

Abstract
Genomic screens of doxorubicin toxicity in S. cerevisiae have identified numerous mutants in amino acid and carbon metabolism which express increased doxorubicin sensitivity. This work examines the effect of amino acid metabolism on doxorubicin toxicity. S. cerevisiae were treated with doxorubicin in combination with a variety of amino acid supplements. Strains of S. cerevisiae with mutations in pathways utilizing aspartate and other metabolites were examined for sensitivity to doxorubicin. S. cerevisiae cultures exposed to doxorubicin in minimal media showed significantly more toxicity than cultures exposed in rich media. Supplementing minimal media with aspartate, glutamate or alanine reduced doxorubicin toxicity. Cell cycle response was assessed by examining the budding pattern of treated cells. Cultures exposed to doxorubicin in minimal media arrested growth with no apparent cell cycle progression. Aspartate supplementation allowed cultures exposed to doxorubicin in minimal media to arrest after one division with a budding pattern and survival comparable to cultures exposed in rich media. Aspartate provides less protection from doxorubicin in cells mutant in either mitochondrial citrate synthase (CIT1) or NADH oxidase (NDI1), suggesting aspartate reduces doxorubicin toxicity by facilitating mitochondrial function. These data suggest glycolysis becomes less active and mitochondrial respiration more active following doxorubicin exposure.
Keywords: anaplerosis, citrate synthase, Doxorubicin, electron transport, growth arrest, mitochondria, stress response
Introduction
Daunorubicin is an anthracycline compound originally isolated from a soil bacteria, Streptomyces peucetius, near Apulia, Italy and found to have anticancer activity.1 Doxorubicin was subsequently isolated from a mutant of S. peucetius as a more active variant. Doxorubicin has become a widely used cancer chemotherapy drug and is part of standard therapy for breast cancer, lymphoma, sarcoma, thyroid cancer and many others. Efforts to understand the mechanism of action of doxorubicin began with its discovery. Doxorubicin produces several effects in treated cells. Doxorubicin creates DNA damage by intercalating into DNA, through topoisomerase II inhibition and possibly other means.2 In addition, doxorubicin has redox activity and is able to oxidize NADH, effectively diverting electrons away from electron transport toward oxygen to create superoxide radicals.3 Doxorubicin has been proposed to induce apoptosis through increased ceramide synthesis and proteolytic activation of the transcription factor CREB3L1.4 Doxorubicin can also lead to death by mitotic catastrophe at low doses.5 Understanding how these activities differentially affect normal and cancer cells is critical to improving the therapeutic ratio of doxorubicin.
Genetic approaches to understand the mechanism of doxorubicin cytotoxicity have been very informative. Collections of S. cerevisiae strains harbouring deletions in all non-essential genes have been screened for sensitivity to doxorubicin.6,7 These screens have identified dozens of mutants with increased sensitivity to doxorubicin. Deletion mutation of 5–10% of non-essential genes (over 400) in S. cerevisiae leads to enhanced doxorubicin toxicity, suggesting the response to doxorubicin requires numerous cellular activities. Mutants lacking DNA repair activities are sensitive to doxorubicin, consistent with its DNA damaging activities. Several other activities are also important in providing doxorubicin resistance. For example, strains with mutations in a number of genes with activity in central carbohydrate and amino acid metabolism are also sensitive to doxorubicin.
Altered metabolism is a hallmark of cancer.8 Cancer cells have relatively high rates of aerobic glycolysis. NADH, superoxide and amino acid metabolism are also altered in cancer. Since metabolism is altered in cancer, and full resistance to doxorubicin depends on many metabolic functions, understanding the metabolic response to doxorubicin could improve cancer therapy. How central carbon and amino acid metabolism operate to provide doxorubicin protection is currently unclear. This work examines the effects of doxorubicin in S. cerevisiae with a variety of metabolic defects and in a variety of media conditions. We present data that aspartate, by providing carbon to the tricarboxylic acid cycle, decreases doxorubicin toxicity. This observation could lead to altered clinical applications of doxorubicin that will increase its cytotoxicity on cancer cells while decreasing off target effects such as cardiotoxicity.
Results
Amino acids can reduce doxorubicin toxicity
S. cerevisiae is more sensitive to doxorubicin during treatment in minimal media compared to rich media (Fig. 1A). Survival was determined as the number of colony forming units per ml. at a given time relative to the number of colony forming units per ml. in the same culture at the start of doxorubicin exposure. The nutritional status of the culture has a significant influence on doxorubicin sensitivity. For example, at 6 hours, survival is 91% for cells cultured in rich media compared to 31% for cells cultured in minimal media.
Figure 1.
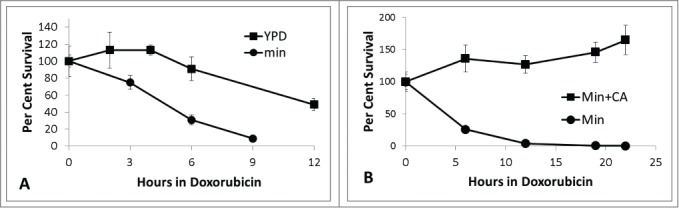
Time course sensitivity to doxorubicin. A. BY4741 was grown in either minimal media (circles) or rich YPD (squares) media and exposed to doxorubicin (150 micromolar) as described in Methods. At various times, aliquots of the culture were removed, diluted and plated to YPD plates to determine the number of colony forming units per ml. Survival is shown as the number of colony forming units per ml at time x relative to the number of colony forming units at time zero. B. BY4741 was grown in minimal media alone (circles) or minimal media supplemented with 0.4% casamino acids, CA, (squares). All platings were performed in triplicate. Error bars represent +/− one standard deviation. For some timepoints, error bars are smaller than the marker.
Mutations in several genes coding for enzymes involved in amino acid metabolism lead to doxorubicin sensitivity.6,7 To further define the role of amino acid metabolism in mediating doxorubicin sensitivity, cell survival was examined following exposure to doxorubicin in minimal media alone or supplemented with casamino acids (Fig. 1B). The casamino acid supplement was pretreated with norite to remove any nucleic acid or other contaminants.9 Figure 1B shows substantial protection from doxorubicin in minimal media supplemented with 0.4% casamino acids. Parental strain BY4741 cells were exposed to doxorubicin in minimal media alone or with various single amino acid supplements to determine the protection afforded by individual amino acids. Amino acids were provided at 3 times the minimum concentration needed to meet auxotrophic requirements (see Methods section) to ensure an abundant supply for growth and/or repair. Figure 2 demonstrates some amino acids are able to provide protection from doxorubicin toxicity. Aspartate, asparagine, glutamate, glutamine, alanine, serine, lysine and methionine all provide significant resistance to doxorubicin toxicity. Adding aspartate (2.3mM) increases cell survival by greater than fold4- after 6 hours of doxorubicin exposure. Similar results were seen after 3 hours doxorubicin exposure (data not shown).
Figure 2.
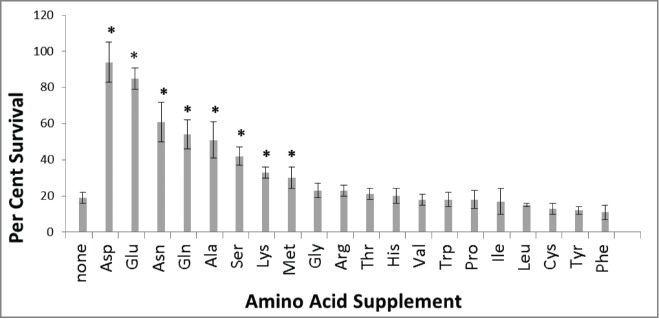
Some amino acids afford doxorubicin resistance. BY4741 cells were grown in minimal media alone or with single amino acid supplementation indicated. Cells were exposed to 150 micromolar doxorubicin for 6 hours as described in Methods. Survival with amino acids was compared to no supplementation using Students t-test. Cultures supplemented with amino acids affording significant protection at the p < 0.05 level are marked with an asterisk. All platings were done in triplicate. Error bars represent +/− one standard deviation.
Figure 3A shows changes in viability with increasing time of doxorubicin exposure for cultures of BY4741 in minimal media with or without 2.3 mM aspartate. The aspartate supplemented culture exhibited significantly less doxorubicin toxicity at each time point, confirming the result shown in Figure 2. Cell survival at 9 hrs in minimal media was increased 8-fold by adding aspartate. Aspartate provides protection from doxorubicin toxicity following longer exposures as well (Fig. 3B). Aspartate protection from doxorubicin toxicity is dose dependent, with higher concentrations of aspartate providing greater protection from doxorubicin (Fig. 3C). Aspartate may provide protection simply by interacting with doxorubicin directly, or by some means other than altered metabolism. D-aspartate participates in fewer metabolic reactions than the proteinogenic L-aspartate used above, but shares identical chemical properties such as pKa. Unlike L-aspartate (Fig. 3C), D-aspartate has a very limited ability to protect cells from doxorubicin toxicity (Fig. 3D). D-aspartate provides limited protection at 23.0 mM, whereas L-aspartate provides comparable protection at 0.23 mM. The specificity and dose dependence of protection afforded by L-aspartate suggest a metabolic mechanism for protection, rather than a non-specific interaction.
Figure 3.
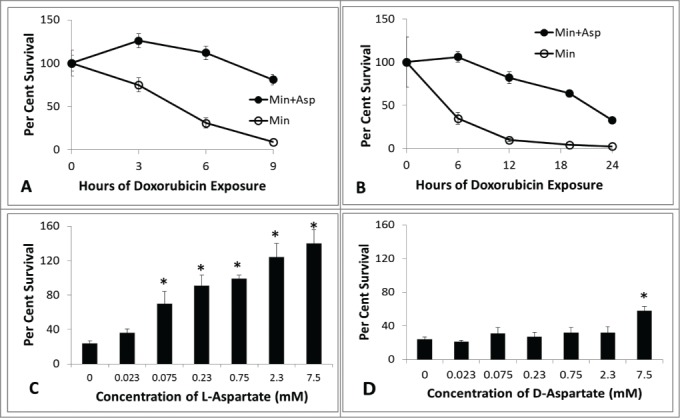
Time course of doxorubicin sensitivity in minimal media with and without aspartate. (A) BY4741 cells were grown in minimal media with (closed circles) and without (open circles) 2.3 mM aspartate and exposed to 150 micromolar doxorubicin. Survival at various times was determined by measuring the number of viable colony forming units per ml. relative to initial value at time 0 hour. (B) Cultures were established as in A, platings were done at longer intervals. (C) Cultures of BY4741 with increasing concentrations of L-aspartate were established. Platings were done for each culture at time 0, then again at 6 hours of exposure to 150 micomolar doxorubicin. (D) Cultures of BY4741 were established as in (C), with the exception of supplementing with D-aspartate at the indicated concentrations. Platings were done in triplicate for each time-point (+/− one standard deviation). For (A and B), the aspartate culture is significantly resistant to doxorubicin (p < 0.05) at each non-zero time-point (Student t-test). For (C and D), an asterisk marks the supplemented cultures with significant resistance relative to non-supplemented culture (Student t-test).
Aspartate reduces doxorubicin toxicity by facilitating the TCA cycle
The amino acids showing the greatest protection from doxorubicin toxicity are closely related to tricarboxylic acid cycle (TCA) substrates. For example, aspartate can be deaminated to produce oxaloacetate by L-aspartate transaminase enzymes Aat1 (mitochondrial) or Aat2 (cytoplasmic). Figure 4A shows supplementing cultures with increasing levels of oxaloacetic acid provides increasing protection from doxorubicin, again suggesting the possibility of aspartate influencing doxorubicin toxicity by contributing to carbohydrate metabolism. Oxaloacetic acid and acetyl-CoA are substrates for citrate synthase (CS) to produce citrate. This reaction can occur as part of the TCA cycle in mitochondria, but can also occur in the cytoplasm and peroxisome. Different isoforms of citrate synthase are located in different cellular compartments. If aspartate influences doxorubicin toxicity by altering carbon metabolism, then citrate synthase may be necessary for aspartate mediated doxorubicin resistance. Figure 4 shows changes in viability for cultures of parental, cit1, cit2 and cit3 mutant strains grown in minimal media with and without aspartate. Aspartate provides doxorubicin resistance independent of CIT2 and CIT3. However, a functional CIT1 is necessary to provide most of the aspartate-mediated doxorubicin protection seen in parental cells. cit1 mutants, unlike the parental culture, show similar sensitivity to doxorubicin with and without aspartate (Fig. 4B). The protection afforded by oxaloacetate and the sensitivity of cit1 mutants strongly implicate TCA cycle activity in providing aspartate mediated resistance to doxorubicin.
Figure 4.
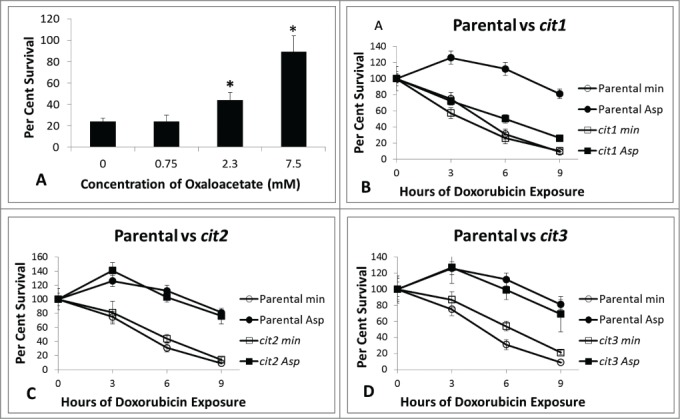
Survival for BY4741 with oxaloacetate and survival for citrate synthase mutants. Cultures of BY4741 with supplemented with varying concentrations of oxaloacetate were established. Doxorubicin was added to 150 micromolar and aliquots of the cultures were removed, diluted and plated to YPD plates at 0 hours and 6 hours to determine the number of colony forming units per ml. Survival is shown as the proportion of colony forming units per ml. at 6 hours divided by the colony forming units per ml at time 0 hour. Supplemented cultures with significant resistance relative to non-supplemented culture as determined by Student t-test are marked with an asterisk. Experimental conditions were identical with those in Figure 3 with the exception of a different supplement. B-D: Cultures were exposed to 150 micromolar doxorubicin in minimal media with and without 2.3 mM aspartate as described in Methods and Figure 1. Figure 4B shows cit1 mutants, 4C cit2 mutants and 4D cit3 mutants. Platings were performed in triplicate for each time-point (+/− one standard deviation). cit1 mutants are uniquely unable to utilize aspartate for doxorubicin protection whereas parental, cit2, and cit3 mutants are all protected in the presence of aspartate.
Doxorubicin is thought to act during DNA replication by interfering with proper topoisomerase activity and likely interferes with cell growth and division.2 S. cerevisiae cells divide by budding. The budding appearance of a cell reflects its position in growth and division. Newly formed daughter cells have no bud, but as they grow, a small bud appears, the bud grows in size then is released to form a new daughter cell. Under normal growth conditions non-budded cells are in G1 phase, cells with small buds are beginning S phase, cells with large buds are in G2.10 Parental and cit1 mutants grown in minimal media with and without aspartate were visually inspected using a haemocytometer to determine cell number and budding appearance. The number of total cells and the cells with various budding patterns were counted in aliquots of cultures of different strains either with or without 2.3 mM aspartate. Cells were examined before and at various times during doxorubicin exposure using a haemocytometer. The number of cells with no bud, small buds, large buds and multiple buds were simply counted. This approach can determine changes in total cell number and relative changes in budding type (cell cycle position) over time. Parental BY4741 cultures grown in minimal media with aspartate continue to divide approximately one additional doubling after exposure to doxorubicin. Parental cultures exposed to doxorubicin in minimal media without aspartate show very little increase in cell number. cit1 mutants show little increase in cell number after doxorubicin exposure independent of the presence of aspartate (Fig. 5). Doubling times for cultures grown in minimal media without doxorubicin is 1.8 hours with aspartate and 2.1 hours without aspartate for both BY4741 and cit1 strains.
Figure 5.
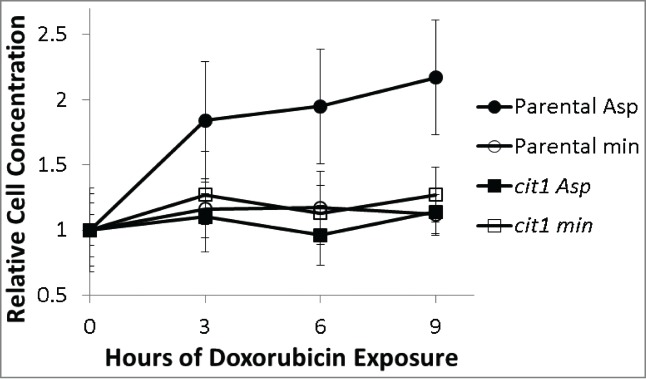
Relative cell concentration changes following 150 micromolar doxorubicin exposure of parental (circles) and cit1 mutants (squares) in minimal media alone (open symbols) or with 2.3 mM aspartate supplementation (closed symbols). Cell concentration was determined by counting aliquots of the doxorubicin treated cultures with a haemocytometer. Three samples were counted for each condition at each time-point. Cell concentration was normalized to the cell concentration determined at time 0 for each culture (+/− one standard deviation). Parental, but not cit1 mutants are able to increase cell concentration when aspartate is present.
TCA cycle activity enhances cell cycle arrest and survival
DNA damage typically leads to cell cycle arrest in G2, where cells have large buds. Topoisomerase II (TOP2) is an essential gene in S. cerevisae and likely one of the main targets of doxorubicin.2 Cells bearing a temperature sensitive allele of top2 arrest with large buds, in G2, at restrictive temperatures.11 Cells treated with radiation sufficient to cause DNA damage and cytotoxicity also arrest in G2 with large buds.12 The percentage of cells at various budding stages does not change significantly for either parental or cit1 cultures grown in minimal media and exposed to doxorubicin. Parental cells exposed to doxorubicin in minimal media with aspartate show an altered distribution, with a decrease in non-budded cells and an increase in the percentage of cells with large or multiple buds (Fig. 6). Parental cells grown in rich (YPD) media show a similar pattern of one doubling in cell number then arrest mainly as cells with large or multiple buds. cit1 mutants do not change distribution during doxorubicin exposure even in the presence of aspartate (Fig. 6). These data suggest aspartate, functioning through TCA cycle activity, allows cells to progress through one division cycle and ultimately arrest later in the cell cycle in G2. These data also show parental cells arrest promptly upon doxorubicin exposure in non-supplemented minimal media. The arrest appears much more abrupt in the absence of aspartate and/or a functional TCA cycle.
Figure 6.
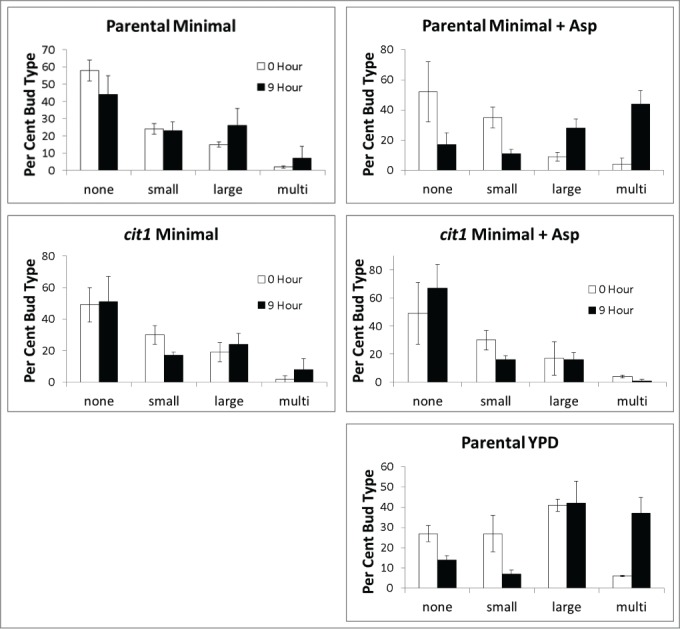
Budding patterns in parental and cit1 cultures following doxorubicin exposure in minimal media with and without aspartate. Cultures described in Figure 5 were further characterized by counting the type of cell and bud, as described in Methods. Analysis was performed before (open bars) and after (solid bars) 9 hours of doxorubicin exposure. A separate culture in complete (rich YPD) media was analyzed in the same way at 0 and 9 hours of doxorubicin exposure. Percentage for each bud type was determined as the number of cells with a particular budding pattern was divided by the total number of cells at the respective time-point. Error bars represent +/− one standard deviation. Budding pattern distribution changes after doxorubicin exposure require aspartate and a functional CIT1.
NADH metabolism influences doxorubicin toxicity
NADH is an important product of the TCA cycle. Cellular content of NADH was determined for BY4741 cells grown in minimal media with and without aspartate, with and without doxorubicin exposure. Levels of NAD and NADH were determined using a commercially available enzymatic cycling reaction coupled to a colorimetric assay. Table 1 shows the ratio of NADH to total NADH+NAD (redox potential) under various conditions. Parental cells grown in the presence of aspartate have a higher ratio of NADH to (NADH+NAD) consistent with the ability of aspartate to contribute to NADH production. Elevated NADH in aspartate grown cells is consistent with increased TCA cycle activity. Doxorubicin treatment was associated with a lower NADH to (NAD+NADH) ratio in aspartate cultures, providing preliminary evidence that NADH metabolism changes upon doxorubicin exposure. The change in NADH to (NAD+NADH) shown in Table 1 is small but consistent over 3 independent cultures for each condition with each culture tested in duplicate.
Table 1.
NAD and NADH levels were determined for parental strain BY4741 grown in minimal media with and without 2.25 mM aspartate in the presence and absence of 150 micromolar doxorubicin. A colorimetric assay was used as described in Methods. Determinations were made in duplicate from 3 independent cultures. The NADH/(NAD+NADH) ratio from different conditions was compared to the ratio from cells grown in minimal medium only using the Student t-test (n = 3). NA is not applicable and NS is not significant, avg. is average and sd is one standard deviation.
| NADH (NAD+NADH) |
|||
|---|---|---|---|
| Media | avg. | sd | t test vs. min |
| min | 0.27 | 0.05 | NA |
| min+dox | 0.29 | 0.03 | NS |
| min+asp | 0.34 | 0.05 | p = 0.02 |
| min+asp+dox | 0.29 | 0.03 | NS |
NADH can be used for several purposes. In broad terms, it can serve as a source of electrons for oxidative phosphorylation or as a cofactor for many different reactions. S. cerevisiae has 2 different isoforms of NADH dehydrogenase. Both forms have an electron transport portion in the mitochondrion. One isoform, Nde1, has a cytoplasmic surface that can directly oxidize cytoplasmic NADH. The other isoform, Ndi1, oxidizes mitochondrial NADH. Parental cells, nde1 and ndi1 mutants were exposed to doxorubicin in minimal media with and without aspartate (Fig. 7). ndi1 mutants grown without aspartate are slightly resistant to doxorubicin compared to the parental strain. Aspartate does not provide additional doxorubicin resistance in the absence of Ndi1. Ndi1 activity, and therefore oxidative phosphorylation, is required for the mechanism by which aspartate affords doxorubicin resistance to the cell.
Figure 7.
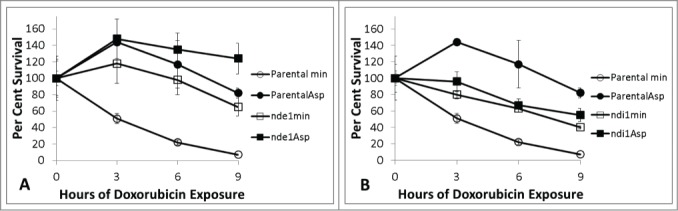
Doxorubicin survival of nde1 (7A) and ndi1 (7B) mutants relative to parental cells in minimal media with and without aspartate treated with 150 micromolar doxorubicin. Parental cultures are represented in circles, mutants in squares. Minimal media alone in open symbols and 2.3mM aspartate supplemented cultures in closed symbols. Platings were performed in triplicate at each time-point. Error bars represent +/− one standard deviation. Aspartate does not afford protection in ndi1 mutants.
nde1 mutants are also resistant to doxorubicin in the absence of aspartate (Fig. 7). nde1 mutants show even more resistance to doxorubicin in the presence of aspartate. nde1 mutants have less capacity to oxidize cytoplasmic NADH. These findings suggest higher levels of cytoplasmic NADH may also decrease doxorubicin toxicity. Nde1 activity does not appear necessary specifically for aspartate mediated doxorubicin resistance, however. The resistance of nde1 mutants in minimal media and the dependence on Ndi1 for aspartate mediated doxorubicin resistance both suggest cytoplasmic and mitochondrial NADH metabolism are important in mediating doxorubicin toxicity. Since both ndi1 and nde1 mutants show doxorubicin resistance in minimal media, NADH dehydrogenase activity may contribute to doxorubicin toxicity, consistent with previous work showing electron transport activity increases doxorubicin toxicity.13
Aspartate import into mitochondria is necessary for doxorubicin protection
Aspartate can facilitate TCA cycle activity in several ways. Aspartate can be deaminated to oxaloacetate either in the cytoplasm by Aat2 or in the mitochondria by Aat1. Transport of either aspartate or oxaloacetate could provide substrates for the TCA cycle. At least 35 transporter proteins active in shuttling metabolites between cytoplasm and mitochondria have been identified.14 The substrates for many but not all of these transport proteins have been described.
Agc1 is a transport protein capable of transporting aspartate and glutamate between cytoplasm and mitochondria. Aspartate provides less protection for agc1 mutants than parental cells (Fig. 8). aat1 mutants also show less aspartate mediated protection (Fig. 8). These results suggest that aspartate mediated doxorubicin protection is partly dependent upon import of aspartate into mitochondria and subsequent deamination to oxaloacetate. The growth rate for parental, agc1 and aat1 strains are comparable in minimal and minimal with aspartate media.
Figure 8.
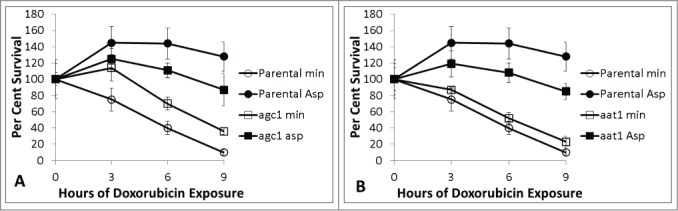
Doxorubicin survival of agc1 (8A) and aat1 (8B) mutants relative to parental cells in minimal media with and without aspartate. Parental cultures are represented in circles, mutants in squares. Minimal media alone in open symbols and 2.3 mM aspartate supplemented cultures in closed symbols. 150 micromolar doxorubicin was used for each experiment. Platings were performed in triplicate for all time-points. Error bars represent +/− one standard deviation. agc1 and aat1 mutants show some, but incomplete, aspartate mediated doxorubicin protection.
One well described pathway for aspartate entry into mitochondria is the aspartate malate shuttle. The malate shuttle includes aspartate deamination to oxaloacetate by Aat2 transaminase in the cytoplasm, then reduction to malate via malate dehydrogenase (Mdh2). Malate is then transported into mitochondria and is available for TCA cycle activity. aat2 and mdh2 mutants and parental strains have comparable sensitivity to doxorubicin, as shown in Figure 9. aat2 mutants are auxotrophic for aspartate and therefore the aat2 mutant could not be reliably assayed in minimal media without aspartate. The data shown in Figure 9 indicate that the aspartate malate shuttle is not necessary for aspartate mediated doxorubicin resistance.
Figure 9.
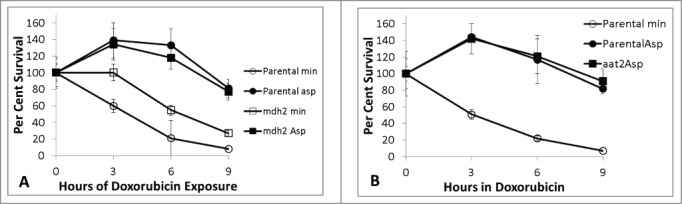
Doxorubicin survival of mdh2 (9A) relative to parental in minimal media with and without aspartate. Figure 9B shows relative survival for aat2 in minimal media with 2.3 mM aspartate only because aat2 is auxotrophic for aspartate under these conditions. 150 micromolar doxorubicin was used for all experiments. Platings were done in triplicate for all time-points. Error bars represent +/− one standard deviation. Aspartate affords protection against doxorubicin independently from the aspartate – malate shuttle system.
Discussion
The results presented herein demonstrate the nutritional status of a cell can dramatically affect doxorubicin toxicity. Cultures supplemented with amino acids closely linked to the TCA cycle are resistant to doxorubicin relative to cultures exposed to doxorubicin in minimal media alone. Aspartate mediated protection is dependent on specific mitochondrial transporter proteins, a functional TCA cycle and electron transport pathways. Cultures exposed in minimal media show little if any cell cycle progression, whereas cultures exposed to doxorubicin in minimal media with aspartate double in cell number ultimately arresting later in the cell cycle, similar to cultures exposed in rich media. The growth and cell cycle progression is dependent on aspartate and a functional CIT1. These findings have significant implications for understanding the mechanism of cellular toxicity of doxorubicin and potentially other DNA damaging agents.
Nutrition and metabolism play significant roles in response to DNA damage. One of the earliest applications of genome wide expression studies in S. cerevisiae examined transcriptional responses to a variety of adverse conditions, including DNA damaging agents.15,16 Exposure to a wide variety of agents induced similar changes. Expression changes in dozens of genes were found, including genes encoding proteins involved in DNA repair, reactive oxygen species detoxification, and carbon and amino acid metabolism. Expression of genes involved in trehalose and glycogen synthesis was induced and expression of genes involved in glycolysis was repressed. Gene expression studies indicate a change in metabolism from glucose consumption to glucose storage. Subsequent work by Kitanovic et al. has shown treatment with the DNA damaging agent methyl methanesulfonate (MMS) inhibits glucose consumption and enhances glycogen and trehalose production.17 In addition, MMS treatment increased citrate synthase activity and led to an increase in the NAD+/NADH ratio reflecting a shift away from glycolysis and toward respiratory metabolism.17 These studies suggest glycolysis activity decreases and respiratory metabolism increases in the wake of DNA damage. The metabolic shift away from glycolysis is part of a larger trend of decreased expression of biosynthetic genes and may be fundamentally important for cell cycle arrest after DNA damage. However, some degree of cell cycle progression may be necessary to allow cells to complete DNA replication and repair for optimal cell cycle arrest. In conditions where glycolysis is inhibited, cells may not be able to generate the energy and biomolecules needed for repair and cell cycle progression. Glycolysis intermediates play a fundamental role in the production of many amino acids and nucleotides.18 If glycolysis stops, the metabolic requirements to complete replication and repair for optimal arrest may be generated by TCA and mitochondrial activity. One example is serine. This amino acid is produced from the glycolytic intermediate phosphoglycerate during fermentative growth when glycolysis is active but is produced from the glyoxylate pathway within the TCA cycle when cells are respiring.19 In the absence of glycolysis, TCA cycle activity would require anaplerotic input from such sources as aspartate, asparagine, glutamate, glutamine and alanine. Our data show cell growth is halted promptly when doxorubicin is added to cultures growing in minimal media. Aspartate, after import into mitochondria and conversion to oxaloacetate, may sufficiently fuel the TCA cycle to produce energy and biomolecules necessary to complete replication and arrest at an optimal position in the cell cycle.
DNA damage poses a complicated threat to cells. Continued growth and division in the presence of DNA damaging agents could magnify and propagate genetic damage leading to mutation and mitotic catastrophe. Arresting growth and division could provide protection against further damage. DNA is significantly more vulnerable to damage during replication when it is single stranded and less protected by histones and other chromatin proteins. Therefore, arrest can provide protection, but cells must arrest at an appropriate place in the cell cycle. Data from this work shows the improved survival seen during doxorubicin exposure in rich media and minimal media with aspartate is associated with one passage through the cell cycle (one doubling of the culture) and arrest late in the cell cycle, likely when replication is complete as signaled by cells with large or multiple buds. Topoisomerase II (TOP2) is likely one target of doxorubicin2 and mutants bearing a temperature sensitive allele of top2 arrest with large buds at the non-permissive temperature.11 The activities of Agc1, Aat1, Cit1 and Ndi1 are necessary for optimal aspartate mediated protection. Together, these activities describe a flow of aspartate into mitochondria, conversion to oxaloacetate, which is then used in the TCA cycle to generate NADH for electron transport. This sequence of events is associated with progression in growth and division, allowing division to occur once (a doubling in cell concentration) then arrest similar to that seen for cells exposed in rich media and with a greater survival. Some aspartate mediated protection against doxorubicin toxicity is seen in agc1 and aat1 mutants, suggesting aspartate may provide additional protection by other means in addition to anapleurosis. Aspartate provides protection for at least 22 hours, equivalent to over 10 doubling times for similar culture without doxorubicin. However, it remains possible that aspartate may allow cells to continue to grow in the presence of doxorubicin allowing greater accumulation of mutations and DNA damage ultimately leading to increased toxicity over a longer time-frame than investigated here. Human cells have activity for mitochondrial aspartate import,20 mitochondrial aspartate aminotransferase,21 mitochondrial citrate synthase,22 and mitochondrial NADH oxidase.23 Better understanding this conserved pathway may lead to more selective cancer treatments using doxorubicin.
Conditions favoring a higher ratio of NADH to total (NADH + NAD) nicotinamide dinucleotide are associated with doxorubicin resistance. Strains mutant for NADH oxidase (ndi1 and nde1) have less ability to oxidize NADH and likely have a higher proportion of NADH to total (NAD + NADH). nde1 and ndi1mutants are resistant to doxorubicin in the absence of aspartate. Parental cells grown in minimal media with aspartate have a higher percentage of reduced to total nicotinamide dinucleotide, and doxorubicin treatment lowered this ratio. The amount of change reported in Table 1 is small, but consistent in 3 independent experiments. The changes in relative NADH levels are likely complex following doxorubicin treatment. The data in Table 1 provide preliminary evidence that NADH metabolism may be important in response to doxorubicin. The ability of doxorubicin to oxidize NADH3 may lower NADH levels during treatment and compromise cellular response and survival.
The data presented in this work show maneuvers that increase the relative abundance of NADH, such as blocking NADH oxidation or increasing TCA cycle activity with anaplerotic amino acids, lead to doxorubicin resistance. These findings may provide insight into doxorubicin cardiotoxicity. Cardiac myocytes respond to doxorubicin treatment by increasing expression of genes related to the TCA cycle and complex I, presumably to compensate for NADH oxidation by doxorubicin.24 TCA cycle intermediates are present in low abundance and turn-over is rapid.25 Cardiac myocytes would likely need to increase TCA cycle intermediates to increase TCA cycle activity. Anaplerotic substrates such as pyruvate, alanine, aspartate, glutamate and others can decrease the toxicity of other forms of cardiac injury.26 If the supply of anaplerotic substrates is not sufficient to meet cardiac needs during doxorubicin exposure, muscle fibers could be catabolized to provide anaplerotic amino acids including aspartate, glutamate, alanine and others. Muscle fiber catabolism could lead to the degradation characteristic of doxorubicin cardiomyopathy.27 The possibility that doxorubicin induced cardiac myopathy results from muscle protein degradation to provide anaplerotic amino acids may warrant further investigation.
Increased mitochondrial activity has been associated with increased doxorubicin toxicity in S. cerevisiae. Specifically, Kule et al.13 have shown cells growing under respiratory conditions with an intact electron transport chain show greater doxorubicin toxicity than fermenting cells or cells without electron transport capabilities. Respiration may increase oxidative stress and therefore increase doxorubicin toxicity. Glutathione depletion also increases oxidative stress and enhances doxorubicin toxicity in S. cerevisiae.28 nde1 and ndi1 mutants examined in this work have decreased electron transport ability and decreased doxorubicin toxicity in non-supplemented minimal media. All these observations suggest increased mitochondrial activity, specifically electron transport and oxidative phosphorylation, increase doxorubicin toxicity. However, our data suggests aspartate enhances mitochondrial TCA cycle activity to decrease doxorubicin toxicity. These observations lead to an apparent conflict, where mitochondrial activity both increases and decreases doxorubicin toxicity. This apparent conflict may be reconciled by considering different effects of mitochondrial activity at different times during doxorubicin exposure. The benefit of mitochondrial activity reported here is limited to cells grown in nutrient deficient media supplemented with aspartate and limited to a brief period of time; a few hours after initial exposure. Cell survival over longer periods of time subsequently declines at similar rates in cultures with and without aspartate (Fig. 4). This suggests the mitochondrial activity enabled by aspartate provides only transient protection during initial exposure. Aspartate was supplied in significant excess, making consumption of aspartate during doxorubicin exposure possible but unlikely. Mitochondria provide many functions for the cell. Electron transport and oxidative phosphorylation provides energy. The TCA cycle provides not only reducing equivalents for electron transport but also a number of key metabolites necessary for biomolecule syntheses.29 The TCA cycle may be an even more important source for biomolecular precursors when glycolysis is slowed or inhibited, as occurs following DNA damage.17 Mitochondrial activity of the TCA cycle may be necessary for the initial response to doxorubicin to supply precursors necessary to complete DNA replication and arrest in a protected, ideal phase of the cell cycle thereby decreasing doxorubicin toxicity. The majority of arrest appears to occur within 3 hours after adding doxorubicin to minimal media with aspartate. Over longer periods of time, increased mitochondrial activity of electron transport and oxidative phosphorylation may increase oxidative stress and increase doxorubicin toxicity. Doxorubicin may trigger increased mitochondrial TCA cycle synthetic activity to decrease toxicity but subsequent increased mitochondrial electron transport and oxidative phosphorylation activity may then increase cytotoxicity. The dual function of mitochondria may explain why aspartate can enhance mitochondrial TCA cycle activity to decrease doxorubicin toxicity while agents such as troglitazone that amplify mitochondrial electron transport activity enhance the toxicity of doxorubicin.30 If doxorubicin causes an increase in mitochondrial activity as an early response, and subsequently produces mitochondrial damage through superoxide production, cells would need to adjust energy and biomolecule production away from mitochondrial sources. This is rather consistent with our prior observation in cardiac tissue from rats treated subacutely with doxorubicin wherein both gene expression and metabolic flux studies indicate a drug-induced reprogramming of cellular metabolism from aerobic fatty acid oxidation to anaerobic glycolysis, with a corresponding stimulation of mitochondrial biogenesis in affected cardiac tissue.31,32 Doxorubicin may incite a complex series of metabolic changes and adaptations.
Given the stark differences in energy metabolism and mitochondrial activity between normal and cancer cells, the initial exposure may provide a window of time to selectively enhance doxorubicin toxicity in cancer cells or provide doxorubicin protection in normal cells. Cancer cells with enhanced aerobic glycolysis activity may be less able or unable to shift to mitochondrial activity to achieve optimal arrest. Along these lines, starvation has been shown to widen the therapeutic ratio of doxorubicin and other cancer treatments.33 One consequence of starvation is to shift metabolism to increase TCA cycle and mitochondrial activity. The benefit of starvation in cancer treatment may in part lie with increasing mitochondrial activity in normal cells allowing a more adept arrest response, whereas cancer cells are less able to increase mitochondrial activity during starvation.
Aspartate plays a role in many synthetic pathways. It is a precursor for other amino acids such as threonine, serine, methionine and others. Aspartate is also a precursor in purine and pyrimidine synthesis. Mutants in these synthetic pathways were also identified in genetic searches for doxorubicin sensitive strains.6,7 Hartman has shown aspartate contributes to hydroxyurea resistance by facilitating nucleotide synthesis.34 The data presented here show aspartate provides protection to cultures exposed to doxorubicin in minimal media largely through conversion to oxaloacetate and subsequent metabolism by Cit1. The importance of TCA cycle activity in responding to doxorubicin is confirmed by the large scale mutational studies of Xia and Westmoreland.6,7 Mutations in TCA cycle participants ACO1, ACO2 and IDH1 also lead to increased sensitivity to doxorubicin.6,7 The requirement of aspartate for both synthetic pathways and NADH production suggests response to doxorubicin requires a careful balance of arrest, repair and growth activities. The data presented here in combination with the system wide genetic data from S. cerevisiae show the response to doxorubicin involves a large coordinated effort between metabolic changes and DNA repair and replication for optimal survival.
Material and Methods
Strains
All strains were obtained through Open Biosystems. The parental strain used is BY4741 (Mata, ura3, leu2, his3, met15). Mutant strains were obtained from Open Biosystems in the same background with the gene under study deleted and replaced by the selectable neomycin resistance gene.35 Strains were maintained on glucose media (YPD) but checked periodically on glycerol media (YPG) to ensure cultures being tested maintained mitochondrial function.
Media
YPD is rich media replete with nutrients. YPD liquid and YPD plates were prepared as described in the Cold Spring Harbor Methods in Yeast Genetics.36 YPD contains 1% yeast extract, 2% peptone and 2% glucose. Two% agar is added to make plates. YPG media contains 1% yeast extract, 2% peptone and 3% glycerol. Minimal media contained 2% glucose, 5 g./L ammonium sulfate, 6.7 g/L yeast nitrogen base (Difco), 50mM phosphate buffer composed of NaH2PO4 and K2HPO4 brought to pH 5.5. The parental strain BY4741 is auxotrophic for histidine, uracil, methionine and leucine and these were supplemented for all minimal media experiments. Amino acid supplementation provided 3-fold the minimum recommended concentrations for each amino acid based on standard media recommendations.36 The final concentrations for amino acid supplementation unless otherwise indicated were: 60 µg/milliliter for arginine, histidine, proline, methionine, cysteine and glycine, 90 µg/milliliter for lysine, tyrosine and isoleucine, 120 µg/milliliter for tryptophan, 150 µg/milliliter for phenylalanine, 300 µg/milliliter for aspartate, alanine, glutamate, glutamine, leucine, 450 µg/milliliter for valine, 600 µg/milliliter for asparagine, threonine, and 1200 µg/milliliter for serine. For aspartate, 300 µg/milliliter is 2.3 mM.
Drug treatment
Doxorubicin was obtained from Sigma (http://www.sigmaaldrich.com/catalog/product/sigma/44583?lang=enandregion=US). Stock solutions were prepared at 15 mM in water and diluted to the indicated concentrations in cultures. Cultures were started from a single colony, grown overnight, diluted 1:500 into the same media, then outgrown again for 6–20 hours. Cultures for doxorubicin sensitivity testing ranged from 1 × 106 to 6 × 106 cells per ml. during outgrowth, then diluted to 1 × 105 cells per ml. during doxorubicin exposure.
Survival assay and calculations: Survival was measured using a time course of doxorubicin exposure to a culture and measuring the concentration of viable cells per ml. over time. Cultures were grown and diluted as described. Fresh drug was added and cells diluted and plated to determine initial colony forming units per ml. At various times, aliquots were removed, diluted and plated. The volume of culture plated was chosen to provide approximately 100 colonies per plate. All platings were done in triplicate to YPD plates. Survival was determined as the number of colony forming units per ml. at time x relative to the initial number of colony forming units per ml. This approach determines the global behavior of the culture, including growth inhibition as well as loss of viability. For example, an increase in the number of viable cells per ml. (growth of the culture) gives a relative survival greater than 100%. Survival curves are shown with error bars depicting one standard deviation. Doxorubicin response experiments were performed a minimum of 3 independent exposure cultures; representative experiments are shown in the figures.
Cell counting studies
Cultures were established, grown, diluted and exposed to doxorubicin as described above, except the initial cell concentration was 3 × 105. Aliquots were withdrawn at time periods and inspected on a haemocytometer. Cells were counted as non-budded, small budded if the bud was less than half the size of the parent, large budded if the bud was greater than half the size of the parent or multi-budded if more than one bud was present. Total cell number was determined. The number of cells with each budding pattern was also determined.
NAD and NADH determination was performed as described by Ocampo, et al. using BioVision NAD/NADH colorimetric determination kit (K337–100, http://www.biovision.com/nad-nadh-quantitation-colorimetric-kit-2853.html).37 Cells were grown as described above and exposed to doxorubicin at a cell concentration of 1 × 106 cells / ml and doxorubicin concentration of 150 micromolar for 3 hours. Cells were then collected, washed, treated with zymolyase and mixed with extraction buffer from the kit.37 Manufacturer's instructions were followed. Contaminating enzyme activity was removed by passing the extraction supernatant through BioVision columns (catalog # 1997–25, http://www.biovision.com/10k-spin-column-5277.html). Reactions were run in a 96 well plate and absorbance at 450nm was measured. A standard curve using known quantities of NAD and NADH was established for each experimental run. Measurements were made in duplicate for each sample. The results of 3 independent treated culture determinations are presented.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgment
The authors wish to express gratitude and appreciation to Dr. Lester Drewes for facilitating collaboration between the University of Minnesota, Duluth and Essentia Health.
Funding
This work was supported by Essentia Health and the Essentia Health Foundation; the Duluth Medical Research Institute; the Whiteside Institute for Clinical Research and the Moe Cancer Research Foundation.
References
- 1.Di Marco A, Cassinelli G, Arcamone F. The discovery of daunorubicin. Cancer Treat Rep 1981; 65 Suppl 4:3-8; PMID:7049379. [PubMed] [Google Scholar]
- 2.Patel S, Sprung AU, Keller BA, Heaton VJ, Fisher LM. Identification of yeast DNA topoisomerase II mutants resistant to the antitumor drug doxorubicin: implications for the mechanisms of doxorubicin action and cytotoxicity. Mol Pharmacol 1997 Oct; 52(4):658-66. [DOI] [PubMed] [Google Scholar]
- 3.Wallace KB. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol Toxicol 2003 Sep; 93(3):105-15; http://dx.doi.org/ 10.1034/j.1600-0773.2003.930301.x. [DOI] [PubMed] [Google Scholar]
- 4.Denard B, Lee C, Ye J. Doxorubicin blocks proliferation of cancer cells through proteolytic activation of CREB3L1. Elife 2012 Dec 18; 1:e00090; http://dx.doi.org/ 10.7554/eLife.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eom YW, Kim MA, Park SS, Goo MJ, Kwon HJ, Sohn S, Kim WH, Yoon G, Choi KS. Two distinct modes of cell death induced by doxorubicin: apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. Oncogene 2005 Jul 14; 24(30):4765-77; http://dx.doi.org/ 10.1038/sj.onc.1208627. [DOI] [PubMed] [Google Scholar]
- 6.Xia L1, Jaafar L, Cashikar A, Flores-Rozas H. Identification of genes required for protection from doxorubicin by a genome-wide screen in Saccharomyces cerevisiae. Cancer Res 2007 Dec 1; 67(23):11411-8; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Westmoreland TJ, Wickramasekara SM, Guo AY, Selim AL, Winsor TS, Greenleaf AL, Blackwell KL, Olson JA Jr, Marks JR, Bennett CB. Comparative genome-wide screening identifies a conserved doxorubicin repair network that is diploid specific in Saccharomyces cerevisiae. PLoS One 2009 Jun 8; 4(6):e5830; http://dx.doi.org/ 10.1371/journal.pone.0005830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011. March 4; 144(5):646-74. [DOI] [PubMed] [Google Scholar]
- 9.Dornfeld K, Johnson M. AP endonuclease deficiency results in extreme sensitivity to thymidine deprivation. Nucleic Acids Res 2005 Nov 27; 33(20):6644-53; http://dx.doi.org/ 10.1093/nar/gki975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howell AS, Lew DJ. Morphogenesis and the cell cycle. Genetics 2012 Jan; 190(1):51-77; http://dx.doi.org/ 10.1534/genetics.111.128314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DiNardo S, Voelkel K, Sternglanz R. DNA topoisomerase II mutant of Saccharomyces cerevisiae: topoisomerase II is required for segregation of daughter molecules at the termination of DNA replication. Proc Natl Acad Sci U S A 1984 May; 81(9):2616-20; http://dx.doi.org/ 10.1073/pnas.81.9.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mercier G, Denis Y, Marc P, Picard L, Dutreix M. Transcriptional induction of repair genes during slowing of replication in irradiated Saccharomyces cerevisiae. Mutat Res 2001 Dec 19; 487(3-4):157-72; http://dx.doi.org/ 10.1016/S0921-8777(01)00116-1. [DOI] [PubMed] [Google Scholar]
- 13.Kule C, Ondrejickova O, Verner K. Doxorubicin, daunorubicin, and mitoxantrone cytotoxicity in yeast. Mol Pharmacol 1994 Dec; 46(6):1234-40. [PubMed] [Google Scholar]
- 14.Palmieri F, Agrimi G, Blanco E, Castegna A, Di Noia MA, Iacobazzi V, Lasorsa FM, Marobbio CM, Palmieri L, Scarcia P, et al.. Identification of mitochondrial carriers in Saccharomyces cerevisiae by transport assay of reconstituted recombinant proteins. Biochim Biophys Acta 2006 Sep-Oct; 1757(9-10):1249-62; http://dx.doi.org/ 10.1016/j.bbabio.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 15.Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 2000 Dec; 11(12):4241-57; http://dx.doi.org/ 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jelinsky SA, Samson LD. Global response of Saccharomyces cerevisiae to an alkylating agent. Proc Natl Acad Sci U S A 1999 Feb 16; 96(4):1486-91; http://dx.doi.org/ 10.1073/pnas.96.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitanovic A, Walther T, Loret MO, Holzwarth J, Kitanovic I, Bonowski F, Van Bui N, Francois JM, Wölfl S. Metabolic response to MMS-mediated DNA damage in Saccharomyces cerevisiae is dependent on the glucose concentration in the medium. FEMS Yeast Res 2009 Jun; 9(4):535-51; http://dx.doi.org/ 10.1111/j.1567-1364.2009.00505.x. [DOI] [PubMed] [Google Scholar]
- 18.Ljungdahl PO, Daignan-Fornier B. Regulation of amino acid, nucleotide, and phosphate metabolism in Saccharomyces cerevisiae. Genetics 2012 Mar; 190(3):885-929; http://dx.doi.org/ 10.1534/genetics.111.133306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Albers E, Laizé V, Blomberg A, Hohmann S, Gustafsson L. Ser3p (Yer081wp) and Ser33p (Yil074cp) are phosphoglycerate dehydrogenases in Saccharomyces cerevisiae. J Biol Chem 2003 Mar 21; 278(12):10264-72; http://dx.doi.org/ 10.1074/jbc.M211692200. [DOI] [PubMed] [Google Scholar]
- 20.Palmieri L, Pardo B, Lasorsa FM, del Arco A, Kobayashi K, Iijima M, Runswick MJ, Walker JE, Saheki T, Satrustegui J, et al.. Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J 2001; 20:5060-9; PMID:11566871; http://dx.doi.org/ 10.1093/emboj/20.18.5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fleisher GA, Potter CS, Wakim KG. Separation of 2 glutamic-oxalacetic transaminases by paper electrophoresis Proc Soc Exp Biol Med 1960 Jan; 103:229-31; http://dx.doi.org/ 10.3181/00379727-103-25469. [DOI] [PubMed] [Google Scholar]
- 22.Reisch AS, Elpeleg O. Biochemical assays for mitochondrial activity: assays of TCA cycle enzymes and PDHc. Methods Cell Biol 2007; 80:199-222; PMID:17445696; http://dx.doi.org/ 10.1016/S0091-679X(06)80010-5. [DOI] [PubMed] [Google Scholar]
- 23.Santidrian AF, Matsuno-Yagi A, Ritland M, Seo BB, LeBoeuf SE, Gay LJ, Yagi T, Felding-Habermann B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest 2013 Mar; 123(3):1068-81; http://dx.doi.org/ 10.1172/JCI64264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tokarska-Schlattner M, Lucchinetti E, Zaugg M, Kay L, Gratia S, Guzun R, Saks V, Schlattner U. Early effects of doxorubicin in perfused heart: transcriptional profiling reveals inhibition of cellular stress response genes. Am J Physiol Regul Integr Comp Physiol 2010 Apr; 298(4):R1075-88; http://dx.doi.org/ 10.1152/ajpregu.00360.2009. [DOI] [PubMed] [Google Scholar]
- 25.Des Rosiers C, Labarthe F, Lloyd SG, Chatham JC. Cardiac anaplerosis in health and disease: food for thought. Cardiovasc Res 2011 May 1; 90(2):210-9; http://dx.doi.org/ 10.1093/cvr/cvr055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olson AK, Hyyti OM, Cohen GA, Ning XH, Sadilek M, Isern N, Portman MA. Superior cardiac function via anaplerotic pyruvate in the immature swine heart after cardiopulmonary bypass and reperfusion. Am J Physiol Heart Circ Physiol 2008 Dec; 295(6):H2315-20; http://dx.doi.org/ 10.1152/ajpheart.00739.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geisberg C, Pentassuglia L, Sawyer DB. Cardiac side effects of anticancer treatments: new mechanistic insights. Curr Heart Fail Rep 2012 Sep; 9(3):211-8; http://dx.doi.org/ 10.1007/s11897-012-0098-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buschini A, Poli P, Rossi C. Saccharomyces cerevisiae as an eukaryotic cell model to assess cytotoxicity and genotoxicity of three anticancer anthraquinones Mutagenesis. 2003. January; 18(1):25-36. [DOI] [PubMed] [Google Scholar]
- 29.Scheffler IE. Mitochondria Second Ed. Hoboken, NJ: John Wiley and Sons; c2008. Chapter 6 Metabolic Pathways Inside Mitochondria, p. 298-344. [Google Scholar]
- 30.Skildum A, Dornfeld K, Wallace K. Mitochondrial amplification selectively increases doxorubicin sensitivity in breast cancer cells with acquired antiestrogen resistance. Breast Cancer Res Treat 2011 Oct; 129(3):785-97; http://dx.doi.org/ 10.1007/s10549-010-1268-2. [DOI] [PubMed] [Google Scholar]
- 31.Berthiaume JM, Wallace KB. Persistent alterations to the gene expression profile of the heart subsequent to chronic doxorubicin treatment. Cardiovasc Toxicol 2007; 7:178-191; PMID:17901561; http://dx.doi.org/ 10.1007/s12012-007-0026-0. [DOI] [PubMed] [Google Scholar]
- 32.Carvalho RA, Sousa RPB, Cadete VJJ, Lopaschuk GD, Palmeira CMM, Bjork JA, Wallace KB. Metabolic remodelling associated with subchronic doxorubicin cardiomyopathy. Toxicology 2010; 270:92-98; PMID:20132857; http://dx.doi.org/ 10.1016/j.tox.2010.01.019. [DOI] [PubMed] [Google Scholar]
- 33.Lee C, Safdie FM, Raffaghello L, Wei M, Madia F, Parrella E, Hwang D, Cohen P, Bianchi G, Longo VD. Reduced levels of IGF-I mediate differential protection of normal and cancer cells in response to fasting and improve chemotherapeutic index. Cancer Res 2010 Feb 15; 70(4):1564-72; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartman JL., 4th Buffering of deoxyribonucleotide pool homeostasis by threonine metabolism. Proc Natl Acad Sci U S A 2007 Jul 10; 104(28):11700-5; http://dx.doi.org/ 10.1073/pnas.0705212104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giaever G, Nislow C. The yeast deletion collection: a decade of functional genomics. Genetics 2014 Jun; 197(2):451-65; http://dx.doi.org/ 10.1534/genetics.114.161620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burke D, Dawson D, Stearns T. Methods in Yeast Genetics, Appendix A, Media, pp 171-182 Cold Spring Harbor Laboratory Press, Plainview, NY: 2000. [Google Scholar]
- 37.Ocampo A, Liu J, Barrientos A. NAD+ salvage pathway proteins suppress proteotoxicity in yeast models of neurodegeneration by promoting the clearance of misfolded/oligomerized proteins. Hum Mol Genet 2013 May 1; 22(9):1699-708; http://dx.doi.org/ 10.1093/hmg/ddt016. [DOI] [PMC free article] [PubMed] [Google Scholar]