

Abstract
Cyclin dependent kinase 5 (Cdk5) is a determinant of PARP inhibitor and ionizing radiation (IR) sensitivity. Here we show that Cdk5-depleted (Cdk5-shRNA) HeLa cells show higher sensitivity to S-phase irradiation, chronic hydroxyurea exposure, and 5-fluorouracil and 6-thioguanine treatment, with hydroxyurea and IR sensitivity also seen in Cdk5-depleted U2OS cells. As Cdk5 is not directly implicated in DNA strand break repair we investigated in detail its proposed role in the intra-S checkpoint activation. While Cdk5-shRNA HeLa cells showed altered basal S-phase dynamics with slower replication velocity and fewer active origins per DNA megabase, checkpoint activation was impaired after a hydroxyurea block. Cdk5 depletion was associated with reduced priming phosphorylations of RPA32 serines 29 and 33 and SMC1-Serine 966 phosphorylation, lower levels of RPA serine 4 and 8 phosphorylation and DNA damage measured using the alkaline Comet assay, gamma-H2AX signal intensity, RPA and Rad51 foci, and sister chromatid exchanges resulting in impaired intra-S checkpoint activation and subsequently higher numbers of chromatin bridges. In vitro kinase assays coupled with mass spectrometry demonstrated that Cdk5 can carry out the RPA32 priming phosphorylations on serines 23, 29, and 33 necessary for this checkpoint activation. In addition we found an association between lower Cdk5 levels and longer metastasis free survival in breast cancer patients and survival in Cdk5-depleted breast tumor cells after treatment with IR and a PARP inhibitor. Taken together, these results show that Cdk5 is necessary for basal replication and replication stress checkpoint activation and highlight clinical opportunities to enhance tumor cell killing.
Keywords: Cdk5, hydroxyurea, ionizing radiation, PARP-1, replication stress
Introduction
Cyclin dependent kinase 5 (Cdk5), a proline directed serine/threonine kinase, modulates many different physiological process including migration,1 survival and synaptic functions2 in neurons. A link between Cdk5, DNA repair and cell cycle regulation has also been shown with Cdk5 phosphorylating Ape1,3 ATM and STAT3.4-6 In non-neuronal cells the absence of Cdk5 was associated with a sensitization to campthotecin, cisplatin, poly(ADP) ribose polymerase (PARP) inhibitors7,8 and ionizing irradiation (IR)8 but not to methyl methane sulphonate or neocarzinostatin.8 Thus although a high level of Cdk5 expression has been associated with tumor progression and metastasis,9-12 the sensitivity of Cdk5 depleted cells would suggest that the low Cdk5 levels found in some tumors could be exploited for therapeutic benefit.
Previous studies from Turner et al. showed that Cdk5 is required for an efficient intra-S checkpoint in response to DNA damage.7 Cdk5 is however not directly required for either DNA double strand break (DSB)7 or single strand break (SSB) repair8 suggesting that Cdk5 is necessary for an earlier step in the replicative stress response. One of the key steps is the priming phosphorylation of replicating protein A (RPA) subunit RPA32 on serines 23 (S23), 29 (S29) and 33 (S33) by cyclin-Cdk complexes and ATR followed by the hyper-phosphorylation on serines 4 (S4) and 8 (S8) that is mostly dependent on DNA-PK.13 This sequential modification of RPA32 is necessary for checkpoint activation and DNA repair. Indeed the reduced hyper-phosphorylation at RPA-S4S8 has a direct consequence on Rad52 and Rad51 loading and recombination repair after replicative stress.14-16
In the present study we sought to further characterize the impact of the absence of Cdk5 on cell survival, and determine cell cycle dynamics under basal growth conditions and in response to replicative stress. In addition we have assessed whether Cdk5 levels in breast tumors are associated with metastasis free survival (MFS) and using an in vitro approach examined the impact of Cdk5 depletion on cell survival in 2 breast tumor models after treatment with IR and a PARP inhibitor.
Results
The depletion of Cdk5 expression results in lower cell survival and altered S-phase dynamics
The S-phase radioresistance, evaluated by the ratio of the surviving fraction after exposure to 2 Gy (SF2) for unsynchronised cells vs synchronized cells, was significantly lower in HeLa cells in which Cdk5 was stably depleted (Cdk5-shRNA) compared to Control cells8 (ratio 1.5 ± 0.16 for Control cells vs 1.06 ± 0.20 for Cdk5-shRNA cells, P = 0.004) (Fig. 1A and E).
Figure 1.

Clonogenic cell survival of Control and Cdk5 deficient cell lines to increasing doses of (A) 137Cs gamma rays (B) Hydroxyurea (HU) (C) 5-fluorouracil (5-FU) and (D) 6-thioguanine (6-TG). (A) Asynchronous or synchronized in S-phase (double thymidine block) cells were irradiated and colonies were allowed to grow for 10–15 days. (B) Asynchronous cells were exposed to increasing concentrations of HU present in the culture medium until colony fixation or to (C) 5-FU or (D) 6-TG for 24 h followed by fresh medium and colony growth. Data represents the combined mean ±SD from at least 2 independent experiments using 2 different HeLa Cdk5 clones for each experiment in triplicate for all conditions. (**P < 0.01; ***P < 0.001; Unpaired t-test). (E) Representative western blot showing the depletion of Cdk5 protein in the 2 Cdk5-shRNA cell lines used compared to the 2 Control clones. Ku80 was used as a gel loading control.
The Cdk5-shRNA HeLa cells also showed an increased sensitivity to chronic hydroxyurea (HU) exposure, and 5-fluorouracil (5-FU) and 6 thioguanine (6-TG) treatment (Fig. 1B-D), all agents that disrupt replication. In order to assess whether a similar phenotype was seen in another cell model we used the same shRNA expression system to stably deplete Cdk5 in U2OS cells and found that asynchronous Cdk5-depleted U2OS cells were more sensitive to the cell killing effects of HU and IR (Fig. S1A and B).
The depletion of Cdk5 in the HeLa cell model on cell growth and replication was further characterized and found to be associated with a slower basal rate of cell proliferation (Fig. S2A) and S-phase (Fig. S2B). The underlying causes were a significantly slower replication velocity in the Cdk5-shRNA cells compared to Control cells (median velocity 1.06 ± 0.03 Kb/min for Control and 0.87 ± 0.02 Kb/min for Cdk5-shRNA cells) as assessed by DNA combing (Fig. 2A) and fewer active origins per megabase of DNA (Fig. 2B). These data show for the first time that Cdk5 plays an active role in the regulation of replication dynamics under basal growth conditions.
Figure 2.
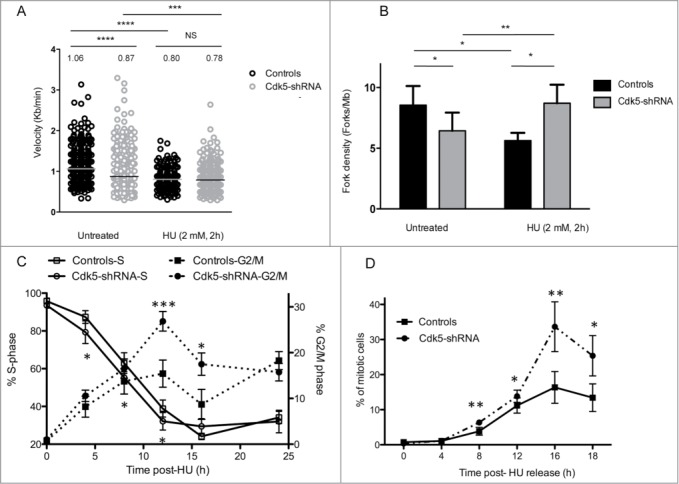
Cdk5-shRNA cells show a faster progression through S and G2 after exposure to HU. (A) Replication fork speed distribution in Control and Cdk5-shRNA cells in treated (HU 2mM, 2 h) or untreated cells. 100 to 250 DNA fibers were scored per condition. The numbers correspond to the median (shown as a horizontal line) replication speed. P values are indicated (NS - not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P <0.0001, Mann-Withney test). Data are based on 2 independent experiments for each Cdk5-shRNA clone, mean values of the 4 experiments have been calculated. (B) Increased fork density in Cdk5-shRNA cells after HU (2 mM, 2 h) treatment: Control and Cdk5-shRNA cell lines were treated or not, then labeled with successive pulses of IdU and CldU for 30 min each. Fork density was determined as the number of forks per Mb in the S-phase DNA population. More than 100 Mb was measured per condition. Data are the combined means ± SD from 2 independent experiments for each Cdk5 clone, mean values of the 4 experiments have been calculated. (C) Cells were treated with HU (2 mM) for 24 h, released into fresh medium (0 h corresponds to 24 h HU treatment) then pulse labeled with BrdU (10 µM, 15 min) at different times post release before collection and the percentage of Controls and Cdk5-shRNA cells in S and G2/M phases were determined by FACS analysis. Data are means ± SD from 3 independent experiments for 2 different HeLa Cdk5 clones. (*P < 0.05; **P < 0.01; ***P < 0.001; Unpaired t-test). (D) Mitotic entry after HU release. Control and Cdk5 cells were treated with 2 mM HU for 24 h and released into medium containing Nocodazole (50 µg/µl). The percentage of mitotic cells was analyzed by FACS by phospho-H3 content. Data are means ± SD from 2 independent experiments for 2 different HeLa Cdk5 clones. (*P < 0.05; **P < 0.01; ***P < 0.001; Unpaired t-test).
Cdk5 is necessary for full activation of checkpoint signaling
In the light of the survival profiles of the Cdk5-depleted cells to agents that generate replicative stress and in order to investigate Cdk5's role in the activation of the intra-S phase checkpoint in more detail, we treated cells with HU that inhibits ribonucleotide reductase causing a rapid depletion of the dNTP pool resulting in a slowing of fork rate progression17 and the activation of ATR-dependent DNA replication checkpoints.18 In contrast to basal growth conditions, after the induction of replicative stress Cdk5-shRNA cells show a faster recovery than Control cells (Fig. 2C). The proportion of cells remaining in the S-phase 8 h after HU release was higher in Control compared to Cdk5-shRNA cells (P = 0.016). This deficiency was also reflected in the statistically higher proportion of Cdk5-shRNA cells in G2/M (P = 0.02) seen from 8 h to 16 h after HU release (Fig. 2C) and a doubling of the number of mitotic cells (P = 0.0015) (Fig. 2D) 16 h after HU release compared to Control cells. The observation that a similar cell cycle profile was observed when cells were treated with aphidicolin, that disrupts replication by inhibiting DNA polymerases19 (Fig. S3A), rules out the possibility that the differences between the Cdk5-shRNA and Control cells are related to dNTP pool sizes. These results support a role for Cdk5 in the activation of the intra-S checkpoint as previously reported.7
Two parameters that ensure the activation of the intra-S checkpoint are the replication fork velocity and fork density which are essential to maintain replication fork stability and block the initiation of latent origins.20-22 As expected,17 HU treatment reduced the fork speed in Control cells (Fig. 2A) from 1.06 Kb/min in untreated cells to 0.80 Kb/min after HU treatment and resulted in a decrease in fork density from 8.4 to 5.6 forks/Mb in HU treated Control cells (Fig. 2B). HU treatment however had a negligible effect on the already slower fork speed in Cdk5-shRNA cells with the velocities seen comparable to those in HU treated Control cells (0.87 Kb/min in untreated Cdk5-shRNA cells, 0.78 Kb/min in HU treated Cdk5-shRNA cells and 0.80 Kb/min in HU treated Control cells). In contrast a significant increase in fork density after HU treatment in the absence of Cdk5, from 6.4 in untreated cells to 8.7 forks/Mb, was observed. These results suggest that Cdk5 is required to inhibit the firing of late origins during replicative stress, providing an explanation as to why Cdk5-shRNA cells recover faster from a HU-block than Control cells.
Impaired RPA32 phosphorylation in Cdk5-shRNA cells after replicative stress
One of the first signals of replication stress is the accumulation of ssDNA at the replication fork that arises from the uncoupling of the replicative helicases and DNA polymerases.23,24 Once formed this ssDNA is coated by RPA and acts as a signaling platform for the recruitment of the ATR-ATRIP complex, the activation of ATR and the subsequent phosphorylation of down-stream effector proteins required for the activation of the DNA replication damage checkpoint.25,26 In order to analyze the consequences of Cdk5 depletion on this cascade of ATR-dependent signaling events, we next assessed the early steps during and up to 24 h after an HU block. Six h after the start of the block a clear reduction in the level of the phosphorylation of Chk1-S317 was seen in the Cdk5-shRNA cells providing an explanation for the higher fork density observed compared to the Control cells (Fig. 3A) and by 24 h significantly lower maximum levels of SMC1-S966 and RPA32-S33 and -S29 phosphorylation were seen in the Cdk5-shRNA cells compared to the Control cells (Fig. 3B). Reduced RPA32-S4S8 and -S33 and SMC1-S966 phosphorylation was also observed 24 h after aphidicolin (2.5 µg/ml) treatment in the Cdk5-shRNA HeLa cells (Fig. S3B–C). However no clear differences in the dephosphorylation patterns of these proteins between the Controls and the Cdk5-shRNA HeLa cells were noted after release from the HU block.
Figure 3.
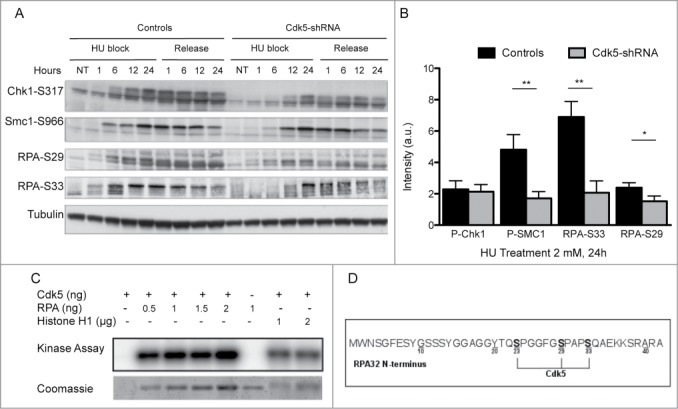
Decreased ATR and Cdks-dependent phosphorylation of Replication Protein A upon HU treatment in Cdk5 depleted cells. (A) Representative western blot analysis of phospho-SMC1-S966, phospho-Chk1-S317 and phospho-RPA-S29 and S33 in protein extracts from Control and Cdk5-shRNA cells during a 24 h HU block (2 mM) and over the following 24 h after release from the block. Total Tubulin was used as a loading control. (B) Quantification data are ratios of intensities of treated to non-treated cellular extracts after normalization with tubulin intensity, data are means ± SD from 2–3 independent experiments using 2 different HeLa Cdk5-shRNA clones have been calculated. (*P < 0.05; **P < 0.01; Unpaired t-test). (C) In vitro kinase assay showing the phosphorylation of purified RPA32 protein by Cdk5/p25 recombinant kinase after 30 min at 25° C. The image is representative of 3 independent experiments. The phosphorylation of histone H1 by Cdk5 was used as a positive control. (D) Sites of phosphorylation of the RPA protein by Cdk5 in vitro (same experimental settings as in 3C), identified by mass spectrometry: S23, S29 and S33. All three sites were detected in 7 independent experiments (full spectra are shown in Fig. S3).
The lower in vivo levels of RPA32 phosphorylation in Cdk5-shRNA cells led us to investigate whether Cdk5 could phosphorylate RPA32 on the same sites as reported for Cdk1 and Cdk227-29 and ATR.30 Using in vitro kinase assays followed by mass spectrometry analysis we found that Cdk5 phosphorylated RPA32 (Fig. 3C) on positions S23, S29 and S33 (Fig. 3D, Fig. S4A–C). Under the same experimental conditions, active Cdk1 and Cdk2 can also phosphorylate these 3 positions but a significantly lower relative frequency of these events was detected compared to that seen with Cdk5 (Fig. S4D). These results suggest that these 3 priming phosphorylations that normally requires the synergistic action of the cyclin-Cdks and PIKK13 can be carried out by Cdk5.
Depletion of Cdk5 expression impacts on the later steps of the DNA damage response
The altered RPA32 phosphorylation profile in response to HU in the absence of Cdk5 led us to compare the levels of global DNA damage in the HeLa cell model using the alkaline comet assay.31 No significant difference in the level of IR induced DNA damage was noted and identical damage levels remained 1 h post-radiation (Fig. S5A) suggesting that DNA strand break repair per se was not deficient in the Cdk5-shRNA cells. The early signaling events in the DNA DSB response were also compared in the Cdk5-shRNA vs Control cells after treatment of asynchronous cell populations with 2 and 5 Gy of gamma irradiation. We confirmed Turner's observation that Cdk5 was not required for the activation of ATM following gamma irradiation (Fig. S5B) and also confirmed this in S-phase synchronised cell populations (Fig. S5C). In contrast to gamma irradiation that induces DNA damage that directly activates the ATM-Chk2 signaling pathway,32-34 HU stress leads to fork stalling followed by resection that activates the ATR signaling cascade. After a 24 h HU treatment a significantly lower level of DNA damage was seen in Cdk5-shRNA compared to Control cells, as seen by the lower mean tail moment in the alkaline comet assay and at all subsequent time points assessed (Fig. 4A). This result was confirmed using 3 additional endpoints. Firstly the gamma-H2AX signal intensity observed 24 h into the HU block and up to 12 h after release was lower in Cdk5-shRNA cells compared to Control cells (P < 0.001 to <0.05 respectively) (Fig. 4B). Secondly, although the proportion of cells with RPA foci was higher in S-phase Cdk5-shRNA cells under basal growth conditions compared to Control cells, in agreement with the increased spontaneous replication stress in Cdk5-shRNA cells and their reduced S-phase kinetics, a smaller percentage of Cdk5-shRNA cells contained ssDNA as assessed by the analysis of RPA foci after HU treatment compared to Control cells (P ≤ 0.0001) (Fig. 4C). Finally using a BrdU labeling technique under native conditions, we could show a reduce amount of ssDNA in Cdk5-shRNA compared to Controls cells (Fig. 4D). These observations would suggest that the resection step is also compromised in the absence of Cdk5 after a HU block. Such a defect would directly impact on the amount of ssDNA formed and the activation of the ATR signaling cascade, thus reducing the intra-S checkpoint activation under conditions of HU stress.
Figure 4.
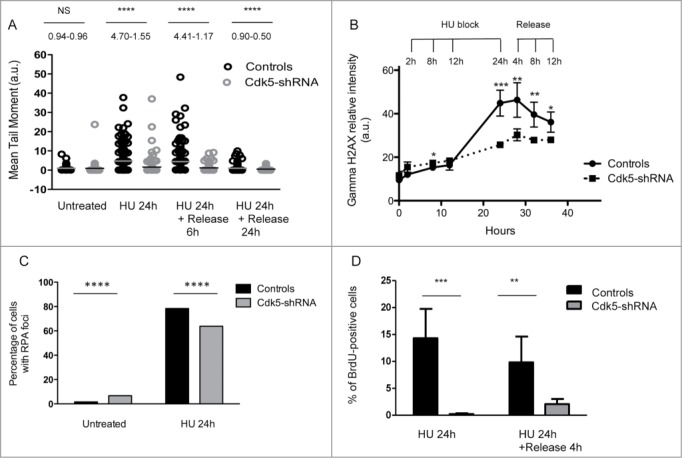
Compromised Cdk5 expression impacts on the processing of DNA Damage after HU treatment. (A) DNA strand break formation and persistence measurement in the Cdk5-shRNA and Control cells using the alkaline Comet assay 24 h after treatment with HU (2 mM). The data correspond to the mean tail moment. Mean values are represented as horizontal lines. 200 cells for 2 different HeLa Cdk5 clones were scored per condition. Mean values for the 2 Cdk5-shRNA clones have been calculated. P values are indicated (NS - not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001) using unpaired t-test. (B) The induction of gamma H2AX was assessed using immune-coupled FACS analysis to quantify total gamma H2AX levels using fluorescence intensity in the cells as a read-out. Cells were collected at the times indicated during HU treatment (2 mM) and after 24 h of treatment when cells were released into a fresh medium containing nocodazole (50 µg/µl). Mean and SD values are presented from 3 independent experiments using 2 different HeLa Cdk5 clones analyzed using an unpaired t-test (*P < 0.05; **P < 0.01; ***P < 0.001). (C) Decreased number of cells with RPA foci in Cdk5-shRNA cells after HU treatment. Cells were released from HU treatment (24 h, 2 mM), and immediately fixed and immunostained for RPA 32 and the number of RPA foci determined. Data are the percentage of cells with more than 5 foci (median ± SE), from 3 independent experiments with a minimum of 300 cells analyzed per experiment. Mean values of the 2 Cdk5-shRNA clones have been calculated. Statistical analysis was carried using an unpaired t-test (****P < 0.0001). (D) Reduced ssDNA in Cdk5-shRNA cells after prolonged exposure to HU. BrdU (10 μM) was incorporated for 2 complete cell cycles before the HU treatment (2 mM for 24 h, followed by a release for 0 min or 4 h in fresh medium) and detected in native condition by immunofluorescence using an anti-BrdU antibody. The graph illustrates the proportion of BrdU-positive cells detected without DNA denaturation, expressed as a percentage of the total cells analyzed. No ssDNA positive cells were detected in HU untreated cells. More than 600 cells per condition were scored; mean values of 3 independent experiments with the 2 Cdk5-shRNA clones and a Control clone ± SD are shown using an unpaired t-test (**P < 0.01; ***P < 0.001).
Prolonged HU exposure leads to fork breakage and DSB formation. The resection of these DSBs can be assessed using as an endpoint the quantification of RPA32-S4S8 phosphorylation and the subsequent accumulation of Rad51 foci formation after HU treatment.35 RPA32-S4S8 phosphorylation as measured in protein extracts of cells treated with HU showed a slower induction and reached significantly lower levels in the Cdk5-shRNA compared to the Control cells (P = 0.0019 at 24 h) (Fig. 5A). An altered profile of RPA32-S4S8 phosphorylation profile was also seen during HU treatment in Cdk5-depleted BT549 breast cancer (Fig. S3D).
Figure 5.
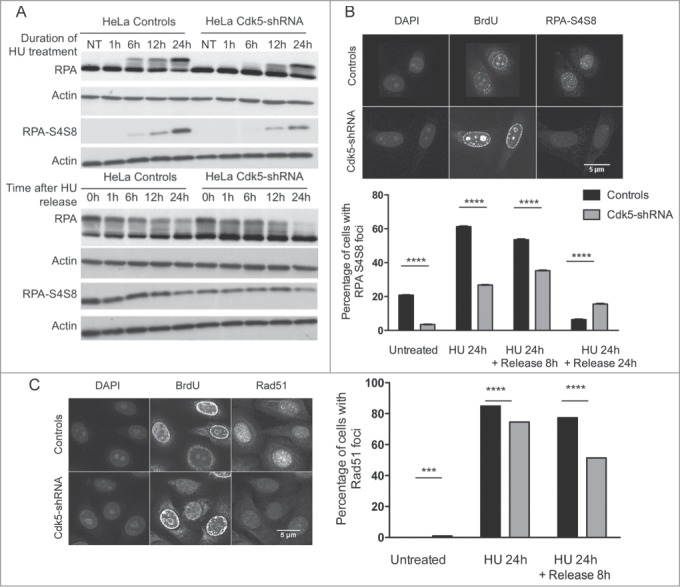
Lower RPA S4S8 phosphorylation and Rad51 foci formation in Cdk5-shRNA cells after HU treatment. (A) Representative western blot of phospho-RPA S4S8 levels in protein extracts from Control and Cdk5-shRNA during HU block (2 mM,) and up to its removal from the culture medium. RPA S4S8 was detected using a phosphospecific antibody. Actin expression was used as a gel loading control. (B) Phospho-RPA foci formation: Cells were released from HU (2 mM, 24 h) at the indicated times, fixed and immunostained for RPA S4S8. The figures are representative of 2 independent experiments with 2 different clones. The mean percentage of cells carrying more than 5 foci are presented, with SEM. Only BrdU positive cells (S-phase) were quantified with 350 to 600 cells analyzed (****P < 0.0001; P < 0.0001, unpaired t-test). (C) Formation and persistence of RAD51 foci in Cdk5-shRNA and Control cells. Cells were treated or not with HU (2 mM, 24 h) and Rad51 foci quantified using a 2D Spinning-Disc/Tirf/Frap system after a pulse labeling with BrdU (10 μM, 15 min) at the indicated times. Data are representative of 4 independent experiments using 2 different Cdk5-shRNA clones with between 600 and 3,000 cells analyzed for each condition and represent the number of BrdU positive cells carrying more than 5 foci, compared by t-test between the different treatment groups (***P < 0.001; ****P < 0.0001).
In addition a statistically lower number of RPA32-S4S8 foci was observed in S-phase Cdk5-shRNA compared to Control HeLa cells after the HU block and also 8 h after release, the time point when DSB repair occurs36 (Fig. 5B). In agreement with this lower number of RPA32-S4S8 foci, S-phase Cdk5-shRNA cells had statistically fewer Rad51 foci after the HU block and 8 h after release (P < 0.0001) (Fig. 5C), indicating a reduction in homologous recombination (HR) activity. The Cdk5-shRNA cells did however show a higher number of Rad51 foci 24 h after release compared to Control cells, indicating the persistence of some unrepaired residual breaks.
In order to investigate the longer term effects of Cdk5 deficiency, we quantified the formation of sister chromatid exchanges (SCEs) which relies on a functional HR pathway in cells after HU treatment (Fig. 6A and B). HU treatment induces a 7-fold increase in SCE numbers in Control cells compared to untreated cells but only a 3-fold increase in Cdk5-shRNA cells. This lower level of HU-induced HR is in agreement with the results from the other endpoints that demonstrate that Cdk5 is necessary for promoting the full and timely activation of the cascade of events initiated in response to replication stress. One additional consequence of replication stress when DNA signaling and repair is reduced, is the accumulation of anaphase chromatin bridges.37 Our results show that, while the level of chromatin bridges is similar in the absence of stress in Control vs Cdk5-shRNA cells, we observe a higher number of chromatin bridges after inducing replication stress in the Cdk5-shRNA cells (Fig. 6C–D). This response profile is similar to that observed in RAD51-deficient cells,38 suggesting that in the absence of Cdk5, the broken forks remain unrepaired, leading to chromosome breaks and chromatin bridge formation that may ultimately lead to cell death, and further suggests that the main sources of DNA damage in the Cdk5-depleted cells after HU stress are replication dependent.
Figure 6.
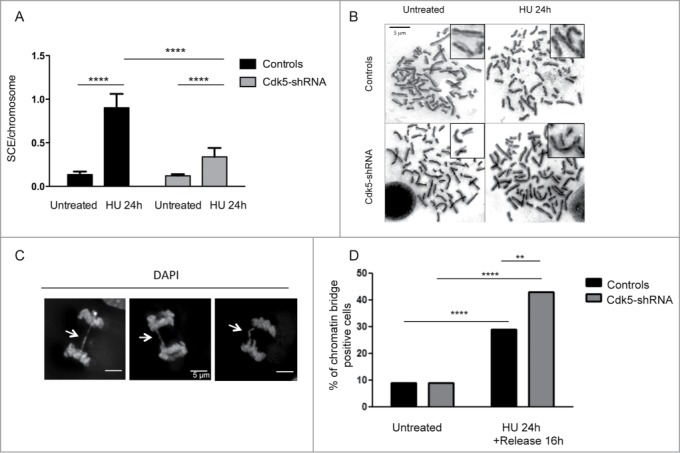
Level of HU-induced SCEs and chromatin bridges in Cdk5-shRNA cells compared to Control cells. (A) Number of SCEs per chromosome in untreated or HU treated (2 mM HU, 24 h) Control or Cdk5-shRNA cells (2 different clones). Between 2372 and 4499 chromosomes from 3 independent experiments were analyzed for each condition, mean values of the 2 Cdk5-shRNA clones were calculated. Error bars represent standard errors of the means. (****P < 0.0001, unpaired t-test). (B) Representative images of SCE in Control and Cdk5-shRNA cells, without and after HU treatment (2 mM, 24 h) Chromosomes were observed with a Leica DMRB microscope at 100x magnification. (C) Representative immunofluorescence deconvoluted z projection images of anaphase cells presenting chromatin bridges (white arrows). DNA was visualized by DAPI staining. Scale bar: 5 µm. (D) Cells were treated or not with HU (2 mM for 24 h), followed by a release for 16 h in a fresh medium. The proportion of chromatin bridges positive cells (DAPI-positive) is expressed as a percentage of the total cells analyzed. At least 80 cells per condition were scored; mean values ± SD of 3 independent experiments with one control and one Cdk5-shRNA clone are shown. **P < 0.01; ****P < 0.0001.
Lower Cdk5 expression is associated with higher MFS in breast cancer patients and increased in vitro sensitivity to cell killing by PARP inhibition and IR
As many cytotoxic anti-cancer treatments target proliferating cells by interfering with DNA replication and Cdk5 appears essential for this response, we investigated whether its expression profile in a panel of 456 breast tumors was associated with clinical outcome in terms of MFS. Low Cdk5 mRNA levels (≤3.8 fold relative to that seen in normal tissues) was associated with longer MFS (P = 0.0078; Fig. 7A). In a sub-set (n = 233) of tumors, Cdk5 protein expression was also evaluated using Reverse Phase Protein Arrays and a similar association between lower Cdk5 expression and longer MFS observed (P = 0.047; data not shown). No association between high Cdk5 expression (>3.8 fold) and age of onset or Scarff Bloom Richardson histological grade were found but high Cdk5 expression was more frequently seen in tumors in which ERBB2 was over-expressed, ERBB2 positive molecular subtypes and in highly proliferating tumors with high KI67 mRNA levels (Table S1).
Figure 7.
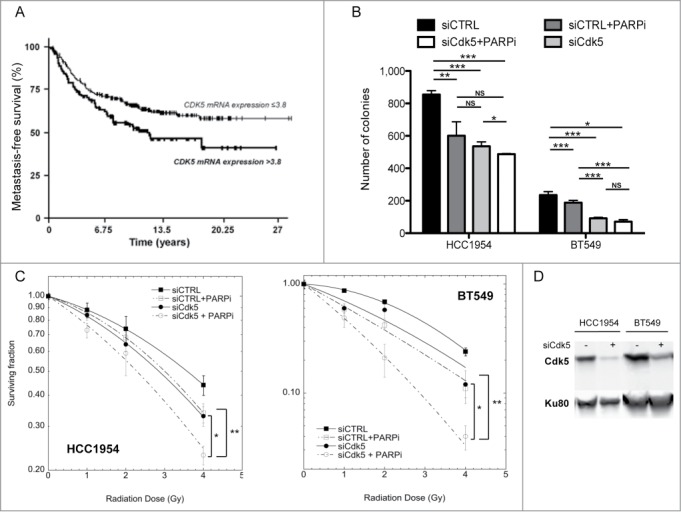
Survival of patients with breast tumors and breast cancer lines depending on Cdk5 expression profile. (A) MFS curves for breast tumor patients with high (NCdk5 > 3.8) and low (NCdk5 ≤ 3.8) tumor expression levels of Cdk5 mRNA. The mean patient follow-up was 9.5 years (range 5 months to 33 years) and 169 patients in the cohort developed metastases. (B) Survival of HCC1954 and BT549 breast tumor cell lines. Cells were transfected either with a siRNA control or a siRNA directed against Cdk5 expression. 72 h later they were plated and left to attach for 4 h before treatment with or without the PARP inhibitor ABT888 (10 μM). 24 h after treatment cell culture medium was changed and colonies grown for 8 days and counted. Data are the mean numbers of colonies counted ± SD from 2 independent experiments each in triplicate (*P < 0.05; **P < 0.01; ***P < 0.001, unpaired t-test). (C) Clonogenic cell survival of HCC1954 (left panel) and BT549 (right panel) breast tumor cells depending on Cdk5 expression after increasing doses of gamma-irradiation in the presence or not of a PARP inhibitor. Cells were transfected with either a control or Cdk5 siRNA as in panel (B) and then the PARP inhibitor ABT888 (10 μM) added 1 h prior to radiation. 24 h after treatment cell culture medium was changed and colonies grown for 8 days and counted. Data are the mean survivals of 2 independent experiments each in triplicate (*P < 0.05; **P < 0.01, unpaired t-test). (D) Representative western blot of breast cancer cell lines after transfection with siRNA control or Cdk5.
In order to investigate whether Cdk5 depletion could impact on cell survival after treatment with IR or a PARP inhibitor, we transfected HCC1954 and BT549 breast tumor cells, chosen from a panel of over 30 breast cancer lines based on their relatively low Cdk5 mRNA levels (Bieche et al., unpublished data), with either a Cdk5 targeting or control siRNA and assessed cell numbers using a colony forming assay. In these 2 cell lines, Cdk5 depletion or PARP inhibition both reduced colony numbers compared to the transfection of the parental line with a control siRNA (P < 0.05 or P < 0.001) (Fig. 7B). In the HCC1954 cell line, the inhibition of PARP activity in the Cdk5 depleted cells resulted in a further reduction (P < 0.05). In contrast in the BT549 cells, the depletion of Cdk5 had a significantly greater impact compared to PARP inhibition and no additional lowering of colonies numbers was noted when PARP activity was inhibited in the Cdk5 depleted cells. In both cell models the depletion of Cdk5 was associated with increased radiation sensitivity (Fig. 7C), as seen in the HeLa and U2OS Cdk5 depleted cells. This radiation sensitivity could be furthered lowered by the combined treatment of IR and PARP inhibition in the Cdk5 depleted cells.
Discussion
Recent studies have demonstrated that Cdk5 is functionally important in different types of cells and in various physiological and pathophysiological contexts39,40 and can impact on cell growth and migration,41,42 apoptosis,43,44 tumor progression,9-12 cell cycle regulation and DNA damage response.7,8,45 Our study has identified a novel role for Cdk5 in S-phase cell cycle progression under basal growth conditions in the absence of genotoxic stress. The observations that the proportion of cells with RPA foci was higher and more RPA32-S4S8 foci/cell were seen in S-phase Cdk5-shRNA cells under basal growth conditions compared to Control cells is in agreement with the increased spontaneous replication stress in Cdk5-shRNA cells and the reduced S-phase kinetics observed under basal conditions. In addition we have shown that Cdk5 is necessary for full checkpoint activation in response to replication stress.
There are a number of reports in the literature that support a role for Cdk5 in basal replication. Cdk5 was identified as a partner of the proliferating cell nuclear antigen, a key replication protein46 and Xu et al.42 recently reported impaired proliferation in Cdk5 depleted breast cancer cells. Nagano and colleagues showed a direct in vivo interaction of Cyclin I and Cdk5 and the involvement of Cyclin I in S-phase cell cycle proliferation.45 Thus it is tempting to speculate that Cdk5 may interact with Cyclin I during the replication process. The observation of higher PAR levels in Cdk5 depleted cells8 could provide an explanation for the slower fork velocity under basal growth conditions. However, as PARG depleted cells provide no evidence for an impact of higher polymer levels on cell cycle progression,47 this is unlikely to be the cause.
In addition to a slower S-phase progression under normal growth conditions, the absence of Cdk5 is associated with a perturbation of S-phase dynamics after treatment with both transient and prolonged HU treatment. The inhibition of ribonucleotide reductase by HU has distinct consequences depending on the treatment time: transient exposures result in the stalling of replication forks requiring an ATR-RPA dependent pathway to stabilize the stalled forks, whereas prolonged HU exposure leads to fork collapse requiring either the initiation of dormant origins or the repair of collapsed forks through HR for replication restart.35,36,48,49 The absence of Cdk5 impacted on both treatment profiles highlighting its central role in replication dynamics. Under transient HU exposure the reduction in fork velocity seen in Control cells was less marked in Cdk5-shRNA cells but was accompanied by a reduced phosphorylation of RPA32 on S33 and S29 in vivo. This is in agreement with observations that RPA phosphorylation can inhibit DNA replication50-52 and suggests that DNA synthesis arrest after HU induced replication stress may be mediated in part by the phosphorylation of RPA32 by Cdk5. The sequential phosphorylation of RPA32 is a key event in checkpoint activation and DNA repair with the priming phosphorylations on S23, S29 and S33 being an essential step.13 It has been reported that S23 and S29 can be phosphorylated in vitro by Cdk1 and Cdk227-29,53 and ATR is considered as the PIKK that phosphorylates S33.54 Our in vitro data suggests that while Cdk1, Cdk2 and Cdk5 can phosphorylate all these serines, Cdk5 shows a higher affinity for the 3 sites. Taken together with the in vivo data, our results confirm Cdk5 as a novel member of the Cdk/PIKK families necessary for the S-phase DNA damage response through a mechanism involving RPA32 phosphorylation.
In terms of the phosphorylation cascades initiated in response to replication stress, Chk1 is a direct target of ATR's kinase activity resulting in its phosphorylation on S317. This phosphorylation event is essential for the inhibition of origin firing through a mechanism involving Cdc25A phosphatase stability.55 In the absence of Cdk5 Chk1-S317 phosphorylation was delayed during the early stages of the HU block, although by 24 h into the HU block no differences between the Control and Cdk5-shRNA depleted cells were seen. These results would suggest that Cdk5 is necessary for the initial step of resection and the priming phosphorylation of RPA32 and suggests that full RPA32 phosphorylation is not necessary for Chk1 activation, in agreement with observations of Sleeth et al.15 The lower levels of these RPA32 priming phosphorylations impact on the hyper-phosphorylation on S4/S8 seen after prolonged HU treatment. This hyper-phosphorylation is a key step in the activation of HR initiating events, and reduced HR events, as measured by Rad51 foci and SCE levels, were seen in the absence of Cdk5. It has been reported that PARP-1 binds to and is activated by stalled replication forks to promote replication fork restart after release from replication blocks.56 However despite the observations that Cdk5-shRNA cells have higher levels of DNA damage induced polymer compared to Control cells,8 we could not find any evidence that the absence of Cdk5 impacts on fork restart (Fig. S6). We also observe less ssDNA formed during HU treatment in Cdk5-shRNA compared to Control cells. The formation of ssDNA is a marker of the functional uncoupling of the replicative helicases and DNA polymerases and this lower level raises the possibility that Cdk5 promotes this process at stalled replication forks, a function that has also been attributed to the helicase MCM.57
Based on the cellular and molecular phenotype of Cdk5-shRNA cells, it is tempting to speculate that the absence of Cdk5 in tumors could be exploited for therapeutic benefit as such tumors are more likely to be sensitive to killing by certain treatments. Cdk5 loss was identified as a determinant of sensitivity to PARP inhibitors.7 Our results support a role for Cdk5 in the activation of the intra-S and also the G2/M checkpoint as previously reported.7 It has been suggested that some determinants of PARP inhibition sensitivity are involved in the G2/M checkpoint such as PLK3, MAPK12/P38g,7,58 thus associating this cell cycle checkpoint to PARP inhibitor sensitivity. Here we suggest that the role of Cdk5 in the signalisation of the replication checkpoint could also impact on PARP inhibition sensitivity.
A better understanding of the gene interactions and functional buffering that occur in tumor cells, together with a measure of the heterogeneity found between different tumors, is important for the development of new therapeutic approaches.59 Over the past few years significant advances have been made to directly target such changes through for instance the use of DNA repair and cell cycle inhibitors. Whether inter-individual differences in tumor Cdk5 expression profiles can be exploited to clinical advantage remains to be fully investigated. Recently Levacque et al.60 reported that Cdk5 expression levels predicts the survival of relapsed multiple myeloma patients to the proteasome inhibitor bortezomib, with individuals with Cdk5 expression in the bottom quartile having significantly higher survival compared with those with Cdk5 in the top quartile. Our observation that significantly longer MFS was associated with breast tumors with lower Cdk5 mRNA expression is in agreement with Liang et al.10 who found in a panel of 108 breast tumors that the over-expression of Cdk5 protein expression was significantly correlated with several poor prognosis markers (ER- (P < 0.001), basal-like (P < 0.001) and high grade of malignancy (P < 0.001). It has to be noted however that differences exist between the 2 studies in some associations which may be due to differences in study sizes, the distribution of the breast tumor subtypes within the cohorts and the criteria for overexpression. As no Cdk5 inhibitors with specificity and selectivity are available to date61 and based on the mechanistic role of Cdk5 in the response to replicative stress, the therapeutic use of PARP inhibitors combined with radiotherapy could be one promising avenue for tumors with low Cdk5 expression. Such an approach would exploit the S-phase cell cycle dependency of PARP inhibition62 and the increased S-phase radiosensitivity associated with the loss of Cdk5 expression. In the 2 breast tumor models tested the radiation sensitivity was increased by PARP inhibitor and further increased when Cdk5 was depleted. While clearly such an approach exploiting replicative stress would require further validation and optimization in more models, it could be applicable to many of the different tumor types in which Cdk5 expression has been shown to be down-regulated.
Materials and Methods
Cell lines and drugs
shRNA design and cloning in pEBVsiRNA vectors carrying a hygromycin B resistance cassette, the establishment and growth of stable knockdown Cdk5-shRNA clones and HeLa cells carrying the pBD650 plasmid that expresses an inefficient shRNA sequence that were used as Controls, was carried out as previously described.8,63 The RNAi targeted sequences for Cdk5 (NM_004935) were nucleotides 703–721 for clone 1499 and nucleotides 455–473 for clone 1500. Both HeLa Cdk5-shRNA clones were used in the different experimental approaches. Cdk5 was depleted in U2OS cells, established from a healthy donor,64 by transfecting the pEBV siRNA vector63,65 targeting Cdk5 nucleotides 703–721. Breast cancer cells BT549 and HCC1954 were obtained from T. DuBois (Transfer Department, Institut Curie, Paris, France). All tissue culture reagents were from Invitrogen and 5-FU (F6627), HU (H8627) and 6-TG (A4882) from Sigma-Aldrich.
Cell growth and colony formation survival assays
To assess cell growth, cells were plated in complete medium (3.105 per 25 cm2 flask/line) and counted each day. For clonogenic survival after treatment with DNA damaging agents, Control (800 cells) and Cdk5-shRNA (1600 cells) cells were plated in 25 cm2 flasks in triplicate, and allowed to adhere for 4 h at 37°C before treatment. Flasks were either irradiated at room temperature using a 137Cs gamma irradiator or treated with increasing concentrations of various drugs added directly to the culture medium. To assess cell survival when irradiated in the S-phase of the cell cycle, cells were synchronized at the G1–S junction by treating them 13 h with 10 µM thymidine (T9250, Sigma-Aldrich), then washed and placed in fresh medium for 9 h and reblocked with 10 µM thymidine for 20 h. Two hours after removal of thymidine, cells were harvested for plating and clonogenic assays. In order to confirm synchronization, cell cycle distribution was monitored by fluorescence activated cell sorting (FACS) analysis using a FACSCalibur cytofluorometer (Becton-Dickinson Biosciences) after bromodeoxyuridine (BrdU) pulse labeling (10 µM, 15 min) as described previously.66 To assess cell survival after the depletion of Cdk5, HCC1954 and BT549 cells were transfected with siRNA (20 µM) directed against Cdk5 (Table S2) or a negative control (siRNA duplex, 5 nmol, SR-CL000–005, Eurogentec) using INTERFERin reagent (409–50, Polyplus transfection) following manufacturer's instructions. 72 h post-transfection 3000 cells were plated in 25 cm2 flasks in triplicate, left to attach for 4 h and treated or not with the PARP inhibitor ABT888, (ALX-270-444-M001, Enzo Life Sciences, 10 µM) for 1 h prior to irradiation. The cell culture medium was changed 23 h later and cells grown under standard culture conditions (37°C and 5% CO2 in a humidified incubator) for 8 days to allow colonies to grow. Colonies were fixed in methanol and stained with 0.05% Coomassie brilliant blue in 3:1:6 ethanol/acetic acid/water and those with more than 50 cells counted.
The colony count relative to mock-irradiated cells (relative cell survival) was adjusted for best fit to the classical linear-quadratic equation (Ln S = −α.D − β.D2) where D is the radiation dose and α and β adjustable parameters characterizing the response, using Kaleidagraph software (Synergy Software). Relative cell survival curves after other treatments were fitted with a linear quadratic fit.
Flow cytometry analysis
To assess the mitotic ratio, cells were treated with HU (24 h, 2 mM), transferred to fresh medium containing 50 µg/µl nocodazole (M1404, Sigma-Aldrich) and analyzed at the indicated times as described66. Phospho-histone H3 immunostaining of mitotic cells and BrdU immunostaining were carried out as described.66 Flow cytometric analysis was performed using a FACSCalibur cytofluorometer (Becton-Dickinson Biosciences). At least 104 events were recorded and data analysis was done with CellQuest Pro software (Becton-Dickinson Biosciences).
Western blotting
Total protein extracts were obtained after collection of cells by scraping in extraction buffer (62.5 mM Tris HCl, 2% SDS 10% Glycerol, 50 mM DTT, 0.01% bromophenol). Protein expression and phosphorylation was analyzed and quantified as described.66 Full details of antibodies used are given in Table S3.
Immunofluorescence protein detection
For the detection of Rad51 and phospho-RPA foci, cells grown on coverslips were pulse-labeled (10 μM BrdU, 30 min), and incubated (1 h) with primary antibodies for Rad51 or RPA or phospho-RPA32 S4S8. After incubation with secondary antibodies cells were fixed (5 min, 1% paraformaldehyde) and stained with anti-BrdU antibody. Coverslips were mounted with Vectashield containing DAPI 1.5 µg/ml (H-1200, Vector Laboratories). Images of fields were acquired automatically on a SPINNING-DISC/TIRF/FRAP system, based on a Nikon Ti Eclipse stand, equipped with a 60 × /1.48 objective and a Yokogawa CSU confocal scanning head. Sets of images were acquired using an Evolve EM-CCD camera (Photometrics), under control of the Metamorph software (Molecular Devices) using its “mosaic scan” option. At least 500 cells were analyzed per experimental condition. The number of foci was quantified using a macro in arbitrary units (A.U.) using Image J software. For phospho-RPAS4S8 foci and Rad51 foci only cells in S-phase were analyzed as determined by BrdU staining intensity.
RPA and Cdk5 kinases Assay
For in vitro kinase assays 1–4 µg purified RPA32 recombinant protein (ab40323, Abcam) was incubated (30 min, 30°C) with 0.5 ng active Cdk5 (14–477, Millipore), Cdk1 (14–450, Millipore) or Cdk2 (14–448, Millipore) in the presence of 10 μCi [γ-32P]-ATP and separated by SDS-PAGE. The phosphorylation of substrates was analyzed by autoradiography. Histone H1 was used a positive control.
Mass spectrometry analysis
Samples were analyzed using a LTQ Orbitrap XL mass spectrometer (MS, Thermo Scientific) coupled to a nano-Liquid Chromatography system (LC, Ultimate 3000, dionex). Details of the separation and MS parameters are provided in the Supplementary Experimental procedures.
Sister Chromatid Exchange Assays
The measurement of Sister Chromatid Exchanges (SCEs) was carried out as previously described.38 In brief cells were grown on coverslips in the presence or not of HU (2 mM) and 5-BrdU (10 µM). After 46 h, colchicine (C9754, Sigma-Aldrich) was added to a final concentration of 0.1 mg/ml for 1 h. Cells were then incubated in a hypotonic solution (1:6 (vol/vol, FBS/distilled water) and fixed with methanol/acetic acid dilutions. After staining with 10 mg/ml Hoechst 33258 (Sigma-Aldrich) in distilled water for 20 min, cells were rinsed with 2 x SSC (Euromedex) and exposed to UV light at 365 nm at a distance of 10 cm for 105 min, rinsed in distilled water, stained with 2% Giemsa solution (VWR) for 16 min, rinsed in distilled water, dried, and mounted. Chromosomes were observed with a Leica DMRB microscope at 100x magnification. Metaphases were captured with a SONY DXC 930 P camera, and SCEs counted manually by 2 observers blind to the knowledge of cell line status.
Molecular Combing
Molecular combing was performed as previously described67 with some modifications to the protocol. Briefly asynchronous cell populations were labeled with Iododeoxyuridine (IdU) and chlorodeoxyuridine (CldU) (30 min pulse labeling of first 20 µM IdU then 100 µM CldU). Agarose plugs were digested by proteinase K (48 h at a dose of 1 mg/ml) then β-agarase (48 h at 42°C). Chromatin fibers were combed and incubated with rat anti-BrdU antibody (1:25, OBT0030, clone BU1/75 ICR1; AbD Serotec) and FITC-conjugated mouse anti-BrdU (1:5, 347583, clone B44; BD Biosciences) for 1 h at 37°C. After a short wash step (0.5 M NaCl, 20 mM Tris at pH 7.8, and 0.5% Tween 20), the fibers were incubated with goat anti-mouse Alexa 488 (1:50, A-11029; Invitrogen) and goat anti-rat Alexa 594 (1:50, A-11007; Invitrogen) for 1 h. DNA counter-staining was performed by using anti-DNA antibody (1:20, MAB3034, clone 16–19; Millipore) and the following 2 secondary antibodies: rabbit anti mouse 350 (AlexaFluor) and goat anti rabbit 350 (AlexaFluor). For each condition, at least 200 fibers were analyzed. Images were acquired with a Leica DM RXA microscope equipped with a motorized XY stage, using an X40 PlanApo N.A. 1.25 objective and a CoolSNAP HQ interline CCD camera (Photo-metrics). For each slide, a mosaic of 10×10 partly overlapping images was collected with a Metamorph software (Molecular Devices) routine developed in-house. Image collections were assembled into a mosaic with the ‘Stitching 2D/3D’ plugin32 (available from http://fly.mpi-cbg.de/˜preibisch/software.html) for ImageJ software (Rasband, W.S., ImageJ, US National Institutes of Health, http://rsb.info.nih.gov/ij/, 1997–2009).
Comet assay
Cells were either grown on coverslips, treated in situ with 2 mM HU and collected by trypsinization, and then suspended in 0.5% low melting point agarose and transferred onto a microscope slide precoated with agarose or treated directly with gamma-radiation on the agarose slides. Comets were performed in alkaline conditions as described previously.68 The tail moment, defined as the product of the percentage of DNA in the tail and the displacement between the head and the tail of the comet, was measured for more than 200 cells for each experimental condition in 2 independent experiments.
Gamma-H2AX measurement by FACS
HU-induced gamma-H2AX intensity was assessed as described.69 Mean gamma-H2AX intensity was calculated at each time-point for each cell line in 3 independent experiments.
Single-stranded DNA labeling by BrdU immunodetection
The experiment was performed as described.35 Briefly, cells on glass coverslips were grown in the culture medium containing 10 μM BrdU (Sigma) for 40 h, corresponding to 2 cell cycles. BrdU were removed with PBS washes and fresh medium with or without 2 mM HU was added, for 24 h. For the release point, cells were washed with PBS and fresh medium was added for 4 h. To finally detect non denaturated single-stranded DNA (ssDNA), the coverslips were processed for immunofluorescence detection using an anti-BrdU antibody. Images were acquired with a Leica DM RXA microscope equipped with a motorized XY stage, using an X40 PlanApo N.A. 1.25 objective and a CoolSNAP HQ interline CCD camera (Photo-metrics). For each slide, a mosaic of 10×10 partly overlapping images was collected with a Metamorph software (Molecular Devices) routine developed in-house. And more than 600 cells per condition were scored as BrdU positive or not.
Chromatin bridge quantification
Controls and Cdk5-shRNA cells grown on coverslips were treated or not with HU (2 mM for 24 h), followed by a release for 16 h in a fresh medium. Immunofluorescence staining and analysis were performed as previously described.70 Nuclear DNA was detected by mounting slides in Prolong Gold antifade reagent supplemented with DAPI (Invitrogen). Cell images were acquired with a 3-D deconvolution imaging system consisting of a Leica DM RXA microscope equipped with a piezoelectric translator (PIFOC; PI) placed at the base of a 63× PlanApo N.A. 1.4 objective, and a CoolSNAP HQ interline CCD camera (Photometrics). Stacks of conventional fluorescence images were collected automatically at a Z-distance of 0.2 mm (Metamorph software; Molecular Devices). Images are presented as maximum intensity projections, generated with ImageJ software, from stacks deconvolved with an extension of Metamorph software.71
Patients and Biological Samples
Primary unilateral non-metastatic breast carcinoma samples were obtained from 456 patients (mean age 61.7 years, range 31–91) who had surgery at Institut Curie's René Huguenin Hospital (Saint-Cloud, France) between 1978 and 2008 in accordance with national law and institutional guidelines under a protocol approved by the local Ethics Committee. Samples were immediately stored in liquid nitrogen until RNA extraction and considered suitable for inclusion if the proportion of tumor cells was more than 70% as assessed by histology.
All individuals met the following criteria: availability of complete clinical, histological and biological; no radiotherapy or chemotherapy before surgery; full follow-up. 291 patients (63.8%) underwent modified radical mastectomy and 165 (36.2%) had breast-conserving surgery with loco-regional radiotherapy. Patients underwent physical examinations and routine chest radiography every 3 months for 2 years, then annually. Mammograms were done annually.
Ten specimens of adjacent normal breast tissue from 10 breast cancer patients or normal breast tissue from women undergoing cosmetic breast surgery were used as sources of RNA for comparative purposes.
RNA extraction and Quantitative Real-time PCR
Total RNA was extracted from breast samples using the acid-phenol guanidinum method as described elsewhere.72 Primers for TBP (Genbank accession NM_003194), encoding the TATA box-binding protein and used as an endogenous RNA control, and the Cdk5 gene are given in Table S2. The cDNA synthesis, PCR reaction conditions and quantification of transcript levels were carried out as previously.73 Results, expressed as N-fold differences in Cdk5 gene expression relative to the TBP gene, termed “NCdk5”, were determined by the formula: NCdk5 = 2ΔCtsample, where ΔCt value of the sample was determined by subtracting the average Ct value of the Cdk5 gene from the average Ct value of the TBP gene. The NCdk5 values of the samples were subsequently normalized such that the median of the 10 normal breast tissue NCdk5 values was 1.
Statistics
To compare data from Control and Cdk5-shRNA cells and for combing analysis a Mann-Whitney test was used. The comparison between Cdk5 mRNA expression levels and clinical parameters was assessed using nonparametric tests: the Mann-Whitney U test (between 1 qualitative parameter and 1 quantitative parameter) and the Spearman rank correlation test (between 2 quantitative parameters).To visualize the efficacy of Cdk5 mRNA levels to discriminate between patients that developed/did not develop metastases in the absence of an arbitrary cut-off value, data were summarized in a receiver operating characteristic curve.74 The area under the curve (AUC) was calculated as a single measure to discriminate efficacy. MFS was determined as the interval between initial diagnosis and detection of the first relapse metastasis. Survival distributions were estimated by the Kaplan-Meier method,75 and the significance of differences between survival rates was ascertained using the log-rank test.76 All others data presented, were analyzed using unpaired T-tests using Prism Graph Pad 5.0b software (GraphPad Software Inc.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Sarah Lambert and Patricia Kannouche for helpful discussions, Christophe Roulin, Frederic Pouzoulet, Géraldine Buhagiar and Stéphane Koundrioukoff (Institut Curie) for technical advice and Claire Grosu, Inés Ahmed and Anne Reynaud for technical help. The gift of RPA-S23 and RPA-S29 antibodies by Heinz Nasheuer and James Borowiec is gratefully acknowledged.
Supplemental Material
Supplemental data for this article can be accessed on publisher's website.
Funding
Inserm U612's research is supported by Inserm and Institut Curie and is part of the Comprehensive Cancer Centre “SIRIC” program (INCa 2011–189). SC received a PhD fellowship from the French Ministry of Research. We acknowledge the PICT-IBiSA [Institut Curie, Orsay] member of the France-BioImaging national research infrastructure and the Fondation pour la Recherche Médicale (FRM N° DGE20111123020).
References
- 1.Xie Z, Samuels BA, Tsai LH. Cyclin-dependent kinase 5 permits efficient cytoskeletal remodeling–a hypothesis on neuronal migration. Cerebral cortex 2006; 16 Suppl 1:i64-8; PMID:16766709; http://dx.doi.org/ 10.1093/cercor/bhj170 [DOI] [PubMed] [Google Scholar]
- 2.Contreras-Vallejos E, Utreras E, Gonzalez-Billault C. Going out of the brain: non-nervous system physiological and pathological functions of Cdk5. Cell Signal 2012; 24:44-52; PMID:21924349; http://dx.doi.org/ 10.1016/j.cellsig.2011.08.022 [DOI] [PubMed] [Google Scholar]
- 3.Huang E, Qu D, Zhang Y, Venderova K, Haque ME, Rousseaux MW, Slack RS, Woulfe JM, Park DS. The role of Cdk5-mediated apurinic/apyrimidinic endonuclease 1 phosphorylation in neuronal death. Nat Cell Biol 2010; 12:563-71; PMID:20473298; http://dx.doi.org/ 10.1038/ncb2058 [DOI] [PubMed] [Google Scholar]
- 4.Hsu F-N, Chen M-C, Lin K-C, Peng Y-T, Li P-C, Lin E, Chiang M-C, Hsieh J-T, Lin H. Cyclin-dependent kinase 5 modulates STAT3 and androgen receptor activation through phosphorylation of Ser727 on STAT3 in prostate cancer cells. Am J Physiol Endocrinol Metabol 2013; 305:E975-E86; PMID:23941877; http://dx.doi.org/ 10.1152/ajpendo.00615.2012 [DOI] [PubMed] [Google Scholar]
- 5.Huang E, Qu D, Zhang Y, Venderova K, Haque ME, Rousseaux MWC, Slack RS, Woulfe JM, Park DS. The role of Cdk5-mediated apurinic/apyrimidinic endonuclease 1 phosphorylation in neuronal death. Nat Cell Biol 2010; 12:563-71; PMID:20473298; http://dx.doi.org/ 10.1038/ncb2058 [DOI] [PubMed] [Google Scholar]
- 6.Tian B, Yang Q, Mao Z. Phosphorylation of ATM by Cdk5 mediates DNA damage signalling and regulates neuronal death. Nat Cell Biol 2009; 11:211-8; PMID:19151707; http://dx.doi.org/ 10.1038/ncb1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R, Rayter S, Tutt AN, Ashworth A. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J 2008; 27:1368-77; PMID:18388863; http://dx.doi.org/ 10.1038/emboj.2008.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolin C, Boudra MT, Fernet M, Vaslin L, Pennaneach V, Zaremba T, Biard D, Cordelieres FP, Favaudon V, Megnin-Chanet F, et al.. The impact of cyclin-dependent kinase 5 depletion on poly(ADP-ribose) polymerase activity and responses to radiation. Cell Mol Life Sci 2012; 69:951-62; PMID:21922195; http://dx.doi.org/ 10.1007/s00018-011-0811-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eggers JP, Grandgenett PM, Collisson EC, Lewallen ME, Tremayne J, Singh PK, Swanson BJ, Andersen JM, Caffrey TC, High RR, et al.. Cyclin-dependent kinase 5 is amplified and overexpressed in pancreatic cancer and activated by mutant K-Ras. Clin Cancer Res 2011; 17:6140-50; PMID:21825040; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang Q, Li L, Zhang J, Lei Y, Wang L, Liu DX, Feng J, Hou P, Yao R, Zhang Y, et al.. CDK5 is essential for TGF-beta1-induced epithelial-mesenchymal transition and breast cancer progression. Sci Rep 2013; 3:2932; PMID:24121667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu JL, Wang XY, Huang BX, Zhu F, Zhang RG, Wu G. Expression of CDK5/p35 in resected patients with non-small cell lung cancer: relation to prognosis. Med Oncol 2011; 28:673-8; PMID:20354813; http://dx.doi.org/ 10.1007/s12032-010-9510-7 [DOI] [PubMed] [Google Scholar]
- 12.Strock CJ, Park JI, Nakakura EK, Bova GS, Isaacs JT, Ball DW, Nelkin BD. Cyclin-dependent kinase 5 activity controls cell motility and metastatic potential of prostate cancer cells. Cancer Res 2006; 66:7509-15; PMID:16885348; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3048 [DOI] [PubMed] [Google Scholar]
- 13.Anantha RW, Vassin VM, Borowiec JA. Sequential and Synergistic Modification of Human RPA Stimulates Chromosomal DNA Repair. J Biol Chem 2007; 282:35910-23; PMID:17928296; http://dx.doi.org/ 10.1074/jbc.M704645200 [DOI] [PubMed] [Google Scholar]
- 14.Shi W, Feng Z, Zhang J, Gonzalez-Suarez I, Vanderwaal RP, Wu X, Powell SN, Roti JLR, Gonzalo S, Zhang J. The role of RPA2 phosphorylation in homologous recombination in response to replication arrest. Carcinogenesis 2010; 31:994-1002; PMID:20130019; http://dx.doi.org/ 10.1093/carcin/bgq035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sleeth KM, Sorensen CS, Issaeva N, Dziegielewski J, Bartek J, Helleday T. RPA mediates recombination repair during replication stress and is displaced from DNA by checkpoint signalling in human cells. J Mol Biol 2007; 373:38-47; PMID:17765923; http://dx.doi.org/ 10.1016/j.jmb.2007.07.068 [DOI] [PubMed] [Google Scholar]
- 16.Wu X, Yang Z, Liu Y, Zou Y. Preferential localization of hyperphosphorylated replication protein A to double-strand break repair and checkpoint complexes upon DNA damage. Biochem J 2005; 391:473-80; PMID:15929725; http://dx.doi.org/ 10.1042/BJ20051020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alvino GM, Collingwood D, Murphy JM, Delrow J, Brewer BJ, Raghuraman MK. Replication in hydroxyurea: it's a matter of time. Mol Cell Biol 2007; 27:6396-406; PMID:17636020; http://dx.doi.org/ 10.1128/MCB.00719-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tourriere H, Pasero P. Maintenance of fork integrity at damaged DNA and natural pause sites. DNA Repair (Amst) 2007; 6:900-13; PMID:17379579; http://dx.doi.org/ 10.1016/j.dnarep.2007.02.004 [DOI] [PubMed] [Google Scholar]
- 19.Hustedt N, Gasser SM, Shimada K. Replication checkpoint: tuning and coordination of replication forks in s phase. Genes (Basel) 2013; 4:388-434; PMID:24705211; http://dx.doi.org/ 10.3390/genes4030388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ge XQ, Blow JJ. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J Cell Biol 2010; 191:1285-97; PMID:21173116; http://dx.doi.org/ 10.1083/jcb.201007074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lambert S, Carr AM. Checkpoint responses to replication fork barriers. Biochimie 2005; 87:591-602; PMID:15989976; http://dx.doi.org/ 10.1016/j.biochi.2004.10.020 [DOI] [PubMed] [Google Scholar]
- 22.Maya-Mendoza A, Petermann E, Gillespie DA, Caldecott KW, Jackson DA. Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J 2007; 26:2719-31; PMID:17491592; http://dx.doi.org/ 10.1038/sj.emboj.7601714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Magdalou I, Lopez BS, Pasero P, Lambert SA. The causes of replication stress and their consequences on genome stability and cell fate. Semin Cell Dev Biol 2014; 30:154-64; PMID:24818779; http://dx.doi.org/ 10.1016/j.semcdb.2014.04.035 [DOI] [PubMed] [Google Scholar]
- 24.Paulsen RD, Cimprich KA. The ATR pathway: fine-tuning the fork. DNA Repair (Amst) 2007; 6:953-66; PMID:17531546; http://dx.doi.org/ 10.1016/j.dnarep.2007.02.015 [DOI] [PubMed] [Google Scholar]
- 25.Branzei D, Foiani M. The checkpoint response to replication stress. DNA Repair (Amst) 2009; 8:1038-46; PMID:19482564; http://dx.doi.org/ 10.1016/j.dnarep.2009.04.014 [DOI] [PubMed] [Google Scholar]
- 26.Xu X, Vaithiyalingam S, Glick GG, Mordes DA, Chazin WJ, Cortez D. The basic cleft of RPA70N binds multiple checkpoint proteins, including RAD9, to regulate ATR signaling. Mol Cell Biol 2008; 28:7345-53; PMID:18936170; http://dx.doi.org/ 10.1128/MCB.01079-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dutta A, Stillman B. cdc2 family kinases phosphorylate a human cell DNA replication factor, RPA, and activate DNA replication. EMBO J 1992; 11:2189-99; PMID:1318195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang F, Newport JW. Distinct roles of cdk2 and cdc2 in RP-A phosphorylation during the cell cycle. J Cell Sci 1993; 106:983-94; PMID:8308077 [DOI] [PubMed] [Google Scholar]
- 29.Niu H, Erdjument-Bromage H, Pan ZQ, Lee SH, Tempst P, Hurwitz J. Mapping of amino acid residues in the p34 subunit of human single-stranded DNA-binding protein phosphorylated by DNA-dependent protein kinase and Cdc2 kinase in vitro. J Biol Chem 1997; 272:12634-41; PMID:9139719; http://dx.doi.org/ 10.1074/jbc.272.19.12634 [DOI] [PubMed] [Google Scholar]
- 30.Olson E, Nievera CJ, Klimovich V, Fanning E, Wu X. RPA2 is a direct downstream target for ATR to regulate the S-phase checkpoint. J Biol Chem 2006; 281:39517-33; PMID:17035231; http://dx.doi.org/ 10.1074/jbc.M605121200 [DOI] [PubMed] [Google Scholar]
- 31.Azqueta A, Collins AR. The essential comet assay: a comprehensive guide to measuring DNA damage and repair. Arch Toxicol 2013; 87:949-68; PMID:; http://dx.doi.org/16474843 10.1007/s00204-013-1070-0 [DOI] [PubMed] [Google Scholar]
- 32.Adams KE, Medhurst AL, Dart DA, Lakin ND. Recruitment of ATR to sites of ionising radiation-induced DNA damage requires ATM and components of the MRN protein complex. Oncogene 2006; 25:3894-904; PMID:16474843; http://dx.doi.org/ 10.1038/sj.onc.1209426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Myers JS, Cortez D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J Biol Chem 2006; 281:9346-50; PMID:16431910; http://dx.doi.org/ 10.1074/jbc.M513265200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shiotani B, Zou L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol Cell 2009; 33:547-58; PMID:19285939; http://dx.doi.org/ 10.1016/j.molcel.2009.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Illuzzi G, Fouquerel E, Ame JC, Noll A, Rehmet K, Nasheuer HP, Dantzer F, Schreiber V. PARG is dispensable for recovery from transient replicative stress but required to prevent detrimental accumulation of poly(ADP-ribose) upon prolonged replicative stress. Nucleic Acids Res 2014; 42:7776-92; PMID:24906880; http://dx.doi.org/ 10.1093/nar/gku505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-Stalled Replication Forks Become Progressively Inactivated and Require Two Different RAD51-Mediated Pathways for Restart and Repair. Molecular Cell 2010; 37:492-502; PMID:20188668; http://dx.doi.org/ 10.1016/j.molcel.2010.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gelot C, Magdalou I, Lopez BS. Replication stress in Mammalian cells and its consequences for mitosis. Genes (Basel) 2015; 6:267-98; PMID:26010955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lahkim Bennani-Belhaj K, Rouzeau S, Buhagiar-Labarchede G, Chabosseau P, Onclercq-Delic R, Bayart E, Cordelieres F, Couturier J, Amor-Gueret M. The Bloom syndrome protein limits the lethality associated with RAD51 deficiency. Mol Cancer Res 2010; 8:385-94; PMID:20215422; http://dx.doi.org/ 10.1158/1541-7786.MCR-09-0534 [DOI] [PubMed] [Google Scholar]
- 39.Arif A. Extraneuronal activities and regulatory mechanisms of the atypical cyclin-dependent kinase Cdk5. Biochem Pharmacol 2012; 84:985-93; PMID:22795893; http://dx.doi.org/ 10.1016/j.bcp.2012.06.027 [DOI] [PubMed] [Google Scholar]
- 40.Liebl J, Furst R, Vollmar AM, Zahler S. Twice switched at birth: cell cycle-independent roles of the “neuron-specific” cyclin-dependent kinase 5 (Cdk5) in non-neuronal cells. Cell Signal 2011; 23:1698-707; PMID:21741478; http://dx.doi.org/ 10.1016/j.cellsig.2011.06.020 [DOI] [PubMed] [Google Scholar]
- 41.Liebl J, Weitensteiner SB, Vereb G, Takacs L, Furst R, Vollmar AM, Zahler S. Cyclin-dependent kinase 5 regulates endothelial cell migration and angiogenesis. J Biol Chem 2010; 285:35932-43; PMID:20826806; http://dx.doi.org/ 10.1074/jbc.M110.126177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu S, Li X, Gong Z, Wang W, Li Y, Nair BC, Piao H, Yang K, Wu G, Chen J. Proteomic Analysis of the Human CDK Family Reveals a Novel CDK5 Complex Involved in Cell Growth and Migration. Mol Cell Proteomics 2014; PMID:25096995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ajay AK, Upadhyay AK, Singh S, Vijayakumar MV, Kumari R, Pandey V, Boppana R, Bhat MK. Cdk5 phosphorylates non-genotoxically overexpressed p53 following inhibition of PP2A to induce cell cycle arrest/apoptosis and inhibits tumor progression. Mol Cancer 2010; 9:204; PMID:20673369; http://dx.doi.org/ 10.1186/1476-4598-9-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Upadhyay AK, Ajay AK, Singh S, Bhat MK. Cell cycle regulatory protein 5 (Cdk5) is a novel downstream target of ERK in carboplatin induced death of breast cancer cells. Curr Cancer Drug Targets 2008; 8:741-52; PMID:19075597; http://dx.doi.org/ 10.2174/156800908786733405 [DOI] [PubMed] [Google Scholar]
- 45.Nagano T, Hashimoto T, Nakashima A, Hisanaga S, Kikkawa U, Kamada S. Cyclin I is involved in the regulation of cell cycle progression. Cell Cycle 2013; 12:2617-24; PMID:23907122; http://dx.doi.org/ 10.4161/cc.25623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loor G, Zhang SJ, Zhang P, Toomey NL, Lee MY. Identification of DNA replication and cell cycle proteins that interact with PCNA. Nucleic Acids Res 1997; 25:5041-6; PMID:9396813; http://dx.doi.org/ 10.1093/nar/25.24.5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ame JC, Fouquerel E, Gauthier LR, Biard D, Boussin FD, Dantzer F, de Murcia G, Schreiber V. Radiation-induced mitotic catastrophe in PARG-deficient cells. J Cell Sci 2009; 122:1990-2002; PMID:19454480; http://dx.doi.org/ 10.1242/jcs.039115 [DOI] [PubMed] [Google Scholar]
- 48.Bolderson E, Petermann E, Croft L, Suraweera A, Pandita RK, Pandita TK, Helleday T, Khanna KK, Richard DJ. Human single-stranded DNA binding protein 1 (hSSB1/NABP2) is required for the stability and repair of stalled replication forks. Nucleic Acids Res 2014; 42:6326-36; PMID:24753408; http://dx.doi.org/ 10.1093/nar/gku276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petermann E, Helleday T. Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol 2010; 11:683-7; PMID:20842177; http://dx.doi.org/ 10.1038/nrm2974 [DOI] [PubMed] [Google Scholar]
- 50.Carty MP, Zernik-Kobak M, McGrath S, Dixon K. UV light-induced DNA synthesis arrest in HeLa cells is associated with changes in phosphorylation of human single-stranded DNA-binding protein. EMBO J 1994; 13:2114-23; PMID:8187764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oakley GG, Patrick SM. Replication protein A: directing traffic at the intersection of replication and repair. Front Biosci (Landmark Ed). 2010; 15:883-900; PMID:20515732; http://dx.doi.org/ 10.2741/3652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patrick SM, Oakley GG, Dixon K, Turchi JJ. DNA damage induced hyperphosphorylation of replication protein A. 2. Characterization of DNA binding activity, protein interactions, and activity in DNA replication and repair. Biochemistry 2005; 44:8438-48; PMID:15938633; http://dx.doi.org/ 10.1021/bi048057b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Treuner K, Findeisen M, Strausfeld U, Knippers R. Phosphorylation of replication protein A middle subunit (RPA32) leads to a disassembly of the RPA heterotrimer. J Biol Chem 1999; 274:15556-61; PMID:10336450; http://dx.doi.org/ 10.1074/jbc.274.22.15556 [DOI] [PubMed] [Google Scholar]
- 54.Vassin VM, Anantha RW, Sokolova E, Kanner S, Borowiec JA. Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J Cell Sci 2009; 122:4070-80; PMID:19843584; http://dx.doi.org/ 10.1242/jcs.053702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones RM, Petermann E. Replication fork dynamics and the DNA damage response. Biochem J 2012; 443:13-26; PMID:22417748; http://dx.doi.org/ 10.1042/BJ20112100 [DOI] [PubMed] [Google Scholar]
- 56.Bryant HE, Petermann E, Schultz N, Jemth A-S, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J 2009; 28:2601-15; PMID:19629035; http://dx.doi.org/ 10.1038/emboj.2009.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev 2005; 19:1040-52; PMID:15833913; http://dx.doi.org/ 10.1101/gad.1301205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bahassi M, Hennigan RF, Myer DL, Stambrook PJ. Cdc25C phosphorylation on serine 191 by Plk3 promotes its nuclear translocation. Oncogene 2004; 23:2658-63; PMID:14968113; http://dx.doi.org/ 10.1038/sj.onc.1207425 [DOI] [PubMed] [Google Scholar]
- 59.Ashworth A, Lord CJ, Reis-Filho JS. Genetic interactions in cancer progression and treatment. Cell 2011; 145:30-8; PMID:21458666; http://dx.doi.org/ 10.1016/j.cell.2011.03.020 [DOI] [PubMed] [Google Scholar]
- 60.Levacque Z, Rosales JL, Lee KY. Level of cdk5 expression predicts the survival of relapsed multiple myeloma patients. Cell Cycle 2012; 11:4093-5; PMID:22987154; http://dx.doi.org/ 10.4161/cc.21886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cuny GD. Kinase inhibitors as potential therapeutics for acute and chronic neurodegenerative conditions. Curr Pharm Design 2009; 15:3919-39; PMID:19751204; http://dx.doi.org/ 10.2174/138161209789649330 [DOI] [PubMed] [Google Scholar]
- 62.Godon C, Cordelieres FP, Biard D, Giocanti N, Megnin-Chanet F, Hall J, Favaudon V. PARP inhibition versus PARP-1 silencing: different outcomes in terms of single-strand break repair and radiation susceptibility. Nucleic Acids Res 2008; 36:4454-64; PMID:18603595; http://dx.doi.org/ 10.1093/nar/gkn403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Biard DS. Untangling the relationships between DNA repair pathways by silencing more than 20 DNA repair genes in human stable clones. Nucleic Acids Res 2007; 35:3535-50; PMID:17483520; http://dx.doi.org/ 10.1093/nar/gkm195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Le Chalony C, Hoffschir F, Gauthier LR, Gross J, Biard DS, Boussin FD, Pennaneach V. Partial complementation of a DNA ligase I deficiency by DNA ligase III and its impact on cell survival and telomere stability in mammalian cells. Cell Mol Life Sci 2012; 69:2933-49; PMID:22460582; http://dx.doi.org/ 10.1007/s00018-012-0975-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Biard DS, Despras E, Sarasin A, Angulo JF. Development of new EBV-based vectors for stable expression of small interfering RNA to mimick human syndromes: application to NER gene silencing. Mol Cancer Res 2005; 3:519-29; PMID:16179499; http://dx.doi.org/ 10.1158/1541-7786.MCR-05-0044 [DOI] [PubMed] [Google Scholar]
- 66.Fernet M, Megnin-Chanet F, Hall J, Favaudon V. Control of the G2/M checkpoints after exposure to low doses of ionising radiation: implications for hyper-radiosensitivity. DNA Repair (Amst) 2010; 9:48-57; PMID:19926348; http://dx.doi.org/ 10.1016/j.dnarep.2009.10.006 [DOI] [PubMed] [Google Scholar]
- 67.Michalet X, Ekong R, Fougerousse F, Rousseaux S, Schurra C, Hornigold N, van Slegtenhorst M, Wolfe J, Povey S, Beckmann JS, et al.. Dynamic molecular combing: stretching the whole human genome for high-resolution studies. Science 1997; 277:1518-23; PMID:9278517; http://dx.doi.org/ 10.1126/science.277.5331.1518 [DOI] [PubMed] [Google Scholar]
- 68.Rimmele P, Komatsu J, Hupe P, Roulin C, Barillot E, Dutreix M, Conseiller E, Bensimon A, Moreau-Gachelin F, Guillouf C. Spi-1/PU.1 oncogene accelerates DNA replication fork elongation and promotes genetic instability in the absence of DNA breakage. Cancer Res 2010; 70:6757-66; PMID:20660370; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-4691 [DOI] [PubMed] [Google Scholar]
- 69.Haddy N, Tartier L, Koscielny S, Adjadj E, Rubino C, Brugieres L, Pacquement H, Diallo I, de Vathaire F, Averbeck D, et al.. Repair of ionizing radiation-induced DNA damage and risk of second cancer in childhood cancer survivors. Carcinogenesis 2014; 35:1745-9; PMID:24670918; http://dx.doi.org/ 10.1093/carcin/bgu077 [DOI] [PubMed] [Google Scholar]
- 70.Rouzeau S, Cordelieres FP, Buhagiar-Labarchede G, Hurbain I, Onclercq-Delic R, Gemble S, Magnaghi-Jaulin L, Jaulin C, Amor-Gueret M. Bloom's syndrome and PICH helicases cooperate with topoisomerase IIalpha in centromere disjunction before anaphase. PLoS One 2012; 7:e33905; PMID:22563370; http://dx.doi.org/ 10.1371/journal.pone.0033905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Savino TM, Gebrane-Younes J, De Mey J, Sibarita JB, Hernandez-Verdun D. Nucleolar assembly of the rRNA processing machinery in living cells. J Cell Biol 2001; 153:1097-110; PMID:11381093; http://dx.doi.org/ 10.1083/jcb.153.5.1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bieche I, Pennaneach V, Driouch K, Vacher S, Zaremba T, Susini A, Lidereau R, Hall J. Variations in the mRNA expression of poly(ADP-ribose) polymerases, poly(ADP-ribose) glycohydrolase and ADP-ribosylhydrolase 3 in breast tumors and impact on clinical outcome. Inter J Cancer Journal international du cancer 2013; 133:2791-800; PMID:23736962 [DOI] [PubMed] [Google Scholar]
- 73.Bieche I, Parfait B, Le Doussal V, Olivi M, Rio MC, Lidereau R, Vidaud M. Identification of CGA as a novel estrogen receptor-responsive gene in breast cancer: an outstanding candidate marker to predict the response to endocrine therapy. Cancer Res 2001; 61:1652-8; PMID:11245479 [PubMed] [Google Scholar]
- 74.Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982; 143:29-36; PMID:7063747; http://dx.doi.org/ 10.1148/radiology.143.1.7063747 [DOI] [PubMed] [Google Scholar]
- 75.Kaplan EL MP. Nonparametric estimation from incomplete observations. J Am Statist Association 1958; 53:457-81; http://dx.doi.org/ 10.1080/01621459.1958.10501452 [DOI] [Google Scholar]
- 76.Peto R, Pike MC, Armitage P, Breslow NE, Cox DR, Howard SV, Mantel N, McPherson K, Peto J, Smith PG. Design and analysis of randomized clinical trials requiring prolonged observation of each patient. II. analysis and examples. Br J Cancer 1977; 35:1-39; PMID:831755; http://dx.doi.org/ 10.1038/bjc.1977.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.