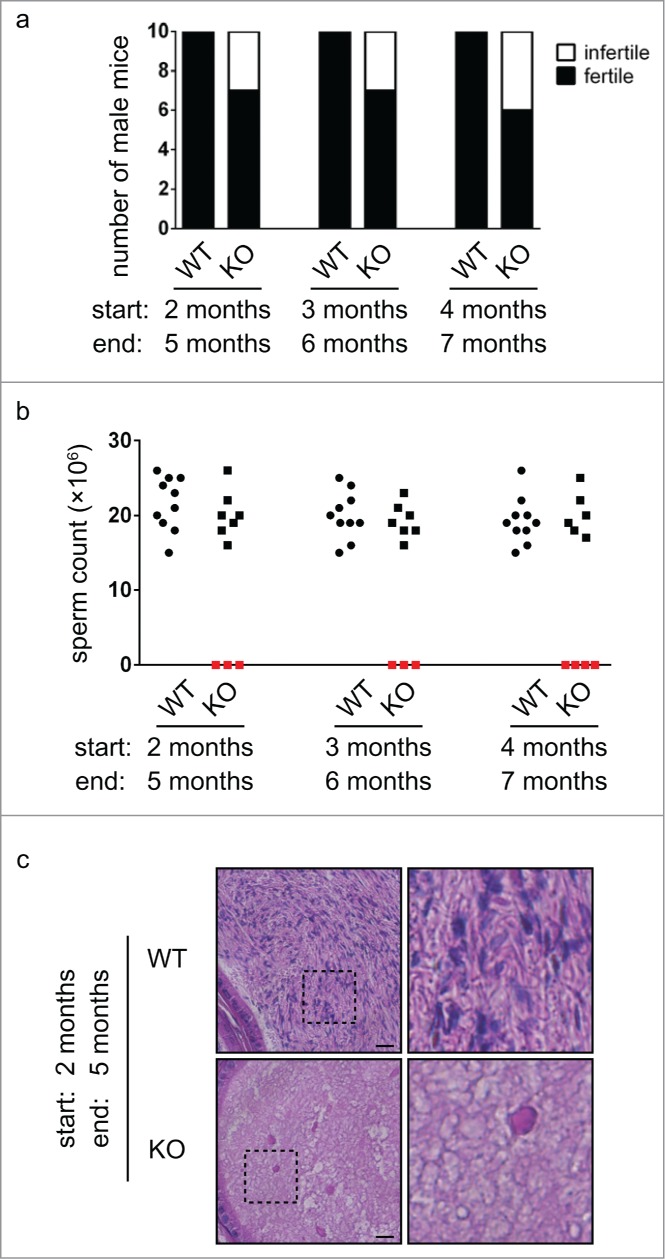
A large portion of Chfr knockout male mice are infertile. (a) Wild type (WT) and Chfr knockout (KO) male mice were mated with WT female mice. Male mice of 3 different ages were used. The numbers of fertile and infertile mice are shown. The age at the start and the end of mating are marked. (b) Sperm were harvested from epididymides at the end of mating. Sperm counts are shown. (c) Hematoxylin and eosin staining of epididymis sections are shown. Higher magnifications of selected areas are shown on right. Scale bar, 50 µM.
To explore the reasons for the infertility of Chfr knockout male mice, we analyzed the testes of these mice. We harvested testes from Chfr knockout male mice of different ages and found that many of them had dramatically smaller testes than those from wild type mice (Fig. 2a). The small testes were observed in Chfr knockout male mice as young as 3 weeks old (Fig. 2b). In agreement with the small testes, little sperm were recovered from the epididymides of these mice even at 12 weeks (Fig. 2c). These suggested that the defective testis development was likely responsible for the absence of sperm in epididymides and infertility of Chfr knockout male mice. To gain more insights into the defect in testes, we focused on Chfr knockout male mice with small testes and analyzed germ cells in these mice. We first studied the testis sections of wild type and Chfr knockout male mice of 12 weeks old. In wild type male mice, developing spermatids at all stages could be observed in the testes. However, only sertoli cells could be found in the testes of Chfr knockout male mice, and no germ cells could be identified (Fig. 2d-e). In more than 80% of seminiferous tubules, there were large vacuoles indicative of tissue degeneration due to germ cell loss (Fig. 2d, f). The complete absence of germ cells in the testes of Chfr knockout male mice suggested that germ cell prodution, but not the structure of testes, was responsible for the infertility of these mice. The defect occurred at very early stages of spermatogenesis, which might involve the maintenance of spermatogonial stem cells that generated all male germ cells.
Figure 2.

Germ cells are completely lost in small testes from Chfr knockout male mice at 12 weeks. (a) Typical pictures of normal testes from WT male mice and small testes from Chfr knockout male mice are shown. (b) The weight of normal testes from WT male mice and small testes from Chfr knockout male mice are shown. (c) Sperm were harvested from epididymides of mice shown in (b), and sperm counts are shown. (d) Testis sections of 12-week-old WT and Chfr knockout male mice were stained with periodic acid schiff (PAS)-hematoxylin. Typical pictures are shown. Scale bar, 50 µM. Asterisk: large vacuoles. (e-f) Seminiferous tubules with germ cells (e) and with large vacuoles (f) are summarized. Mean and standard deviation are shown.
To further clarify the defective early spermatogenesis in Chfr knockout male mice, we studied the testis sections of younger mice that were 6 weeks old. In wild type male mice of this age, the first few waves of spermatogenesis had completed and sperm could be found in epididymides (Fig. 2c). In agreement with this observation, mature sperm and developing spermatids were readily identified in the testes of wild type mice (Fig. 3a-e). Interestingly, although no sperm could be identified in the epididymides of Chfr knockout male mice (Fig. 2c), developing spermatids could be observed in around 80% of seminiferous tubules in the testes (Fig. 3f-j, p). This suggested that germ cells were still produced in these mice. However, some defects were observed. First, few mature sperm were observed in Chfr knockout male mice (Fig. 3f-h), suggesting that spermatogenesis was delayed. Second, in the testis sections of wild type mice, multiple layers of germ cells at different developmental stages could be observed in one seminiferous tubule due to the presence of multiple waves of spermatogenesis (Fig. 3a-e). In Chfr knockout male mice, only around 15% of seminiferous tubules had 2 layers of germ cells (Fig. 3g-j, q) and most of them had only one layer of germ cells (Fig. 3f, k-m, q). Around 20% of seminiferous tubules contained sertoli cells only (Fig. 3n-o, p) and most of them had large vacuoles indicative of tissue degeneration due to germ cell loss (Fig. 3k-o, r). This observation suggested that germ cell production was abolished after the first few waves, which were likely caused by exhaustion of spermatogonial stem cells.
Figure 3.

Germ cells are decreased in small testes from Chfr knockout male mice at 6 weeks. Testis sections of 12-week-old male mice were stained with periodic acid schiff (PAS)-hematoxylin. Typical pictures of normal testes from WT male mice (a-e) and small testes from Chfr knockout male mice (f-o) are shown. Scale bar, 50 µM. Asterisk: large vacuoles. (p-r) Seminiferous tubules with germ cells (p), with multiple layers of germs cells (q), and with large vacuoles (r) are summarized. Mean and standard deviation are shown.
To examine if the defect started earlier in development, we further characterized spermatogenesis in younger Chfr knockout male mice. At 4 weeks, the first round of spermatogenesis had not completed, since little sperm could be found in epididymides in wild type mice (Fig. 2c). In the testes of wild type male mice, germ cells from the first wave of spermatogenesis had become elongated spermatids (Fig. 4a). Multiple waves of spermatogenesis were present in the testes since germ cells at different stages could be identified and multiple layers of germ cells could be found (Fig. 4a-d). In the testes of Chfr knockout male mice of 4 weeks old, the earliest germ cells from the first wave of spermatogenesis only developed to round spermatids (Fig. 4e). This suggested that there was a delay in germ cell development in these mice. Similar as 6-week-old Chfr knockout male mice, only around 15% of seminiferous tubules contained multiple layers of germ cells of different stages (Fig. 4e, j). In most seminiferous tubules, only one layer of germ cells was identified (Fig. 4f-g, j). Around 15% of seminiferous tubules contained sertoli cells only (Fig. 4h-i) and most of them contained large vacuoles indicative of tissue degeneration due to germ cell loss (Fig. 4g-i). These observations suggested the loss of spermatogonial stem cells could begin within 4 weeks.
Figure 4.

Spermatogenesis in Chfr knockout male mice with small testes are delayed at 3 and 4 weeks. Testis sections of 3 and 4-week-old male mice were stained with periodic acid schiff (PAS)-hematoxylin. Typical pictures of normal testes from WT male mice (a-d for 4 weeks and k-m for 3 weeks) and small testes from Chfr knockout male mice (e-h for 4 weeks and n-p for 3 weeks) are shown. Scale bar, 50 µM. Asterisk: large vacuoles. (i-j) For 4-week-old male mice, seminiferous tubules with multiple layers of germs cells (i) and with large vacuoles (j) are summarized. Mean and standard deviation are shown.
We further extended our study to mice of 3 weeks old, the earliest time point when small testes were found in Chfr knockout male mice. In wild type mice of 3 weeks old, round spermatids could be observed in testes, suggesting that the first wave of spermatogenesis had passed meiosis (Fig. 4k-m). In Chfr knockout male mice of 3 weeks old, no round spermatids could be observed, suggesting that the delay in spermatogenesis occurred before or at meiosis (Fig. 4n-p). Unlike mice older than 4 weeks, there was no sign of significant germ cell loss in the testes. No vacuoles were observed in seminiferous tubules either. Therefore, 3 weeks could be the starting point of germ cell loss. Indeed, significant apoptosis could be observed in testes of Chfr knockout male mice as early as in 3 weeks and around 75% of seminiferous tubules contained apoptotic cells (Fig. 5a-b). The apoptosis did not occur in cells undergoing meiosis or in round spermatids, but occurred in premeiotic cells that are close to sertoli cells at the basement membranes of seminiferous tubules, which were likely spermatogonial stem cells.
Figure 5.

Apoptosis occur in germ cells from Chfr knockout male mice. (a-b) Apoptosis was detected in testis sections of 3 and 4-week-old male mice. Apoptotic cells are marked by arrow heads. Seminiferous tubules with apoptotic cells are summarized. Scale bar, 50 µM. (c) Levels of ATM, phosphorylated ATM at serine 1981, Chk2, and phosphorylated Chk2 are shown in WT and Chfr knockout MEFs with or without ionizing radiation (IR). (d) Levels of ATM, phosphorylated ATM at serine 1981, Chk2, and phosphorylated Chk2 are shown in 3-week-old WT and Chfr knockout testes. N, normal size testes. S, small testes. Quantification was performed. Mean and standard deviation are shown.
We next explored the underlying mechanism of the infertility phenotype in Chfr knockout male mice. We have previously shown in somatic cells that CHFR is important for DNA damage repair through functioning synergistically with RNF8 to activate ATM.18 Here we showed that in mouse embryonic fibroblast (MEF) derived from Chfr knockout mice, ionizing-induced ATM activation (marked by Serine 1981 phosphorylation) was reduced, which is accompanied by reduced phosphorylation of ATM's downstream target Chk2 (Fig. 5c). This suggested that CHFR deficiency alone was important for efficient DNA damage-induced ATM activation and downstream signaling pathways. We went on to examine if this was the case in testes. Interestingly, activated ATM could be detected in the absence of exogenous DNA damage in testes of 3-week-old mice (Fig. 5d). Consistently, the amount of activated ATM as well as Chk2 phosphorylation were mildly decreased in the normal size testes and further decreased in the small testes of Chfr knockout male mice (Fig. 5d). It has been shown that ATM is required for long-term maintenance of spermatogonial stem cells.15 Therefore, the loss of premeiotic germ cells in Chfr knockout male mice might be caused, at least in part, by inefficient ATM activation in testes.
Discussion
In this study, we have found that around 30% of Chfr knockout male mice are infertile. They have dramatically small testes and produce no mature sperm. Apoptosis of premeiotic germ cells starts at 3 weeks old, which leads to gradual loss of germ cells. Within 12 weeks, all germ cells are absent and only sertoli cells are left. Large vacuoles indicative of tissue degeneration due to germ cell loss are also present in seminiferous tubules of adult mice. These observations suggest that CHFR is important for the viability of spermatogonial stem cells in adults, which are essential for sustained germ cell production.
It has been shown previously that DNA damage response proteins, such as ATM, RAD18, and RNF168, are important for the viability of spermatogonial stem cells in adults.14-17 Since spermatogonial stem cells give rise to all germ cells and pass the genetic information to the offspring, it is essential that genomic integrity is carefully maintained in these cells, which requires robust DNA damage response.21 When certain DNA damage response fails to repair the DNA lesions, apoptosis is activated to eliminate the cells to prevent the damaged or mutated DNA to be passed on to the offspring. Only a few DNA damage response proteins including CHFR are known to be important for the viability of spermatogonial stem cells in adults. It will be interesting to characterize if other DNA damage response proteins are also involved in this process.
We have shown previously that CHFR functions with RNF8 to activate ATM in response to DNA damage.18 Here we show that CHFR is required for intact ATM activation in testes in the absence of exogenous DNA damage. The constitutively activated ATM in testes is likely caused by the massive double-stranded breaks generated by SPO11 during meiotic prophase. It has been shown that CHFR is important for the first wave of protein ubiquitination at DNA damage sites.18,19 Similar mechanisms could exist in testes as well. Interestingly, the maintenance of spermatogonial stem cells in adults is defective in both Chfr and Atm knockout male mice. Therefore, it is likely that the loss of premeiotic germ cells in Chfr knockout male mice is partially due to decreased ATM activation in spermatogonial stem cells. It is worth noticing that the defect is more severe in Chfr knockout male mice than in Atm knockout male mice. It is possible that CHFR regulates other pathways in spermatogonial stem cells in addition to those that activate ATM.
Besides the defect in maintaining sustained germ cell production, we have found that spermatogenesis is delayed in Chfr knockout male mice. It is possible that the decreased ATM activation affects the speed of meiotic progression. ATM is important for initiating DNA double-strand break repair in meiotic phase. As a result, meiosis in Atm knockout mice arrests before pachynema.22 But in Chfr knockout male mice, there is no sign of meiotic arrest, as post-meiotic haploid round spermatids and elongating spermatids could be identified. It is possible that the remaining activated ATM in the testes of Chfr knockout male mice is sufficient for initiating DNA double-strand break repair in meiotic phase, but the decrease in ATM activation affects the speed of DNA double-strand break repair and slows down meiotic progression. During mitosis, CHFR ubiquitinates Aurora A and PLK1 and promotes their degradation in order to precisely control the levels of these 2 proteins.20 It is also possible that the deficiency of CHFR slows down mitotic progression of premeiotic germ cells.
In summary, we have shown that CHFR is important for maintaining sustained germ cell production and preventing germ cell loss. It still remains unknown why only 30% of the Chfr knockout male mice have complete germ cells loss that results in infertility. Similar incomplete penetrance of the infertility phenotype has been observed in Rad51c knockout mice.23 Nevertheless, we have uncovered a unique role of CHFR during male fertility.
Material and Methods
Mice
Chfr knockout mice were generated previously and housed in Unit for Laboratory Animal Medicine (ULAM) in University of Michigan. All mice work was approved by University Committee on Use and Care of Animals (UCUCA) of University of Michigan.
Fertility analysis
Each male mouse was housed with 2 8-week-old wild type C57BL6/J female mice for 3 months. Pregnancy of the female mice was checked twice a week. Female mice were taken away from the cage once they were pregnant. Three months after initial mating, sperm were collected from male mice by dissecting and cutting epididymides in Research Vitro Fert medium (Cook, Australia).
Histology and TUNEL assay
Epididymides and testes were harvested, fixed in Bouin's solution (Sigma), dehydrated, and embedded in paraffin. Five μm sections were cut. Epididymis sections were stained with hematoxylin and eosin. Testis sections were stained with periodic acid schiff (PAS)-hematoxylin. DeadEnd Colorimetric TUNEL System (Promega) was used to detect apoptotic cells.
Ionizing radiation, immunoprecipitation and Western blotting
Chfr knockout mouse embryonic fibroblast (MEF) was described previously. Ionizing radiation (IR) was performed using JL Spepherd 137Cs radiation source. Thirty minutes after 20 Gy IR, cells were harvested. For harvesting germ cells, testes were incubated in collagenase solution (1 mg/ml) at 37 °C for 10 minutes. Cells from testes and MEFs were pelleted and lysed in NETN 300 (50 mM Tris-HCl pH8.0, 300 mM NaCl, 1 mM EDTA, and 0.5% NP-40) at 4°C for 10 minutes. The lysate was diluted using the same volume of ddH2O. Immunoprecipitation and Western blotting were performed according to standard procedures. Chk2 phosphorylation was detected by Chk2 immunoprecipitation followed by anti-pSQ/TQ antibody blotting. Anti-ATM, anti-pATM-S1981, anti-Chk2, and anti-pSQ/TQ antibodies were from Cell Signaling Technology.
Statistical methods
Twenty cross sections of seminiferous tubules were analyzed for each testis section. Three mice were used for each analysis. Western blotting from 3 experiments was quantified using Image J. Mean and standard deviation were plotted in all figures.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was funded by Zhejiang Provincial Natural Science Foundation of China (LR15H040001 to LY), National Natural Science Foundation of China (81471494 to LY), and National Institutes of Health (CA130899, CA132755, CA187209, and GM108647 to XY). XY is a recipient of Era of Hope Scholar Award from the Department of Defense and a research scholar of Leukemia and Lymphoma Society.
References
- 1.Wosnitzer M, Goldstein M, Hardy MP. Review of Azoospermia. Spermatogenesis 2014; 4:e28218; PMID:25105055; http://dx.doi.org/ 10.4161/spmg.28218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu LY, Yu X. Double-strand break repair on sex chromosomes: challenges during male meiotic prophase. Cell Cycle 2015; 14:516-525; PMID:25565522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Romanienko PJ, Camerini-Otero RD. The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol Cell 2000; 6:975-987; PMID:11106738; http://dx.doi.org/ 10.1016/S1097-2765(00)00097-6 [DOI] [PubMed] [Google Scholar]
- 4.Baudat F, Manova K, Yuen JP, Jasin M, Keeney S. Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol Cell 2000; 6:989-998; PMID:11106739; http://dx.doi.org/ 10.1016/S1097-2765(00)00098-8 [DOI] [PubMed] [Google Scholar]
- 5.Lu LY, Xiong Y, Kuang H, Korakavi G, Yu X. Regulation of the DNA damage response on male meiotic sex chromosomes. Nat Commun 2013; 4:2105; PMID:23812044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernandez-Capetillo O, Mahadevaiah SK, Celeste A, Romanienko PJ, Camerini-Otero RD, Bonner WM, Manova K, Burgoyne P, Nussenzweig A. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev Cell 2003; 4:497-508; PMID:12689589; http://dx.doi.org/ 10.1016/S1534-5807(03)00093-5 [DOI] [PubMed] [Google Scholar]
- 7.Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell 2006; 21:187-200; PMID:16427009; http://dx.doi.org/ 10.1016/j.molcel.2005.11.025 [DOI] [PubMed] [Google Scholar]
- 8.Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Gen Dev 1996; 10:2411-2422; PMID:8843194; http://dx.doi.org/ 10.1101/gad.10.19.2411 [DOI] [PubMed] [Google Scholar]
- 9.Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, et al.. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 1996; 86:159-171; PMID:8689683; http://dx.doi.org/ 10.1016/S0092-8674(00)80086-0 [DOI] [PubMed] [Google Scholar]
- 10.Marcon L, Boissonneault G. Transient DNA strand breaks during mouse and human spermiogenesis new insights in stage specificity and link to chromatin remodeling. Biol Reprod 2004; 70:910-918; PMID:14645105; http://dx.doi.org/ 10.1095/biolreprod.103.022541 [DOI] [PubMed] [Google Scholar]
- 11.Lu LY, Wu J, Ye L, Gavrilina GB, Saunders TL, Yu X. RNF8-dependent histone modifications regulate nucleosome removal during spermatogenesis. Dev Cell 2010; 18:371-384; PMID:20153262; http://dx.doi.org/ 10.1016/j.devcel.2010.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roest HP, van Klaveren J, de Wit J, van Gurp CG, Koken MH, Vermey M, van Roijen JH, Hoogerbrugge JW, Vreeburg JT, Baarends WM, et al.. Inactivation of the HR6B ubiquitin-conjugating DNA repair enzyme in mice causes male sterility associated with chromatin modification. Cell 1996; 86:799-810; PMID:8797826; http://dx.doi.org/ 10.1016/S0092-8674(00)80154-3 [DOI] [PubMed] [Google Scholar]
- 13.Kanatsu-Shinohara M, Shinohara T. Spermatogonial stem cell self-renewal and development. Ann Rev Cell Dev Biol 2013; 29:163-187; PMID:24099084; http://dx.doi.org/ 10.1146/annurev-cellbio-101512-122353 [DOI] [PubMed] [Google Scholar]
- 14.Takubo K, Hirao A, Ohmura M, Azuma M, Arai F, Nagamatsu G, Suda T. Premeiotic germ cell defect in seminiferous tubules of Atm-null testis. Biochem Biophys Res Commun 2006; 351:993-998; PMID:17097049; http://dx.doi.org/ 10.1016/j.bbrc.2006.10.145 [DOI] [PubMed] [Google Scholar]
- 15.Takubo K, Ohmura M, Azuma M, Nagamatsu G, Yamada W, Arai F, Hirao A, Suda T. Stem cell defects in ATM-deficient undifferentiated spermatogonia through DNA damage-induced cell-cycle arrest. Cell stem cell 2008; 2:170-182; PMID:18371438; http://dx.doi.org/ 10.1016/j.stem.2007.10.023 [DOI] [PubMed] [Google Scholar]
- 16.Sun J, Yomogida K, Sakao S, Yamamoto H, Yoshida K, Watanabe K, Morita T, Araki K, Yamamura K, Tateishi S. Rad18 is required for long-term maintenance of spermatogenesis in mouse testes. Mech Dev 2009; 126:173-183; PMID:19068231; http://dx.doi.org/ 10.1016/j.mod.2008.11.004 [DOI] [PubMed] [Google Scholar]
- 17.Bohgaki T, Bohgaki M, Cardoso R, Panier S, Zeegers D, Li L, Stewart GS, Sanchez O, Hande MP, Durocher D, et al.. Genomic instability, defective spermatogenesis, immunodeficiency, and cancer in a mouse model of the RIDDLE syndrome. PLoS Genet 2011; 7:e1001381; PMID:21552324; http://dx.doi.org/ 10.1371/journal.pgen.1001381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu J, Chen Y, Lu LY, Wu Y, Paulsen MT, Ljungman M, Ferguson DO, Yu X. Chfr and RNF8 synergistically regulate ATM activation. Nat Struct Mol Biol 2011; 18:761-768; PMID:21706008; http://dx.doi.org/ 10.1038/nsmb.2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu C, Wu J, Paudyal SC, You Z, Yu X. CHFR is important for the first wave of ubiquitination at DNA damage sites. Nucleic Acids Res 2013; 41:1698-1710; PMID:23268447; http://dx.doi.org/ 10.1093/nar/gks1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu X, Minter-Dykhouse K, Malureanu L, Zhao WM, Zhang D, Merkle CJ, Ward IM, Saya H, Fang G, van Deursen J, et al.. Chfr is required for tumor suppression and Aurora A regulation. Nat Genet 2005; 37:401-406; PMID:15793587; http://dx.doi.org/ 10.1038/ng1538 [DOI] [PubMed] [Google Scholar]
- 21.Rube CE, Zhang S, Miebach N, Fricke A, Rube C. Protecting the heritable genome: DNA damage response mechanisms in spermatogonial stem cells. DNA Repair 2011; 10:159-168; PMID:21123119; http://dx.doi.org/ 10.1016/j.dnarep.2010.10.007 [DOI] [PubMed] [Google Scholar]
- 22.Barlow C, Liyanage M, Moens PB, Tarsounas M, Nagashima K, Brown K, Rottinghaus S, Jackson SP, Tagle D, Ried T, et al.. Atm deficiency results in severe meiotic disruption as early as leptonema of prophase I. Development 1998; 125:4007-4017; PMID:9735362 [DOI] [PubMed] [Google Scholar]
- 23.Kuznetsov S, Pellegrini M, Shuda K, Fernandez-Capetillo O, Liu Y, Martin BK, Burkett S, Southon E, Pati D, Tessarollo L, et al.. RAD51C deficiency in mice results in early prophase I arrest in males and sister chromatid separation at metaphase II in females. J Cell Biol 2007; 176:581-592; PMID:17312021; http://dx.doi.org/ 10.1083/jcb.200608130 [DOI] [PMC free article] [PubMed] [Google Scholar]