

In non-small-cell lung cancer (NSCLC)—the leading cause of cancer death worldwide—mutations in epidermal growth factor receptor (EGFR), a receptor tyrosine kinase (RTK), are found in about 14%. Treatment with EGFR tyrosine kinase inhibitors (TKIs) had shown promise; however, a recent trial with erlotinib found tumor reduction >90% in only 5% of patients.1 This discrepant response raised the possibility of an inherent, not acquired, resistance in the tumor cells.
RTK ligands in the tumor microenvironment are important determinants of therapeutic responses to anticancer kinase inhibitors.2 Indeed, hepatocyte growth factor-mediated activation of the RTK Met is thought to be the cause of innate resistance to EGFR TKIs in NSCLC cells. On the other hand, intrinsic signaling pathways become paradoxically activated following RTK inhibition, presenting another mechanism that contributes to innate resistance. Because its precise role and mechanism remain unclear, we investigated the molecular machinery by which the Ras/mitogen-activated protein kinase (MAPK) pathway is activated after EGFR inhibition, despite blockade of RTK activity in NSCLC cells (Fig. 1).3
Figure 1.
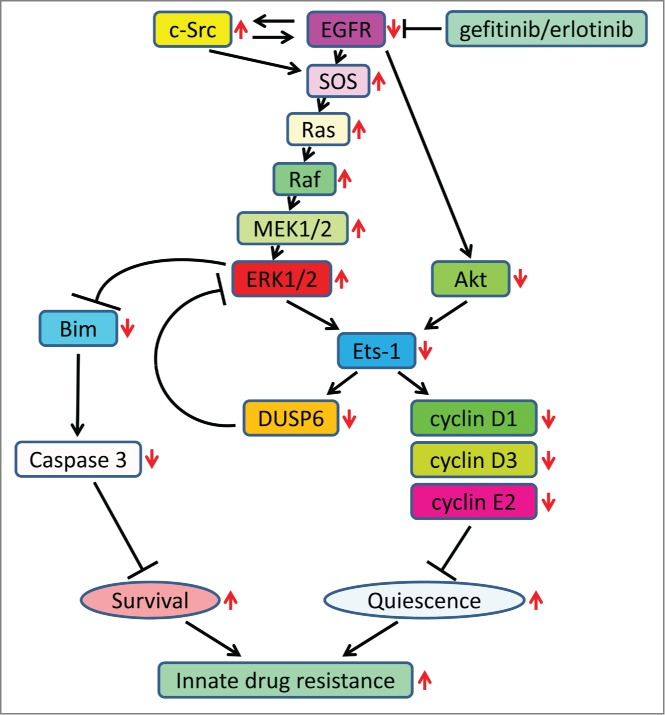
Summary of the molecular mechanism of EGFR inhibition and innate drug resistance in EGFR-mutated NSCLC cells. Increases and decreases in activity/expression of signaling molecules or biological outcomes resulting from EGFR inhibition are indicated by red arrows.
EGFR TKIs decreased both the MAPK and Akt protein kinase pathways for a short time, after which the Ras/MAPK pathway became re-activated. However, the transcriptional targets of the Ras/MAPK pathway were not re-expressed. This is because Akt inhibition by TKIs selectively blocked the transcriptional activation of Ets-1, which inhibited its target gene, dual specificity phosphatase 6 (DUSP6), a negative regulator specific for ERK1/2 (MAPK). As a result, ERK1/2 was paradoxically activated. Furthermore, elevated c-Src stimulated Ras GTP-loading and activated Raf and MEK kinases.
These observations suggested that, to maintain Ets-1 transcription factor in an active state, both ERK1/2 and Akt are essential (Fig. 1). Therefore, in the absence of Akt activity after EGFR inhibition, Ets-1 target genes including DUSP6 and cyclins D1, D3, and E2 remained suppressed—despite high levels of ERK1/2. Reduction of DUSP6 combined with c-Src to renew activation of the Ras/MAPK pathway. Active ERK1/2, though unable to promote transcription of Ets-1 target genes, was able to induce accelerate Bim protein turnover resulting in increased cell survival. Bim is essential for apoptosis and caspase induction in EGFR-mutated NSCLC cells. Thus, we believe our study was the first to define Ets-1 transcription factor as a cause of drug resistance.
We found that Ets-1 resides at the juncture of the Akt and MAPK pathways (Fig. 1) and its inactivation contributed to innate drug resistance to EGFR TKIs. Its synthesis and transactivation of its target genes require Akt and ERK1/2 kinase activities. For this reason, increased activity of ERK1 and ERK2 after EGFR inhibition cannot by itself transactivate Ets-1 target genes without Akt protein kinase. The persistence of Ets-1 inactivation after EGFR inhibition is quiescence and survival of the NSCLC cells. These findings, which revealed a new point of convergence of Akt and MAPK signaling, elucidated a new aspect of the innate resistance to EGFR TKIs in the absence of growth factors, without activation of RTKs, and suggested that Ets-1 could not be a therapeutic target in NSCLC treatment.
We found that addition of a MEK inhibitor enhances programmed cell death by rewiring apoptotic signaling.3 Therefore, to reduce the probability of emergent resistance to EGFR TKIs in NSCLCs, combined TKI and MEK inhibitor treatment should be considered. A recent report from other laboratory has also proposed this novel combined therapy, which is thought to be effective not only in innate resistance, but also in acquired resistance with T790M second-site EGFR mutation.4 Interestingly, Ercan et al. reported that ERK1/2 reactivation accompanied DUSP6 reduction after L858R/T790M EGFR-selective inhibition in original TKI-resistant cells.5 Thus, regardless of the type of drug resistance, our study provides a clear rationale for combined treatment in this population.
Our study also raised critical issues relevant to the MAPK signaling cascade.3 For example, we found that EGFR TKI-induced activation of Ras accompanied phosphorylation of ERK1/2, but not Akt. Therefore, we question the role of Ras as the immediate upstream regulator of the phosphoinositide 3-kinase (PI3K)/Akt pathway and suggest an important role for RTKs in Ras-mediated activation of this pathway. Likewise, we determined that single-agent EGFR inhibitor blocked protein kinase activities of other co-activated RTKs in EGFR-mutated NSCLC cells. This finding casts doubt on a model that suggests that redundant input from different RTKs is necessary to drive and maintain downstream signaling cascades in EGFR-mutated tumor cells.6 Moreover, our findings revealed that c-Src promoted activation of the Ras/MAPK pathway independent of Shc kinase activity in EGFR-mutated NSCLC cells.7 These issues are intriguing and prompt further study.
In summary, to elucidate the discrepant responses to EGFR TKIs among EGFR-mutated NSCLCs, we found that EGFR inhibition evokes innate drug resistance by preventing Akt activity and thus inactivating Ets-1 function. The persistent inactivation of Ets-1 decreases DUSP6 expression, causing paradoxical activation of ERK1/2. Because addition of a MEK inhibitor enhances programmed cell death by rewiring apoptotic signaling, we may reduce the probability of emergent resistance to EGFR TKIs by combined TKI and MEK inhibitor treatment. More studies are warranted to bring this strategy from bench to bedside.
References
- 1.Rosell R, et al.. Lancet Oncol 2012; 13:239-46; PMID:22285168; http://dx.doi.org/ 10.1016/S1470-2045(11)70393-X [DOI] [PubMed] [Google Scholar]
- 2.Wilson TR, et al.. Nature 2012; 487:505-9; PMID:22763448; http://dx.doi.org/ 10.1038/nature11249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phuchareon J, et al.. Proc Natl Acad Sci U S A 2015; 112:E3855-63; PMID:26150526; http://dx.doi.org/ 10.1073/pnas.1510733112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tricker EM, et al.. Cancer Discov 2015; 5:960–971;PMID:26036643; http://cancerdiscovery.aacrjournals.org/content/5/9/960.abstract?etoc [Google Scholar]
- 5.Ercan D, et al.. Cancer Discov 2012; 2:934-47; PMID:22961667; http://dx.doi.org/ 10.1158/2159-8290.CD-12-0103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stommel JM, et al.. Science 2007; 318:287-90; PMID:17872411; http://dx.doi.org/ 10.1126/science.1142946 [DOI] [PubMed] [Google Scholar]
- 7.Batzer AG, et al.. Mol Cell Biol 1994; 14:5192-201; PMID:7518560 [DOI] [PMC free article] [PubMed] [Google Scholar]