

Abstract
Apoptosis plays a critical physiological role in controlling cell number and eliminating damaged, non-functional and transformed cells. Cancerous cells as well as some types of normal cells are often resistant to cell death induced by pro-inflammatory cytokines through death receptors. This potentially allows cancer cells to evade the control from the immune system and to proceed toward a more malignant stage, although the mechanisms of this evasion are not well established. We have recently identified the stress-responsive Sestrin2 protein as a critical regulator of cell viability under stress conditions. Sestrin2 is a member of a small family of antioxidant proteins and inhibitors of mechanistic Target of Rapamycin Complex 1 (mTORC1) kinase. Down-regulation of Sestrin1/2 leads to genetic instability and accelerates the growth of lung adenocarcinoma xenografts. Here we addressed the potential role of Sestrin2 in regulation of cell death induced by TNFR1 and related Fas and TRAIL receptors in lung adenocarcinoma cells. We found that Sestrin2 silencing strongly inhibits cytokine-induced cell death through a mechanism independent of ROS and mTORC1 regulation. We determined that the X-linked inhibitor of apoptosis protein (XIAP) plays a critical role in the control of cytokine-induced cell death by Sestrin2. Thus our study defines a new, previously unrecognized role of Sestrin2 in the regulation of apoptosis.
Keywords: Sesn2, XIAP, death receptors, caspases, apoptosis
Introduction
Carcinogenesis is a process often opposed by a stress and accompanied by acute inflammation, which may cause elimination of cancer cells through induction of apoptosis; however, sustained inflammation is considered to be a promoter of carcinogenesis.1 Many cancer cells acquire resistance to cell death through downregulation of proapoptotic proteins and up-regulation of cell death inhibitors.2 The stress-responsive Sestrin2 (Sesn2) gene belongs to an evolutionary-conserved Sestrin gene family found in most eukaryotes.3-5 Sestrins support cell viability under oxidative and metabolic stress but sensitize cells to DNA-damage.3,6,7 The variability of the Sestrins-mediated responses is associated with several activities of Sestrins such as suppression of reactive oxygen species and inhibition of mechanistic Target of Rapamycin Complex 1 (mTORC1) kinase.6,8,9 The effects of mTORC1 on cell viability can be mediated by regulation of protein synthesis through phosphorylation p70S6K and 4EBP1 proteins or autophagosomal-lysosomal proteolysis via phosphorylation of ULK1 and ATG13 proteins.10-12 Sesn2 might have tumor suppressive function as it is a target of tumor suppressor p53,3 and is inactivated in the majority of human tumors.13 Deficiency of Sesn2 can facilitate transformation and stimulation of growth of lung adenocarcinoma xenografts,8,14,15 althou-gh the precise role of Sesn2 in suppression of carcinogenesis is yet to be established.
The immune system provides an additional level of protection from carcinogenesis by eliminating malignant cells through activation of death receptors (DR) such as Fas, TRAILR1/2 and, possibly, TNFR1. DR belong to the Tumor Necrosis Factor Receptor (TNFR) superfamily of type-I transmembrane proteins containing N-terminal cysteine-rich extracellular domain, transmembrane domain and C-terminus containing 80 amino-acid length peptide called death domain (DD).16,17 After interaction with cognate ligands, DR undergo conformational changes, leading to their oligomerization and recruitment of effector proteins transducing signals from the receptor.18 For example, activated TNFR1 recruits TRADD (TNFR1-associated Death Domain) and RIP1 (receptor interacting protein kinase 1) followed recruitment of FADD (Fas Associated Death Domain) protein via their DD. FADD in turn interacts with pro-caspase 8/10 death effector domain (DED), forming a complex called DISC, where procaspase 8/10 is cleaved and activated which triggers the activation of executive caspases 3, 6 and 7.19-21 Activated caspases also cleave Bid protein, a proapoptotic Bcl2 family member, which translocates to mitochondria and stimulates apoptosome formation and activation of caspase 9, 3, 6 and 7 amplifying the apoptotic cascade.22
TNFR1 also recruits TRAF2 (TNFR-associated factor 2), cIAP1 and cIAP2 (cellular inhibitors of apoptosis 1 and 2) proteins in a TRADD-dependent manner. RIP1 is ubiquitinated by cIAP1/2 following recruitment and activation of TAK and IKK kinases. IKK phosphorylates and stimulates proteosomal degradation of IκBα (inhibitor of κBα) and IκBα-related proteins, which work as inhibitors of NF-κB transcription factor. Once activated, NF-κB translocates to the nucleus and activates the expression of antiapoptotic genes such as cFLIP, cIAP1/2, XIAP, Bcl2, BclXL. For example, cFLIP is a close homolog of caspase 8 lacking its protease activity. When tethered to DISC, cFLIP competes with caspase 8 and inhibits caspase activation.17,23 The IAP family proteins, such as XIAP, cIAP1 and cIAP2, are other critical apoptotic inhibitors. They contain several N-terminal BIR domains and a C-terminal RING domain. While BIR domains may interact with and inhibit the activation of caspases directly, RING domains possess an E3 ubiquitin ligase activity. Despite their structural similarity, the different IAP members inhibit cell death through different although overlapping mechanisms. cIAP1/2 are mostly involved in ubiquitination of TRAF2 followed by NF-κB activation. In contrast, XIAP directly binds caspases 9, 3 and 7 and inhibits their proteolythic activity. The activities of IAPs are also regulated by direct interaction with their natural inhibitor Smac/Diablo which is released from mitochondria after induction of cell death.24 Moreover, IAPs can also be regulated on the level of protein stability. Besides activation of caspases and NF-kB, TNFR1 also stimulates the members of the mitogen-activated protein kinase (MAPK) family, JNK, p38 and ERK, which modulate the cell death response.18
DISC complex also mediates cell death triggered by the other members of the TNFR family: Fas and TRAILR1/2. However, activation of Fas or TRAILR1/2 leads to direct recruitment of FADD and caspase 8 to the receptors causing robust induction of cell death.17,23 Nevertheless, under certain conditions, Fas and TRAILR1/2 can tether TRADD via FADD recruitment, stimulating complex formation with RIP1, TRAF2 and cIAP1/2 proteins and activation of the pro-survival pathway.23
Here we demonstrate that Sesn2 supports DR-induced cell death in lung adenocarcinoma cells by a mechanism independent on ROS and mTORC1. Sesn2 silencing leads to accumulation of IAP family members such as cIAP1/2 and XIAP. We also show that XIAP is responsible for regulation of DR-induced cell death by Sesn2 which controls XIAP stability through regulation of its lysosomal degradation.
Results
Sesn2 silencing suppresses DR-induced apoptosis
Sesn2 is an important modulator of cell death.3,6, 25 To study whether Sesn2 regulates DR-induced cell death in human adenocarcinoma cells, we silenced Sesn2 in H460 and A549 cells with shSesn2 lentiviral vector and treated cells with TNFα in the presence of translation inhibitor cycloheximide (CHX) or, alternatively, transcription inhibitor actinomycin D (ActD). Both CHX and ActD are inhibitors of the pro-survival pathway induced by TNFα which interferes with cell death by TNFα in vitro. Either TNFα+CHX or TNFα+ActD treatment strongly activated cell death in control shLuc-expressing cells as determined by accumulation of apoptotic cells in sub-G1 phase, and Sesn2 silencing significantly inhibited TNFα-induced cell death (Fig. 1A, C, S2A). To exclude the possibility of off-target effects of shSesn2, we treated H460 cells expressing alternative shSesn2–2 with TNFα+CHX and obtained similar results (Fig. S1A, B). Interestingly, Sesn2 silencing did not affect the caspase-dependent cell death induced by staurosporin (Fig. S2B), indicating that Sesn2 is a specific regulator of DR-induced cell death.
Figure 1.

Sesn2 supports TNFα, Fas and TRAIL induced cell death. (A–C) Sesn2 silencing compromises TNFα+cycloheximide (CHX)-induced cell death in lung adenocarcinoma cells. (A) Sesn2-silenced or control (shLuc) H460 cells were treated with TNFα (10 ng/ml)+CHX (10 ug/ml) for the indicated time intervals and number of cells with sub-G1 DNA content stained with propidium iodide (PI) was assessed by flow cytometry. (B) Cells were treated as in (A) for 3 hrs and expression of full-size and cleaved forms of the corresponding proteins were analyzed by immunoblotting. (C) Sesn2-silenced and control A549 cells were treated with TNFα+CHX for 4h and analyzed as in (A). (D) Cells were treated with TNFα+CHX for different time intervals and analyzed as in (B). (E, F) Sesn2 silencing inhibits Fas-induced cell death. (E) Sesn2-silenced or control H460 cells were treated with FasL for 24 hrs and sub-G1 content was analyzed by flow cytometry as in (A). (F) Sesn2-silenced or control H460 cells were treated with activatory anti-Fas antibody and analyzed 30 hrs later as in (A). (G) Sesn2 silencing suppresses TRAIL-induced cell death. Sesn2-silenced or control H460 cells were treated with TRAIL for indicated time intervals and analyzed as in (A).
To study whether Sesn2 is important for apoptosis, we analyzed TNFα-induced activation of caspases examining cleaved caspases 8, 3, 7, and 9 by immunoblotting. We also examined cleavage of PARP and Bid mediated by caspases.22 We observed strong activation of the caspases by TNFα+CHX treatment in control cells although these effects were compromised in Sesn2-silenced cells (Fig. 1B, D), which indicates that Sesn2 is a major regulator of apoptosis. To distinguish Sesn2 effects on apoptosis from other types of cell death, we utilized Annexin V-PI staining, that allows the discrimination between necrosis and apoptosis. We found that most cells were AnnexinV+ (early apoptotic) or AnnexinV+PI+ (late apoptotic) but not AnnexinV-PI+, indicating that apoptosis was the major mechanism of cell death (Fig. S1C, D). Accordingly, we did not observe any inhibitory effects of Sesn2 silencing on necroptosis induced by TNFα+CHX treatment in the presence of a pan-caspase inhibitor Z-VAD-FMK26 (Fig. S2C).
Activated Fas and TRAILR1/2 also induce cell death via DISC formation and caspase activation. To determine the role of Sesn2 in Fas and TRAIL -induced cell death, we treated H460 cells with FasL, an activatory anti-Fas antibodies (Ab) or TRAIL and observed that all of these treatments activated cell death in a Sesn2-dependent manner (Fig. 1E–G).
Sesn2 does not affect expression of NF-κB-regulated genes or mediators of TNFα-induced cell death
Although TNFα induces the apoptotic cascade, it also activates the pro-survival signaling pathways triggering NF-κB-dependent transcriptional activation of anti-apoptotic genes.27,28 To determine whether Sesn2 modulates expression of NF-κB-regulated genes, we analyzed expression of several NF-kB targets in untreated and TNFα-treated cells by quantitative real-time PCR (qPCR). Although many of these genes such as IKBα, IL6, cIAP1, COX2 and A20 were activated by TNFα treatment, we did not observe a difference in their mRNA levels between Sesn2-silenced and control cells. Moreover we did not observe activation of other reported potential NF-κB targets such as BAX, Bcl-XL, BFL-1/A1, BIM and cIAP2 in our experimental conditions and the expression of these genes was not affected by Sesn2 silencing (Fig. 2A, Fig. S3).
Figure 2.

NFκB activity and the expression of critical regulators of TNFR1-induced cell death are not altered in Sesn2-silenced cells (A) Sesn2-silenced and control cells have similar levels of expression of NF-κB regulated genes under normal conditions and after TNFα treatment. (B) Sesn2-silenced or control H460 cells were treated with TNFα and the expression of the corresponding NF-κB-inducible genes was analyzed by qPCR. Sesn2 silencing does not affect the expression of the mediators of TNFα signaling TNFR1, RIP1 and TRADD in H460 and A549 cells. Sesn2-silenced or control cells were analyzed by immunoblotting with the indicated antibody. (C) Sesn2 silencing does not affect the expression of TNFR1, TRADD and RIP1 in untreated and TNFα+CHX treated cells. Sesn2-silenced or control cells were treated with TNFα+CHX and the expression of the indicated proteins was analyzed by immunoblotting. (D) Sesn2 silencing does not affect cFLIPL and cFLIPS expression in response to TNFα+CHX treatment. Sesn2-silenced or control cells were treated with TNFα+CHX for different time intervals and the expression of FLIP and control GAPDH proteins were analyzed by immunoblotting.
As reported earlier, Sesn2 is involved in regulation of protein synthesis or degradation.8,29,30 Considering the possible impact of Sesn2 on the expression of proteins transducing signaling from TNFα toward caspases, we compared the levels of TNFR1, RIP1 and TRADD proteins in control and Sesn2-silenced cells by immunoblotting, and observed no difference in the expression levels of these proteins between these 2 cell lines (Fig. 2B,C). We also measured levels of cFLIPs, the components of DISC and inhibitors of cell death, and did not observe any difference in the expression of long cFLIPL and short cFLIPS isoforms between the 2 cell lines. Also cFLIP was degraded with a similar rate in response to TNFα+CHX treatment in both cell lines (Fig. 2D).16
Sesn2 silencing does not affect activation of MAPK and AKT kinases by TNFα
MAPKs are involved in positive and negative regulation of DR-induced cell death.31,32 To determine whether Sesn2 plays a role in activation of JNK, p38 and MEK by TNFα, we examined their phosphorylation by immunoblotting. As shown in Figure 3A, Sesn2 did not affect the magnitude of JNK, p38 or ERK activation by TNFα, nor the duration of JNK activation. In addition, treatment with the JNK inhibitor SP600125 suppressed TNFα-induced apoptosis in both control and Sesn2-silenced cells in a similar fashion, preserving the ratio in cell death between control and Sesn2-silenced cells (Fig. 3B). We also analyzed phosphorylation of AKT, another regulator of apoptosis and a potential target of Sesn2,7,33 and did not observe any effect of Sesn2 silencing on AKT phosphorylation in untreated or TNFα-treated cells (Fig. 3A). These data indicate that Sesn2 does not play any significant role in regulation of MAPK or AKT signaling in response to TNFα treatment.
Figure 3.
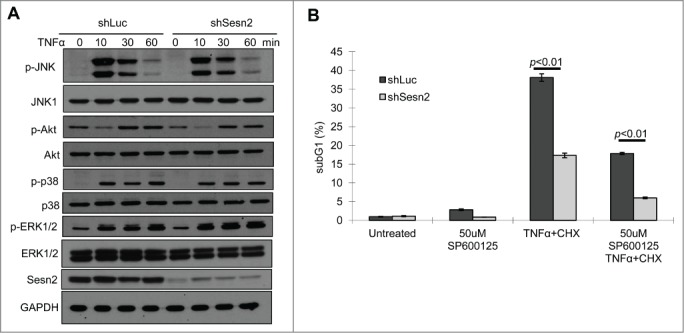
Sesn2 supports TNFα+CHX-induced cell death not via a MAPK-dependent mechanism. (A) Sesn2 is not involved in the regulation of JNK, p38, ERK and AKT phosphorylation. Sesn2-silenced or control H460 cells were treated with TNFα for different time interval and phosphorylation and expression of corresponding proteins were analyzed by immunoblotting. (B) JNK inhibition has similar effect on cell death in control and Sesn2-silenced cells. Sesn2-silenced or control H460 cells were treated with TNFα+CHX in the presence or absence of JNK inhibitor SP600125 for 4 hrs and the sub-G1 population of apoptotic cells was analyzed by flow cytometry.
Sesn2 regulates TNFα-induced cell death in the ROS- and mTORC1-independent manner
We have previously shown that Sestrins suppress ROS accumulation and inhibit mTORC1 activity.6,8,34 Since elevated ROS might support cell death through activation of JNK,32 we analyzed ROS levels in Sesn2-silenced and control cells treated with TNFα. As expected,6,14 Sesn2 knockdown increased ROS levels in untreated cells (Fig. 4A). Treatment with TNFα slightly increased ROS levels and co-incubation with a ROS scavenger N-acetylcysteine (NAC) decreased ROS accumulation in both control and Sesn2-silenced cells (Fig. 4A). To study whether Sesn2 silencing can affect cell death via a ROS-dependent mechanism, we induced cell death by TNFα+CHX in the presence or absence of NAC and found that NAC treatment suppressed cell death in a similar manner in both control and Sesn2-silenced cells, preserving the difference in the levels of cell death induced by TNFα between these 2 cell lines (Fig. 4B).
Figure 4.
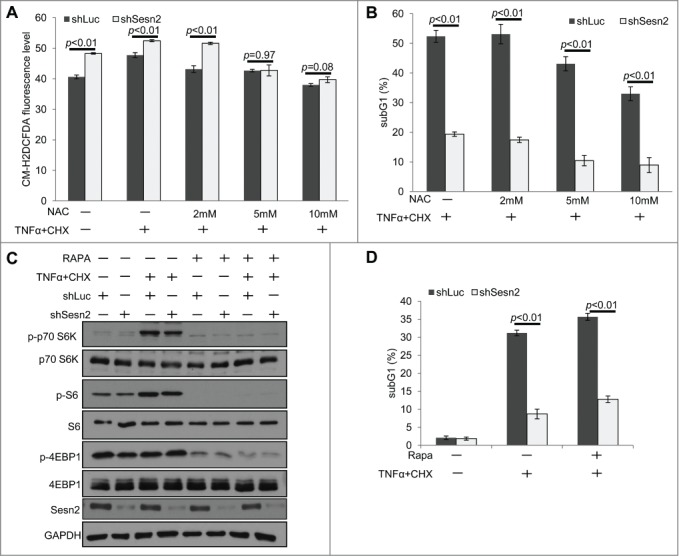
The stimulatory effect of Sesn2 on TNFα+CHX-induced cell death is not mediated by ROS or mTORC1 regulation. (A) Sesn2-silenced cells have increased ROS levels as compared to control cells which can be reversed by NAC treatment. Sesn2-silenced or control H460 cells were treated with TNFα+CHX for 4 hrs in the presence or absence of different concentrations of NAC. Cells were incubated with DCFDA and analyzed by flow cytometry. (B) NAC treatment slightly inhibits cell death in both Sesn2-silenced and control cells, but does not eliminate the difference in cell death between these 2 cell lines. Cells were treated as in (A) and sub-G1 DNA content was analyzed by flow cytometry. (C) Sesn2 silencing does not affect mTORC1 activation by TNFα+CHX treatment. Sesn2-silenced or control cells were treated with TNFα+CHX in the presence or absence of rapamycin and phosphorylation and expression of the indicated proteins were analyzed by immunoblotting. (D) mTORC1 inhibition by rapamycin does not affect the magnitude of TNFα+CHX-induced cell death in Sesn2-silenced and control cells. Sesn2-silenced or control cells were treated with TNFα+CHX in the presence or absence of rapamycin (10 nM) and cell death was determined as in B.
In parallel, we also analyzed the potential impact of Sesn2 on mTORC1 regulation by TNFα. While we observed the activation of mTORC1 in response to TNFα treatment (depicted by increased p70S6K and S6 phosphorylation), there was no difference in p70S6K, S6 and 4EBP1 phosphorylation between Sesn2-silenced and control cells (Fig. 4C). Moreover, treatment with mTORC1 inhibitor rapamycin strongly suppressed phosphorylation of mTORC1 targets (Fig. 4C), but had no significant impact on the TNFα-induced cell death in both Sesn2-silenced and control cells (Fig. 4D). Thus, we concluded that control of ROS or mTORC1 is dispensable for the regulation of TNFα-indu-ced cell death by Sesn2.
IAP proteins accumulate in Sesn2-silenced cells and XIAP mediates the effects of Sesn2 on TNFα-induced apoptosis
The IAP family members are major regulators of DR-induced cell death.35 To study whether Sesn2 can regulate expression of IAP proteins we analyzed their levels by immunoblotting and found that cIAP1/2 and XIAP were accumulated in the Sesn2-silenced cells as compared to control (Fig. 5A). Within the IAP family, XIAP is the most potent regulator of cytokine-induced cell death due to its direct strong inhibitory effect on caspases.24,36 Therefore, we reasoned that XIAP might be responsible for regulation of DR-induced apoptosis by Sesn2. To study whether Sesn2 controls XIAP mRNA levels we analyzed XIAP mRNA levels in Sesn2-silenced and control cells by qPCR, but did not observe any difference between these 2 cell lines (Fig. 5B). Thus the regulation of XIAP expression by Sesn2 is mediated by post-transcriptional mechanisms such as protein synthesis and/or degradation. To study whether XIAP is responsible for regulation of TNFα-induced cell death by Sesn2, we silenced XIAP by shRNA lentivirus (Fig. 5C) and treated cells with TNFα+CHX. Strikingly, XIAP knockdown in shSesn2-silenced H460 cells restored the levels of cell death observed in control cells and eliminated the difference in the levels of cell death between control and Sesn2-silenced cells (Fig. 5D), indicating that XIAP plays a major role in regulation of TNFα-induced cell death by Sesn2.
Figure 5.
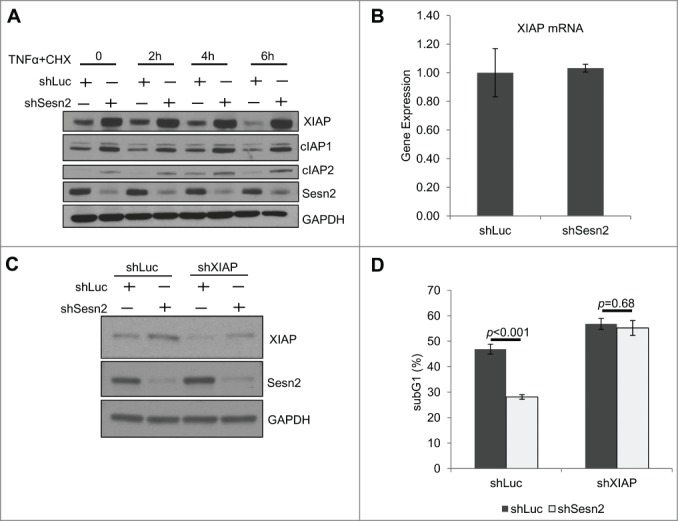
Sesn2 supports TNFα+CHX-induced cell death through regulation of XIAP. (A) Sesn2-silencing causes accumulation of IAP proteins. (A) XIAP, cIAP1 and cIAP2 proteins are accumulated in Sesn2-silenced cells as compared to the control. The levels of the indicated proteins in untreated and TNFα+CHX treated cells were determined by immunoblotting. (B) Sesn2 silencing does not affect XIAP mRNA-expression. RNA from Sesn2-silenced or control H460 cells was isolated with Trizol reagent, converted to cDNA and analyzed by qPCR. (C, D) XIAP knockdown restore the levels of TNFα+CHX-induced cell death in Sesn2-silenced H460 cells. (C) Analysis of XIAP protein expression in XIAP-silenced H460 cells. Sesn2-silenced or control H460 cells were infected with lentiviral vector expressing shXIAP and the proteins were analyzed by immunoblotting. (D) Analysis of the effect of XIAP knockdown on TNFα+CHX-induced cell death. Cells were generated as in C and cell death was determined by the analysis of sub-G1 population by flow cytometry.
Sesn2 controls XIAP through regulation of protein stability
Protein synthesis and/or degradation are the major mechanisms of post-transcriptional control of protein expression. To study the impact of Sesn2 on these processes we pulse-labeled control and Sesn2-silenced cells with 35S methionine-cysteine for 1 hr, removed the radioactivity from the medium and monitored the levels of newly-synthesized XIAP protein at different time intervals after replacement with non-radioactive medium by SDS-PAGE electrophoresis and autoradiography. This allowed us to analyze the contribution of Sesn2 to XIAP synthesis comparing the XIAP levels immediately after 35S labeling, as well as the half-life of the XIAP protein in control and Sesn2-silenced cells. We did not observe any significant difference in the levels of freshly-synthesized Sesn2 protein immediately after pulse chase labeling, suggesting that Sesn2 does not considerably contribute to XIAP protein synthesis (Fig. 6A). However, we found striking differences in XIAP protein levels at different time points after replacement with non-radioactive medium. While the half-life of XIAP was about 5 hrs in control cells, it increased 2-fold in Sesn2-silenced cells. Thus Sesn2 knockdown causes XIAP protein stabilization by suppressing its degradation (Fig. 6A and B).
Figure 6.
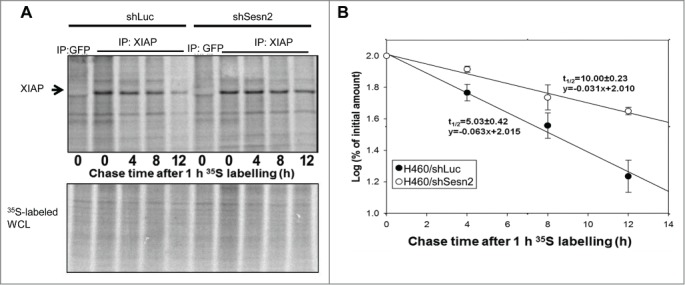
Sesn2 controls XIAP expression via regulation of protein stability. (A, B) XIAP protein is stabilized in Sesn2-silenced cells. (A) 35S pulse-chase labeling experiment demonstrated significant effect of Sesn2 knockdown on the stability of XIAP protein. Sesn2-silenced or control H460 cells were incubated with 35S protein labeling mix for 1 hr followed by incubation with non-radioactive medium for different time intervals. At each time-point the cells were lyzed and XIAP protein was immunoprecipitated with anti-XIAP antibody (anti-GFP antibodies were used as a negative control) and analyzed by SDS-PAGE followed by autoradiography, On the lower panel, the whole cell lysates (WCL) were analyzed by SDS-PAGE electrophoresis. (B) Quantification of the radioactively-labeled proteins in Sesn2-silenced and control cells. Cells were treated as in A and the amount of labeled Sesn2 protein and its half-life were analyzed on a phosphoimager with ImageQuant software.
Sesn2 can regulate XIAP protein degradation through the lysosomal pathway
The majority of intracellular proteins are degraded through the proteosomal and/or lysosomal pathways. To establish the mechanism responsible for the decreased XIAP degradation in shSesn2 cells, we examined how inhibition of either of these pathways affects XIAP degradation in control or Sesn2-silenced cells. We treated cells with either proteosomal inhibitor MG132 or lysosomal inhibitor chloroquine (CQ) and measured XIAP protein levels at different time points after treatment. Both treatments led to accumulation of XIAP protein in control cells indicating that XIAP can be degraded through both mechanisms (Fig. 7A and B). Nevertheless we observed a conspicuous difference in XIAP accumulation in Sesn2-silenced cells treated with either MG132 or CQ. Although the XIAP level was originally higher in Sesn2-silenced cells as compared to the control, XIAP continued to accumulate in response to MG132 treatment with similar dynamic as in the control cells (Fig. 7A), indicating that Sesn2 is not likely to be affecting the rate of XIAP proteosomal degradation. In contrast, although CQ treatment led to accumulation of XIAP in control cells, no additional accumulation of XIAP was observed in Sesn2-silenced cells (Fig. 7B). These data suggest that XIAP lysosomal degradation was already impaired in the Sesn2-silenced cells and inhibition of the lysosomal pathway had no additional effect on XIAP protein levels in Sesn2-silenced cells.
Figure 7.
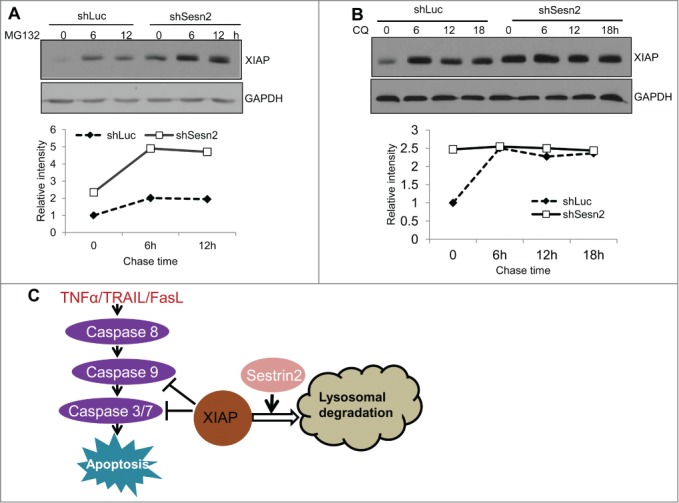
Sesn2 controls XIAP stability through regulation of lysosomal degradation. (A) Inhibition of proteosomal synthesis causes XIAP accumulation in both Sesn2-silenced and control H460 cells with similar rates. Sesn2-silenced or control H460 cells were treated with proteosomal inhibitor MG132 (20 μM) for different time intervals and XIAP and control GAPDH protein levels were analyzed by immunoblotting. (B) Inhibition of lysosomal synthesis does not cause additional XIAP protein accumulation in Sesn2-silenced cells. Sesn2-silenced or control H460 cells were treated with lysosome inhibitor chloroquine (CQ) (50 μM) for the indicated time intervals and XIAP and GAPDH protein levels were examined by immunoblotting. (C) The scheme illustrating regulation of DR-induced cell death by Sesn2. Sesn2 stimulates degradation of XIAP via lysosomal pathway, as result supporting DR-induced cell death.
Total autophagosomal-lysosomal protein degradation can be monitored by p62/SQSTM1 degradation or accumulation of the high-mobility LC3-II form. We measured p62 and LC3-II levels by immunoblotting and did not observe any difference between control and Sesn2-silenced cells, although p62 protein levels were decreased in both cell lines in response to TNFα+CHX (Fig. S4). Therefore, although total autophagy can be stimulated by TNFα treatment, it does not depend on Sesn2. It was also reported previously that Sesn2 can be involved in specific degradation of Keap1 via direct interaction with p62 and Keap1.30 Anticipating potential similarity in the mechanisms of degradation of Keap1 and XIAP, we analyzed whether Sesn2 can interact with p62 and XIAP and whether Sesn2 can affect interaction between p62 and XIAP. We did not observe any interaction between Sesn2 and either p62 or XIAP in H460 cells (data not shown). Although we detected an interaction between p62 and XIAP, such interaction was not affected by the Sesn2 status (Fig. S5).
Discussion
Tumor suppressive mechanisms rely on apoptosis for elimination of pre-malignant and malignant cells.37 The immune system is responsible for surveillance of transformed cells and their elimination through activation of DRs.17,18 Cancer cells, in turn, develop resistance to apoptosis, through disregulation of expression of antiapoptotic and proapoptotic proteins.38 Here, we determined that down-regulation of Sesn2, a major controller of cell viability under different stress conditions,3,6 is a new potential strategy of evasion from DR-induced cell death for adenocarcinoma lung cancer cells. The importance of Sesn2 in modulating cell death prompted us to study its potential impact on regulation of DR-induced cell death. As we demonstrated here, Sesn2 is important for cell death induced by TNFα, Fas and TRAIL in human lung adenocarcinoma H460 and A549 cells. While we observed an inhibition of caspase activation by Sesn2 silencing, it played no role in the control of NF-κB-regulated transcription or regulation of the expression of cell death mediators such as TNFR1, TRADD and RIP1. Sesn2 also had no impact on the activation of JNK, p38 or ERK kinases, ruling out their role in Sesn2-dependent processes. This is a new and previously uncharacterized function of Sesn2, which is not relevant to inhibition of ROS accumulation or mTORC1 activity.
Examining different regulators of DR-induced cell death, we found that Sesn2 silencing caused accumulation of the IAP family members XIAP, cIAP1 and cIAP2 which all can contribute to suppression of cell death by Sesn2 silencing. cIAP1/2 inhibit cell death mostly through ubiquitination and degradation of RIP1 and RIP1-dependent activation of NF-κB, so we compared RIP1 expression and NF-κB activation between Sesn2-silenced and control cells, and were not able to assign the regulation of any of these proteins by Sesn2 to its effects on cell death.24,39 In contrast, XIAP directly binds caspases and inhibits their proteolytic activity and we demonstrated the clear effect of Sesn2 on activation of caspases in response to DR activation. Although we could not completely waive away the role of cIAP1/2 in regulation of DR-induced apoptosis by Sesn2, in the following experiments we focused on the XIAP protein as the most prominent potential mediator of the effects of Sesn2 on cell death (Fig. 7C). Accordingly, we found that XIAP knockdown in Sesn2-silenced cells restored the levels of cell death to those observed in control cells, indicating that XIAP is the major contributor to the suppression of DR-induced cell death by Sesn2 downregulation. Although a direct inhibitory effect of XIAP was demonstrated on caspases 3, 7 and 9, these caspases can amplify activation of caspase 8 via a positive feedback loop,40,41 explaining the inhibition of caspase 8 activation in the Sesn2-silenced cells.
To study the potential mechanism of XIAP regulation by Sesn2, we analyzed its stability by pulse-chase labeling and observed that XIAP half-life was extended 2-fold in Sesn2-silenced cells. Moreover, we found that Sesn2 regulates XIAP levels through control of lysosomal degradation. The potential role of Sesn2 in regulation of the autophagy-lysosomal pathway via mTORC1 inhibition was previously reported,8,42,43 however, we did not see any effects of Sesn2 silencing on the mTORC1 activation by TNFα. The autophagic rate can be monitored by degradation of p62 protein, which works as a cargo for delivery of many proteins to autophagosomes, and by conversion of LC3-I into the processed LC3-II form. We did not observe any difference in the p62 levels between Sesn2-silenced or control TNFα+CHX treated or untreated cells, although TNFα+CHX treatment caused down-regulation of p62 in both cell lines (Fig. S4). Similarly, we did not see any effect of Sesn2 silencing on the expression of LC3-I and LC3-II forms, arguing against a significant role of Sesn2 in regulation of general autophagy in our experimental system.
Nevertheless, Sesn2 can be involved in the specific lysosomal degradation of some proteins, such as PDGFRβ44,45 and Keap1.30 As demonstrated, Sesn2 regulates Keap1 degradation through direct interaction with Keap1 and p62, directing Keap1 to the autophagosomal-lysosmal pathway. Speculating that a similar mechanism for XIAP degradation might be involved, we analyzed whether Sesn2 interacts with either XIAP or p62, but did not observe any noticeable interaction between Sesn2 and any of these proteins. Moreover, although we were able to co-immunoprecipitate XIAP and p62, Sesn2 silencing did not affect interaction between these 2 proteins (Fig. S5). Thus, although p62 can take part in the XIAP lysosomal degradation, we were not able to assign any role of Sesn2 in this process. Although the mechanism of XIAP degradation by Sesn2 is yet to be characterized, we speculate that Sesn2 can label and target XIAP and other IAP members to lysosomes via several potential mechanisms. One of them might involve interaction with and activation of ULK1,46 which plays an important role in early stages of autophagosome biogenesis and could direct some proteins into autophagosomes. Another mechanism might involve the recently characterized GATOR complex as the major Sestrin interactor.9,47 GATOR proteins can be associated with lysosomes48 and could mediate the effects of Sesn2 in the recognition and specific degradation of particular proteins. Other members of the Sestrin family might be also involved in regulation of DR-induced cell death via control of IAP proteins. Although the dramatic effects of Sesn2 inactivation on DR-induced cell death in lung adenocarcinoma cells can be explained by high relative expression of Sesn2 as compared to the other Sestrins members or, alternatively, its higher specificity for XIAP degradation. Nevertheless we anticipate that Sesn1/3 can play a role in the regulation of DR-induced cell death under some experimental conditions, especially in the cell types where these proteins are predominantly expressed.
Tumors are always associated with inflammation and infiltration of immune cells, which play an ambivalent role in carcinogenesis. Despite the potential role of these factors in eliminating malignant cells via activation of cell death, they can also contribute to tumor progression via activation of pathways supporting cell proliferation, angiogenesis and metabolic re-programming. Thus cancer cells that are capable of inhibition of pro-apoptotic machinery induced by DR, and still maintain intact pro-survival pathways, might be selected. One of the most vivid examples is the inactivation of tumor suppressor p53, the important positive regulator of DR-induced cell death, found in the majority of human cancers.13 Another example is the accumulation of the IAP proteins. XIAP and other IAP family members are up-regulated in advanced lung and some other human cancers,24,39, 49,50 although the mechanisms of accumulation of IAP proteins are yet to be characterized. Here we demonstrated that Sesn2 silencing is involved in accumulation of XIAP and other IAP members. Sesn2 and Sesn1 can be downregulated in human cancers due to inactivation of its master regulator p53 found mutated in majority of human cancers,14,34 or due to some other yet to be characterized mechanisms. Accordingly, loss of heterozygosity in Sesn2 locus 1p34 or in Sesn1 locus 6q21 is found in many human cancers5,51 arguing for a potential tumor suppressive function of Sestrins. The ability of Sesn2 to support DR-induced cell death along with its antioxidant and mTORC1-suppressing functions, make this protein a primary candidate as a suppressor of lung carcinogenesis.
Materials and Methods
Cell culture, transfection, infection and treatment
Human lung adenocarcinoma H460 and A549 cells and human embryonic kidney HEK 293T cells were cultured in high-glucose DMEM supplemented with 10% FBS and penicillin/streptomycin. All transfections and infections were performed as described previously.8 Human recombinant TNFα, TRAIL and FasL were from R&D System and reconstituted in PBS with 0.2% BSA. N-Acetyl-L-cysteine (NAC), chloroquine (CQ), MG132, cycloheximide, actinomycin D and rapamycin were from Sigma Aldrich; SP600125 and anisomycin were from Cell Signaling Inc.; Z-VAD-FMK was from Enzo Lifescience For ROS examination cells were incubated with DCFDA (Life Science) for 45 min followed by flow cytometry analysis.
Cell lysis, immunoprecipitation and immunoblot analyses
For immunoblot analysis cells were lysed in RIPA-SDS buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS, and protease/phosphatase inhibitors (Roche)). For immunoprecipitation experiments cells were lysed in NP40 buffer (50mM Tris pH 7.4, 150 mM NaCl, 0.5% NP40, 0.1 mM EDTA, and protease/phosphatase inhibitors). The lysates were incubated with the mix of indicated antibodies and protein A:G-sepharose beads for 3 hrs, then washed 4 times with lysis buffer. The proteins were separated by SDS-PAGE and analyzed by immunoblotting; Quantification was performed by Image J software. The antibodies used were: Sesn2 from Proteintech; FLIP from Enzo Lifescience; PARP, GAPDH, p53, p62/SQSTM1 from Santa Cruz; all others were from Cell Signaling.
Constructs
pLSLPw-shLuc was previously described.8 The sequence for shSesn2 is 5′-GAAGACCCTACTTTCGGAT-3′, shSesn2–2: 5′-GAGATGGAGAGCCG-CTTT-3′, shXIAP: 5′-CCAGAATGGTCAGTACAAA-3′. The primers used for qPCR were: A20: 5′-CTGCCCAGGAATGCTACAGATAC-3′ and 5′-GTGGAACAGCTCGGATTTCAG-3′; COX2: 5′-CACCCATGTCAAAACCGAGG-3′ and 5′-CCGGTGTTGAGCAGTTTTCTC-3′; IκBα: 5′-GATCC-GCCAGGTGAAGGG-3′ and 5′-GCAATTTCTGGCTGGTTGG-3′; IL6: 5′-AATTCGGTACATCCTCGAC-GG-3′ and 5′-GGTTGTTTTCTGCC-AGT-GCC-3′; XIAP: 5′-AGCCAAGGGGAA-TGAAGTGA-3′ and 5′-GGGGA-AGG-GCATTTGAAGAA-3′; GAPDH: 5′-CATGGGTGTGAACCATGAGA-3′ and 5′-CAGTGATGGCATGGACTG-TG-3′.
Analysis of cell death
Cell death was evaluated by analysis of sub-G1 population and Annexin V-propidium iodide (PI) staining. For sub-G1 analysis, cells were fixed with 70% ice-cold ethanol and kept at −20°C overnight. After washing with PBS, cells were incubated with 100 µg/ml RNase A and 40 µg/ml PI at room temperature for 30 min in the dark. Samples were acquired with a BD FACS Calibur and analyzed with CellQuest Pro software. For Annexin V-PI staining, cells were re-suspended in Annexin V Binding Buffer and stained with anti-Annexin V FITC antibody and PI as per manufacturer's recommendations (BD Biosciences).
Quantitative Real-Time PCR
Total RNA was extracted with Trizol reagent (Invitrogen). Purified RNA (1 ug) was converted into cDNA according to the Tetro cDNA synthesis kit (Bioline). qPCR was performed with the iTaq universal SYBR green supermix (Bio-rad). Data were analyzed by the 2-ΔΔCT method for relative quantification. Experimental Ct values were normalized to GAPDH levels and relative mRNA expression was calculated versus a control sample.
Pulse-chase protein labeling and examination of protein stability
Cells (1×106 per 6 cm well) were incubated with 200μCi Sprotein labeling mix (PerkinElmer), containing 35S-labeled methionine and cysteine for 60 min, washed and replaced with normal unlabeled medium contained 3 mM methionine and 1mM cysteine. Cells were harvested and lysed in a buffer containing 50 mM Tris (pH 7.5), 0.5 mM EDTA, 1% SDS, and 1 mM DTT, and sonicated for 10 minutes. The normalized lysates were diluted 1:9 with immunoprecipitation buffer (50 mM Tris 7.5, 300 mM NaCl, 1% NP40, and 1 mM DTT), and immunoprecipitated with equal amount (1.2 μg) of either anti-XIAP (Santa Cruz sc-55552) or control anti-GFP (Santa Cruz sc-9996) monoclonal antibodies followed by SDS-PAGE. The gels were fixed, dried, and the signal intensities were acquired using ImageQuant software.
Statistical analysis
Differences between samples were analyzed by Student's t-test, or one-way Anova according to data distribution. Analyses were performed using GraphPad Prism (GraphPad Software Inc.). P values <0.05 were considered significant. Results are presented as mean ± standard deviation of at least 3 independent experiments.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Drs. Richard Moran, Andrei Ivanov and Jane Roberts for commenting on the manuscript. We also thank Nadusha Pryadilova for everyday support.
Funding
This work was supported by NIH RO1 CA172660 to AB.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010 Mar 19; 140(6):883-99; PMID:20303878; http://dx.doi.org/ 10.1016/j.cell.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011 Mar 4; 144(5):646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 3.Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, Gorodin S, Fishman A, Chajut A, Einat P, et al.. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene 2002 Sep 5; 21(39):6017-31; PMID:12203114; http://dx.doi.org/ 10.1038/sj.onc.1205877 [DOI] [PubMed] [Google Scholar]
- 4.Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA, Ocorr K, Ellisman MH, Bodmer R, Bier E, et al.. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 2010 Mar 5; 327(5970):1223-8; PMID:20203043; http://dx.doi.org/ 10.1126/science.1182228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Budanov AV, Lee JH, Karin M. Stressin' Sestrins take an aging fight. EMBO Mol Med 2010 Oct; 2(10):388-400; PMID:20878915; http://dx.doi.org/ 10.1002/emmm.201000097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 2004 Apr 23; 304(5670):596-600; PMID:15105503; http://dx.doi.org/ 10.1126/science.1095569 [DOI] [PubMed] [Google Scholar]
- 7.Lee JH, Budanov AV, Talukdar S, Park EJ, Park HL, Park HW, Bandyopadhyay G, Li N, Aghajan M, Jang I, et al.. Maintenance of metabolic homeostasis by sestrin2 and sestrin3. Cell Metab 2012 Sep 5; 16(3):311-21; PMID:22958918; http://dx.doi.org/ 10.1016/j.cmet.2012.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008 Aug 8; 134(3):451-60; PMID:18692468; http://dx.doi.org/ 10.1016/j.cell.2008.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parmigiani A, Nourbakhsh A, Ding B, Wang W, Kim YC, Akopiants K, Guan KL, Karin M, Budanov AV. Sestrins Inhibit mTORC1 Kinase Activation through the GATOR Complex. Cell Rep 2014 Nov 6; 9(4):1281-91; PMID:25457612; http://dx.doi.org/ 10.1016/j.celrep.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell 2006 Feb 10; 124(3):471-84; PMID:16469695; http://dx.doi.org/ 10.1016/j.cell.2006.01.016 [DOI] [PubMed] [Google Scholar]
- 11.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev 2004 Aug 15; 18(16):1926-45; PMID:15314020 [DOI] [PubMed] [Google Scholar]
- 12.Dann SG, Selvaraj A, Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med 2007 Jun; 13(6):252-9; PMID:17452018; http://dx.doi.org/ 10.1016/j.molmed.2007.04.002 [DOI] [PubMed] [Google Scholar]
- 13.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997 Feb 7; 88(3):323-31; PMID:9039259; http://dx.doi.org/ 10.1016/S0092-8674(00)81871-1 [DOI] [PubMed] [Google Scholar]
- 14.Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med 2005 Dec; 11(12):1306-13; PMID:16286925; http://dx.doi.org/ 10.1038/nm1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu H, Sun H, Zhang H, Liu J, Fan F, Li Y, Ning X, Sun Y, Dai S, Liu B, et al.. An ShRNA Based Genetic Screen Identified Sesn2 as a Potential Tumor Suppressor in Lung Cancer via Suppression of Akt-mTOR-p70S6K Signaling. PloS one 2015; 10(5):e0124033; PMID:25962159; http://dx.doi.org/ 10.1371/journal.pone.0124033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003 Jul 25; 114(2):181-90; PMID:12887920; http://dx.doi.org/ 10.1016/S0092-8674(03)00521-X [DOI] [PubMed] [Google Scholar]
- 17.Guicciardi ME, Gores GJ. Life and death by death receptors. FASEB J 2009 Jun; 23(6):1625-37; PMID:19141537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 1998 Aug 28; 281(5381):1305-8; PMID:9721089; http://dx.doi.org/ 10.1126/science.281.5381.1305 [DOI] [PubMed] [Google Scholar]
- 19.Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, et al.. A unified model for apical caspase activation. Mol Cell 2003 Feb; 11(2):529-41; PMID:12620239; http://dx.doi.org/ 10.1016/S1097-2765(03)00051-0 [DOI] [PubMed] [Google Scholar]
- 20.Donepudi M, Mac Sweeney A, Briand C, Grutter MG. Insights into the regulatory mechanism for caspase-8 activation. Mol Cell 2003 Feb; 11(2):543-9; PMID:12620240; http://dx.doi.org/ 10.1016/S1097-2765(03)00059-5 [DOI] [PubMed] [Google Scholar]
- 21.Muzio M, Stockwell BR, Stennicke HR, Salvesen GS, Dixit VM. An induced proximity model for caspase-8 activation. J Biol Chem 1998 Jan 30; 273(5):2926-30; PMID:9446604; http://dx.doi.org/ 10.1074/jbc.273.5.2926 [DOI] [PubMed] [Google Scholar]
- 22.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997 Nov 14; 91(4):479-89; PMID:9390557; http://dx.doi.org/ 10.1016/S0092-8674(00)80434-1 [DOI] [PubMed] [Google Scholar]
- 23.Sessler T, Healy S, Samali A, Szegezdi E. Structural determinants of DISC function: new insights into death receptor-mediated apoptosis signalling. Pharmacol Ther 2013 Nov; 140(2):186-99; PMID:23845861; http://dx.doi.org/ 10.1016/j.pharmthera.2013.06.009 [DOI] [PubMed] [Google Scholar]
- 24.Obexer P, Ausserlechner MJ. X-linked inhibitor of apoptosis protein - a critical death resistance regulator and therapeutic target for personalized cancer therapy. Front Oncol 2014; 4:197; PMID:25120954; http://dx.doi.org/ 10.3389/fonc.2014.00197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ben-Sahra I, Dirat B, Laurent K, Puissant A, Auberger P, Budanov A, Tanti JF, Bost F. Sestrin2 integrates Akt and mTOR signaling to protect cells against energetic stress-induced death. Cell Death Differ 2013 Apr; 20(4):611-9; PMID:23238567; http://dx.doi.org/ 10.1038/cdd.2012.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al.. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 2008 May; 4(5):313-21; PMID:18408713; http://dx.doi.org/ 10.1038/nchembio.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS Jr. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998 Sep 11; 281(5383):1680-3; PMID:9733516; http://dx.doi.org/ 10.1126/science.281.5383.1680 [DOI] [PubMed] [Google Scholar]
- 28.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev 2004 Sep 15; 18(18):2195-224; PMID:15371334 [DOI] [PubMed] [Google Scholar]
- 29.Braunstein S, Badura ML, Xi Q, Formenti SC, Schneider RJ. Regulation of protein synthesis by ionizing radiation. Mol Cell Biol 2009 Nov; 29(21):5645-56; PMID:19704005; http://dx.doi.org/ 10.1128/MCB.00711-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN, Lee HE, Kang D, Rhee SG. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab 2013 Jan 8; 17(1):73-84; PMID:23274085; http://dx.doi.org/ 10.1016/j.cmet.2012.12.002 [DOI] [PubMed] [Google Scholar]
- 31.Deng Y, Ren X, Yang L, Lin Y, Wu X. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell 2003 Oct 3; 115(1):61-70; PMID:14532003; http://dx.doi.org/ 10.1016/S0092-8674(03)00757-8 [DOI] [PubMed] [Google Scholar]
- 32.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005 Mar 11; 120(5):649-61; PMID:15766528; http://dx.doi.org/ 10.1016/j.cell.2004.12.041 [DOI] [PubMed] [Google Scholar]
- 33.Zhao B, Shah P, Budanov A, Qiang L, Ming M, Aplin A, Sims DM, He YY. Sestrin2 positively regulates AKT signaling and survival in human squamous cell carcinoma and melanoma cells. J Biol Chem 2014 Nov 6; 289(52):35806-14; PMID:25378405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Budanov AV. Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Antioxid Redox Signal 2011 Sep 15; 15(6):1679-90; PMID:20712410; http://dx.doi.org/ 10.1089/ars.2010.3530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deveraux QL, Reed JC. IAP family proteins–suppressors of apoptosis. Genes Dev 1999 Feb 1; 13(3):239-52; PMID:9990849 [DOI] [PubMed] [Google Scholar]
- 36.Galban S, Duckett CS. XIAP as a ubiquitin ligase in cellular signaling. Cell Death Differ 2010 Jan; 17(1):54-60; PMID:19590513; http://dx.doi.org/ 10.1038/cdd.2009.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature 2004 Nov 18; 432(7015):307-15; PMID:15549092; http://dx.doi.org/ 10.1038/nature03098 [DOI] [PubMed] [Google Scholar]
- 38.Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer 2002 Apr; 2(4):277-88; PMID:12001989; http://dx.doi.org/ 10.1038/nrc776 [DOI] [PubMed] [Google Scholar]
- 39.Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov 2012 Feb; 11(2):109-24; PMID:22293567; http://dx.doi.org/ 10.1038/nrd3627 [DOI] [PubMed] [Google Scholar]
- 40.Ferreira KS, Kreutz C, Macnelly S, Neubert K, Haber A, Bogyo M, Timmer J, Borner C. Caspase-3 feeds back on caspase-8, Bid and XIAP in type I Fas signaling in primary mouse hepatocytes. Apoptosis 2012 May; 17(5):503-15; PMID:22246639; http://dx.doi.org/ 10.1007/s10495-011-0691-0 [DOI] [PubMed] [Google Scholar]
- 41.Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, et al.. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, −3, −6, −7, −8, and −10 in a caspase-9-dependent manner. J Cell Biol 1999 Jan 25; 144(2):281-92; PMID:9922454; http://dx.doi.org/ 10.1083/jcb.144.2.281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maiuri MC, Malik SA, Morselli E, Kepp O, Criollo A, Mouchel PL, Carnuccio R, Kroemer G. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle 2009 May 15; 8(10):1571-6; PMID:19377293; http://dx.doi.org/ 10.4161/cc.8.10.8498 [DOI] [PubMed] [Google Scholar]
- 43.Ishihara M, Urushido M, Hamada K, Matsumoto T, Shimamura Y, Ogata K, Inoue K, Taniguchi Y, Horino T, Fujieda M, et al.. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am J Physiol Renal Physiol 2013 Aug 15; 305(4):F495-509; PMID:23698117; http://dx.doi.org/ 10.1152/ajprenal.00642.2012 [DOI] [PubMed] [Google Scholar]
- 44.Liu SY, Lee YJ, Lee TC. Association of platelet-derived growth factor receptor β accumulation with increased oxidative stress and cellular injury in sestrin 2 silenced human glioblastoma cells. FEBS Lett 2011 Jun 23; 585(12):1853-8; PMID:21536039; http://dx.doi.org/ 10.1016/j.febslet.2011.04.041 [DOI] [PubMed] [Google Scholar]
- 45.Heidler J, Fysikopoulos A, Wempe F, Seimetz M, Bangsow T, Tomasovic A, Veit F, Scheibe S, Pichl A, Weisel F, et al.. Sestrin-2, a repressor of PDGFRbeta signalling, promotes cigarette-smoke-induced pulmonary emphysema in mice and is upregulated in individuals with COPD. Dis Model Mech 2013 Nov; 6(6):1378-87; PMID:24046361; http://dx.doi.org/ 10.1242/dmm.013482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ro SH, Semple IA, Park H, Park H, Park HW, Kim M, Kim JS, Lee JH. Sestrin2 promotes Unc-51-like kinase 1 mediated phosphorylation of p62/sequestosome-1. FEBS J 2014 Sep; 281(17):3816-27; PMID:25040165; http://dx.doi.org/ 10.1111/febs.12905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, Spooner E, Isasa M, Gygi SP, Sabatini DM. The Sestrins Interact with GATOR2 to Negatively Regulate the Amino-Acid-Sensing Pathway Upstream of mTORC1. Cell Rep 2014 Sep 24; PMID:25263562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol 2014 Mar 31; 24(7):400-6; PMID:24698685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu W, Wu Y, Wang L, Gao L, Wang Y, Liu X, Zhang K, Song J, Wang H, Bayer TA, et al.. Protein signature for non-small cell lung cancer prognosis. Am J Cancer Res 2014; 4(3):256-69; PMID:24959380 [PMC free article] [PubMed] [Google Scholar]
- 50.Hofmann HS, Simm A, Hammer A, Silber RE, Bartling B. Expression of inhibitors of apoptosis (IAP) proteins in non-small cell human lung cancer. J Cancer Res Clin Oncol 2002 Oct; 128(10):554-60; PMID:12384799; http://dx.doi.org/ 10.1007/s00432-002-0364-z [DOI] [PubMed] [Google Scholar]
- 51.Lee JH, Budanov AV, Karin M. Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab 2013 Dec 3; 18(6):792-801; PMID:24055102; http://dx.doi.org/ 10.1016/j.cmet.2013.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.