

RNA-Binding Proteins (RBPs) are proteins that bind to double or single stranded RNA molecules. In general, RBPs are thought to play a major role in post-transcriptional control of RNAs, such as: mRNA localization, mRNA stabilization, polyadenylation, splicing and translation. Hundreds of RBPs have been discovered in mammals. However, only a small fraction of these RBPs was mechanistically and functionally characterized thus far, leaving the question of why the cell has invested such a huge amount of resource to produce so many RBPs. One of the answers may come from the recently identified non-coding RNAs; it is now recognized that majority of our genome is used to code for non-coding RNAs (ncRNAs), including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs). It is anticipated that many RBPs will be needed to modulate the biogenesis, modification and functional action of these large amount of ncRNAs in a cell.
Trbp was initially identified as a RBP binding to HIV RNA and has subsequently been reported to participate in numerous cellular and molecular events. In several previous studies, Trbp has been characterized as a key regulator of cell growth and invasion, but the functional outcomes appear distinct, even controversial under different conditions.1-4 Our recent study, using cardiomyocyte-specific Trbp knockout mice demonstrated that loss of Trbp did not affect cardiomyocyte growth or proliferation. Instead, Trbp is required for normal cardiac contraction by regulating fast-and slow-twitch myofiber gene program in the heart. Mechanistically, we uncovered a linear genetic pathway in the heart in which the function of Trbp is mediated by a single miRNA (miR-208a) and a single miR-208a target (Sox6). Our study demonstrated that miR-208a is a primary target of Trbp, as miR-208a is abolished in Trbp mutant hearts. Reintroduction of Trbp into the heart of Trbp mutant mouse restored its level, whereas the Sox6 shRNA, though able to rescue animal surviving and cardiac function, failed to rescue the expression miR-208a. In addition, there is accumulation of pre-miR-208a in Trbp mutant hearts, indicating a requirement of Trbp for the processing of miR-208a. Moreover, normalization of miR-208a expression with miR-208a transgene in Trbp mutant hearts reduces Sox6 mRNA level and fully rescued the cardiac defects. Together, our study directly links Trbp's physiological function to miRNA biogenesis in the heart.5
Previously, Trbp was found to associate with the Dicer complex to modulate the maturation of miRNAs.2 However, answers to whether Trbp directly regulates miRNA biogenesis remain controversial. Melo et al., (2009) reported that Trbp functioned as a tumor suppressor, likely by regulating miRNA expression.3 In contrast, Goodazi et al. (2014) suggested that Trbp could modulate the stability of downstream mRNAs to promote tumor metastasis4. Kim et al. (2014) and Nakamura et al., (2015), found that Trbp acts as a regulator of mRNA translation by repressing PKR activity to facilitate G2/M transition in cultured cells or modulate immuno-metabolism in liver tissue.6,7 Together, these reports suggest that Trbp can regulate a broad spectrum of RNA species, and participate in multiple cellular processes, in temporal and spatial-specific manners (Fig. 1).
Our miRNA profiling studies surprisingly found that only a small fraction of miRNAs was deregulated in Trbp mutant mouse hearts. This observation is consistent with a recent study by Kim et al. demonstrating that Trbp does not affect the global abundance of miRNAs.6 However, these observations are distinct from several prior studies, in which Trbp has been suggested to function as an obligated co-factor of Dicer to regulate miRNA biogenesis globally. Of note, most previous studies on Trbp-regulated miRNA expression were conducted biochemically in vitro, whereas a mouse genetic approach was taken in our study in vivo. Therefore the apparent discrepancy likely lies in the difference in cellular context. Such seemingly inconsistency further underscores the importance of in vivo genetic studies for RNA-binding proteins.
The findings that Trbp only regulated a subset of miRNAs indicated that this RBP has certain target specificity. In the heart, loss-of Trbp led to the dysregulation of only a few miRNAs, among them, miR-208a was abolished. The view that miR-208a is a primary Trbp target in the heart is further supported by the functional rescue of Trbp mutants by this miRNA. However, we still don't know how the target specificity of Trbp is determined. We have examined the effects of Trbp on miRNA expression, and we compared them with previously reported miRNA expression data when Trbp was knocked down in vitro. Yet we were unable to identify any common sequence domain or similar secondary structure among those Trbp-regulated miRNAs. Indeed, one of the challenges for almost all RBPs is to define their RNA binding specificities. We suggest that the recognition of specific pre-miRNAs by Trbp is biological context dependent. The conditional mouse Trbp allele that we have generated will be an invaluable reagent allowing us to further determine how Trbp regulates specific miRNAs in different cells and tissues. These studies will shed new lights into the molecular mechanisms by which RBPs specifically regulate their RNA substrates. Most importantly, investigating and understanding the mole-cular events regulated by RBPs will enable developing specific drug targets to treat human diseases.
Figure 1.
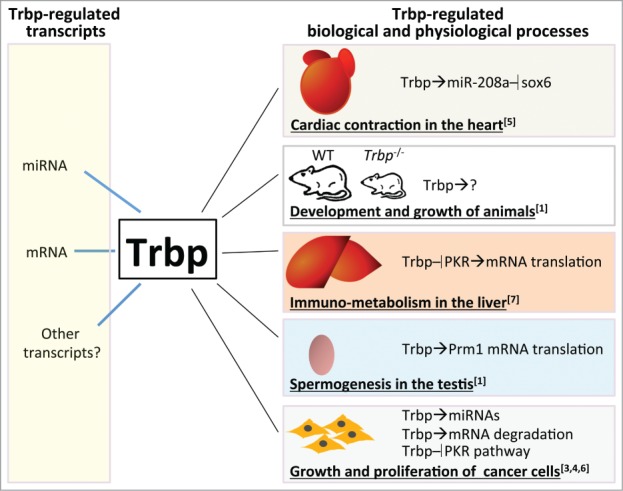
Trbp regulates multiple mRNA and miRNA species and participates in numerous biological and physiological processes. Its functional roles and regulatory targets are highly biological context dependent.
Acknowledgements
We apologize to our colleagues whose work could not be cited because of space constraints.
Funding
Research in the Wang lab is supported by the Muscular Dystrophy Association and the NIH (HL085635, HL116919).
References
- 1.Zhong J, et al.. Nat Genet 1999; PMID:10369260; http://dx.doi.org/ 10.1038/9684 [DOI] [PubMed] [Google Scholar]
- 2.Chendrimada TP, et al.. Nature 2005; PMID:15973356; http://dx.doi.org/ 10.1038/nature03868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Melo SA, et al.. Nat Genet 2009; PMID:19219043; http://dx.doi.org/ 10.1038/ng.317 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4.Goodarzi H, et al.. Nature 2014; PMID:25043050; http://dx.doi.org/ 10.1038/nature13466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding J, et al.. Nat Genet 2015; PMID:26012717; http://dx.doi.org/ 10.1038/ng.3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim Y, et al.. Cell Rep 2014; PMID:25437560; http://dx.doi.org/ 10.1016/j.celrep.2014.09.039 [DOI] [PubMed] [Google Scholar]
- 7.Nakamura T, et al.. Cell Rep 2015; PMID:25843719; http://dx.doi.org/ 10.1016/j.celrep.2015.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]