

Abstract
The role of the tumor suppressor BRCA2 has been shaped over 2 decades thanks to the discovery of its protein and nucleic acid partners, biochemical and structural studies of the protein, and the functional evaluation of germline variants identified in breast cancer patients. Yet, the pathogenic and functional effect of many germline mutations in BRCA2 remains undetermined, and the heterogeneity of BRCA2-associated tumors challenges the identification of causative variants that drive tumorigenesis.
In this review, we propose an overview of the established and emerging interacting partners and functional pathways attributed to BRCA2, and we speculate on how variants altering these functions may contribute to cancer susceptibility.
Introduction
Inherited mutations in the human BRCA2 gene are responsible for about half the cases of early-onset breast cancer.1 The effect of many of these mutations remains unknown, representing an important burden for patient care. A detailed characterization of the function and partners of BRCA2 may help define the mechanisms by which mutations in BRCA2 contribute to pathogenicity.
BRCA2 protein displays no significant sequence similarity to other known human proteins, and little conservation in mammalian evolution. As an example, mouse Brca2 shows only 59% amino acid identity with the human BRCA2.2 Thus, the impact of mutations in BRCA2 function can only be predicted when they are located in regions where the domains are well defined, i.e., the central region and the C-terminus of the protein.
Since it was discovered as a breast cancer predisposition gene,3 BRCA2 has been shown to interact with a large number of proteins, illustrating its functional complexity. Although the function of BRCA2 has been revised multiple times, how its interacting partners influence its function and which of these interactions might be relevant for cancer predisposition has not been discussed in detail.
In this review, we describe the interacting partners and regulatory mechanisms of BRCA2 that have molded our understanding of its function over the last 2 decades and discuss the possible implications for the clinic. Because we focus on the interactions that may have an impact on tumorigenesis, the function of BRCA2 in meiosis is not discussed here. For clarity, we have broken down the text into sections describing the functions of BRCA2 arbitrarily ordered from better- to lesser-known (see Fig. 1).
Figure 1.
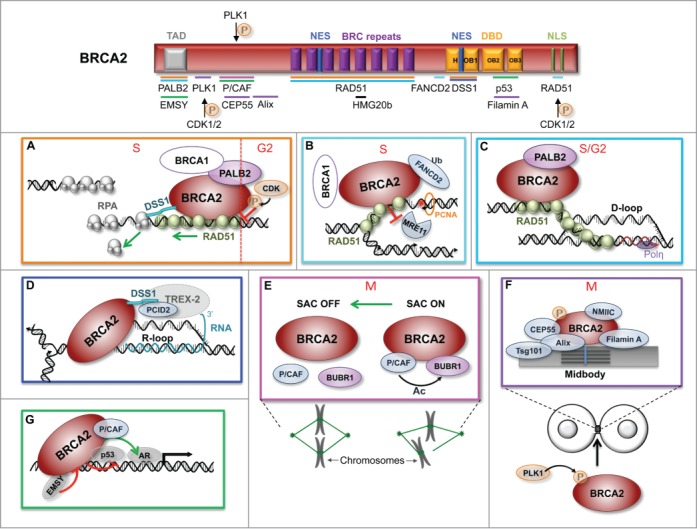
Schematic representation of the structural domains of BRCA2 showing the binding sites for its protein partners. The panels illustrate the established and suggested functions of BRCA2 in: (A) HR-mediated repair. (B) Protection of DNA replication forks. (C) D-loop extension at stalled replication forks. (D) R-loop metabolism. (E, F) Mitosis progression and cytokinesis. (G) Transactivation activity. The phase of the cell cycle is indicated in red. The color-coding of the binding sites in the scheme corresponds to the colors of the panels in which the proteins are involved. DBD, DNA binding domain; H, helical domain; NLS, nuclear localization signal; NES, nuclear export signal; OB, oligonucleotide/oligosaccharide binding fold; SAC, spindle assembly checkpoint; TAD, Transactivation domain; AR, Androgen receptor. Ac, acetylation; P, phosphorylation; Ub, ubiquitination.
DNA double-strand break repair
Brca2 deficiency in mice leads to embryonic lethality causing radiation hypersensitivity4 and gross chromosomal rearrangements (GCRs),5 providing evidence for a role of BRCA2 in the maintenance of genome stability.
BRCA2 was first identified as a DNA repair protein due to its physical interaction with the recombination protein RAD51.6,7 Specifically, BRCA2 and RAD51 act on the homologous recombination (HR) repair pathway, a high-fidelity mechanism to repair DNA double-strand breaks (DSBs). During HR, RAD51 and BRCA2, together with other factors, promote a reaction in which a damaged chromatid pairs with an intact partner resulting in the exchange of genetic information from the latter to the former. This process requires the resection of the DSB to generate a 3′-ssDNA overhang that can be used by the recombination machinery to search for homology. The formation of a displacement loop (D-loop) between resected DNA and the template chromatid is formed to prime DNA synthesis. This structure is then resolved resulting in the repair of the damaged chromatin.
In humans, the principal RAD51 binding site of BRCA2 is located in the BRC repeats, 8 consecutive interspersed motifs in the central region of the primary sequence.6 An additional RAD51 interaction site maps to the C-terminal region.8 Through the BRC repeats, BRCA2 stimulates RAD51 assembly on single-stranded DNA (ssDNA) over double-stranded DNA (dsDNA) and promotes its HR function.9-11
BRCA2 binds DNA through a DNA binding domain (DBD) located in the C-terminal region containing 3 oligonucleotide/oligosaccharide binding folds (OB folds), a helical domain, and a tower domain.12 The DNA binding and RAD51 interacting sites of BRCA2 are sufficient to promote the assembly of RAD51 onto ssDNA and ssDNA/dsDNA junctions.11,13 In addition, consistent with a mediator function, BRCA2 enables the displacement of replication-protein A (RPA) from ssDNA to form an active RAD51 nucleoprotein filament that promotes DNA strand exchange11,14,15 (panel A). These findings established human BRCA2 as a mediator of HR in somatic cells and are consistent with a role in the repair of DSBs but also in daughter strand gap (DSG) repair, a process that repairs DNA lesions generated during replication across a damaged DNA template.16
The DBD of BRCA2 harbors a DSS1 (deleted in split hand/split foot)12 binding site. In the fungus Ustilago maydis, this acidic protein regulates the association of the ortholog of BRCA2 (Brh2) with DNA,17 in agreement with the crystal structure of the mammalian DBD of BRCA2 and DSS1.12 Although in humans the role of DSS1 in HR was described as indirect by enhancing the stability of BRCA218 or its cellular sublocalization19; recently, DSS1 has been shown to promote the loading of BRCA2-RAD51 to RPA-coated ssDNA through its ability to bind RPA and attenuate its affinity for ssDNA, supporting a direct role for DSS1 in HR.14,20 The importance of the interaction of DSS1 with BRCA2 in HR is further supported by the fact that deletion of DSS1 phenocopies BRCA2 deficiency.18,21,22
Besides promoting HR, BRCA2 might also have a role in regulating illegitimate HR. The crystal structure of BRC4-RAD51 places BRC4 at the interface of RAD51 oligomers, and by 'mimicking' this interface it blocks filament formation.23 This structural mechanism supported the findings showing that overexpression of BRC424,25 or full length BRCA226 downregulates HR. Blocking HR could be necessary to suppress recombination in mitosis as it has been suggested.27 Phosphorylation of S3291 at the C-terminal RAD51 binding site of BRCA2 is involved in this switch-off mechanism by inhibiting the interaction with RAD51,8 (see “BRCA2 regulation” section below).
Another factor that promotes HR by interacting with BRCA2 is PALB2 (partner and localizer of BRCA2). PALB2 interaction site maps to the N-terminus of BRCA2.28,29 In vitro, both proteins cooperate in stimulating RAD51-mediated D-loop formation, a critical step in HR.30 The depletion of PALB2 leads to a decrease in HR efficiency, mitomycin C (MMC) hypersensitivity and reduction of the retention of BRCA2 in the nucleus.28 PALB2 forms a complex with BRCA1 and BRCA231 providing an indirect link between the 2 BRCA proteins. As BRCA1 and BRCA2, PALB2 is also considered a breast cancer predisposing gene.32
Inter-strand crosslink repair
Fanconi anemia (FA) is an autosomal recessive inherited syndrome characterized by congenital and developmental abnormalities, bone marrow failure, and a strong cancer predisposition. In around 3-5% of cases, FA is caused by biallelic mutations in BRCA2. The Fanconi anemia pathway, defective in these patients, is involved in repairing inter-strand crosslinks (ICLs), toxic lesions that preclude replication and transcription by inhibiting DNA strand separation.33 Broadly used chemotherapeutic drugs such as cisplatin or MMC induce these types of lesions.33 The finding that BRCA2 gene is identical to FANCD1 and that transient expression of BRCA2 can restore the repair of ICL in BRCA2-deficient cells demonstrated a direct implication of BRCA2 in the FA pathway.34,35
The FA relies on more than 15 FA factors and various DNA repair processes, nucleotide incision, translesion synthesis (TLS), HR and Nucleotide excision repair (NER). ICLs are first processed by an incision flanking the ICL region promoted by FANCD2 ubiquitination, which generates a DSB. TLS restores the DNA template, and HR is required to repair the DSB. To promote the faithful repair of DSB, FA proteins such as FANCD2 and BRCA1 favor HR by inhibiting the binding to DSB ends of factors from the error-prone repair pathway Non Homologous End Joining (NHEJ).36,37
As in DSB repair, the HR step in ICL repair requires the loading of RAD51 (FANCR) onto the resected DNA followed by strand invasion to resolve the DSB. Therefore, HR proteins such as BRCA2 (FANCD1), PALB2 (FANCN), BRCA1 (FANCS), among others38 are essential in this step. Then, the NER excises the remaining DNA adducts and fills the gap.
DNA replication stress
MEFs carrying a truncated form of BRCA2 display chromosomal breaks and GCRs.39 Because these cells were lacking DNA replication intermediates arising from stalled replication forks as measured by 2-D gels,40 the above observations suggested a role of BRCA2 in stabilizing RAD51 nucleoprotein filaments during replication.40 Later on, it was found that the C-terminal RAD51 binding region of BRCA2 can protect the newly synthesized DNA from degradation by the nuclease MRE11 in such context, establishing a BRCA2 DNA-end protection function at stalled forks.41 BRCA1 and FANCD2 also participate with BRCA2 in this process,42 (panel B). Interestingly, the deficiency of these proteins can be overcome by overexpression of RAD51, a feature found in tumors with acquired resistance to chemotherapeutic drugs.43
BRCA2 and PALB2 also interact and stimulate the polymerase Polη (XPV). The three proteins co-localize at replication stress-induced foci upon treatment with hydroxyurea (HU) or aphidicolin (APH), and stimulate DNA synthesis by Polη on D-loop substrates in vitro.44 Together, these findings suggest a role for BRCA2 in both preventing the newly synthesized DNA from nucleolytic degradation at stalled replication forks and in promoting the DNA synthesis necessary to restore the damaged template after DNA strand invasion at replication-dependent DSBs (panel C).
Although a defect in replication restart was not observed in BRCA2 deficient cells in previous works, a recent article shows that together with FANCJ and FANCD2, BRCA2 might be implicated in the suppression of origin firing and replication restart independently of other FA factors.45
Telomere maintenance
Because of its linear structure, the ends of eukaryotic chromosomes are potential targets for the DNA damage repair pathways, which could have dramatic consequences for the cell. Homologous recombination prevents chromosome ends from degradation and fusion, therefore maintaining telomere integrity. By loading RAD51 onto telomeric DNA, BRCA2 facilitates telomere replication and protection. BRCA2 depletion results in the accumulation of fragile telomeres and increase telomere sister chromatid exchange.46
Interestingly, BRCA2- but not BRCA1-deficient tumors show telomere shortening and telomere fragility, suggesting that the absence of this function in BRCA2-mutated cells could promote tumorigenesis.47 This aspect may contribute to the difference in tumor subtypes observed in BRCA1 versus BRCA2-related tumors.
R-loop metabolism
R-loops are RNA-DNA hybrids that result as normal byproducts of transcription. However, when persistent, they can cause the collision between the replication and transcription machineries and can be at the origin of replication fork stalling.48 As in the absence of the R-loop processing enzyme Senataxin,49 cells depleted for BRCA2 but not for RAD51, accumulate R-loops, implicating BRCA2 in processing or preventing such structures50 (panel D). Insight on this role of BRCA2 comes from the BRCA2 interacting protein DSS1 that, together with PCID2 and other factors, constitute the TREX-2 complex. This complex is located at the nuclear pore and is involved in the biogenesis and export of messenger ribonucleoprotein (mRNP). The findings that BRCA2 co-localizes with PCID2 and is recruited to DNA-RNA hybrids in cells, suggest a RAD51-independent role for BRCA2 in preventing or processing R-loops.50
G2/M checkpoint, mitosis and cytokinesis
BRCA2 and PALB2 have been implicated in G2 checkpoint control maintenance after DNA damage as depletion of either leads to premature checkpoint abrogation.51 γ-Irradiated cells overexpressing BRC424 show a G2/M but not G1/S checkpoint defect, supporting the idea of a specific role of BRCA2 in the G2/M checkpoint maintenance upon DNA damage.
BRCA2 interacts with the histone acetyl-transferase P/CAF through the N-terminus.52,53 P/CAF acetylates BUBR1, an essential component of the spindle assembly checkpoint (SAC) in mitosis. BRCA2 has been shown to act as a scaffold protein bringing together P/CAF and BUBR1 for proper SAC activation54 (panel E). Thus, by regulating BUBR1 acetylation, BRCA2 may also participate in the proper segregation of chromosomes. A defect in the G2/M checkpoint or SAC disruption could account for some of the mitotic abnormalities observed in BRCA2-deficient tumors.
Although not without controversy,55 mounting evidence suggests a role for BRCA2 in cytokinesis.56-58 A number of identified interacting partners support this hypothesis; for example, disrupting the interaction of BRCA2 with the kinesin-like coiled-coil high mobility group protein HMG20b (BRAF35) leads to cytokinesis defects; specifically, delayed cell division, which triggers the accumulation of binucleated cells.59 The recruitment of BRCA2 to the midbody is modulated by Polo-Like Kinase 1 (PLK1) phosphorylation at S193, which in turn allows the interaction of BRCA2 with Myosin IIC (NMCII), a molecular motor protein that binds to cytoskeletal actin and regulates cytokinesis.58 The association of BRCA2 with the actin-binding protein Filamin A is also required for BRCA2 localization to the midbody (panel F). Both the S193A substitution or the untimely dissociation of BRCA2-Filamin A lead to cytokinetic defects. In addition to NMCII and Filamin A, the interaction of BRCA2 with other members of the midbody and components of the endosomal sorting complex (ESCRT) such as Alix, Tsg101 and CEP55 is required for proper abscission.57 The function of the midbody is controlled by Aurora B, which also co-immunoprecipitates with BRCA2.56,57 Together, these observations support a role for BRCA2 as a scaffold protein in the midbody that facilitates abscission independently of its role in HR.57,59 This activity may provide an explanation for the numerical chromosomal aberrations observed in BRCA2 associated tumors.60
Transcription control
The third exon of BRCA2 has been shown to be involved in transcriptional activation.61 Although its targets are still not well characterized, this function is regulated by multiple interactions (panel G). For example, EMSY protein associates with the N-terminal region of BRCA2 and represses its transactivation activity. Because EMSY is amplified in sporadic breast cancer and high-grade ovarian tumors, this interaction linked BRCA2 to sporadic breast cancer for the first time.62 BRCA2 is also associated with acetyltransferase activity through interaction with the transcription co-activator protein P/CAF,52 which could be an indirect means for BRCA2 to regulate transcription by the histone modifying activity of P/CAF. BRCA2 cooperates with both BRCA1 and P/CAF to enhance AR (Androgen Receptor) and GRIP1 (coactivator of AR)-mediated transactivation, promoting the anti-proliferative effect of AR.63
In addition, BRCA2 associates with the tumor suppressor and transcription regulator protein p53 through the DBD. Interestingly, overexpression of BRCA2 inhibits the apoptotic and transcriptional activity of p53 reducing the expression levels of its targets genes such as p21 and Bax. Because p53 transactivation deficiency leads to HR suppression, the association of BRCA2 and p53 could play an important role in the regulation of HR.64
BRCA2 regulation
BRCA2 is regulated by several kinases in a cell-cycle dependent manner. Phosphorylation at S3291 by CDKs takes place in G2/M transition of the cell cycle and prevents the interaction of the C-terminus of BRCA2 and RAD51, which in turn promotes disassembly of RAD51 filaments to block HR.8 Interestingly, abrogation of the BRCA2 C-terminal interaction with RAD51 by the S3291A substitution can still restore HR function in an I-SceI meganuclease-induced HR repair assay but not the replication fork protection activity of BRCA2. These findings suggest that the BRCA2 C-terminal interaction with RAD51 is essential for protection of stalled replication forks but is dispensable for the repair of DSB by HR.41
In response to genotoxic stress and subsequent activation of the checkpoint signaling kinase ATR, CDK2 interacts with the kinase from the Hippo tumor suppressor pathway, LATS1, and limits the phosphorylation of S3291 to maintain genome stability during stalled replication.65 The equivalent phosphorylation event in avian cells couples the disassembly of RAD51 foci with the entry into mitosis, consistent with the idea that phosphorylation of S3291 would act as a switch-off mechanism of HR.27 In addition to the C-terminus, CDK1 and CDK2 phosphorylate the BRCA2 N-terminal region at T77 during G2/M phase facilitating the association with the mitotic regulator PLK1. This interaction is important to coordinate the additional phosphorylation of RAD51 by PLK1 which allows its association with stalled replication forks.66
BRCA2 localization to the nucleus depends on 2 nuclear localization signals (NLS) located in the last 156 amino acids of the C-terminus.67 The BRC repeats of BRCA2 harbor a nuclear export signal (NES) required for its centrosome localization,68 while another NES occluded by BRCA2-DSS1 binding, regulating its export to the cytoplasm.19 In addition, in the U. maydis ortholog of BRCA2, Brh2, the Dss1 binding site also regulates the oligomerization state of BRCA2. In the absence of Dss1, Brh2 forms a dimer whereas Dss1 binds to the monomeric form of Brh2 which is proposed to be the active form for HR.69 A recent EM structure of human BRCA2 is consistent with a dimer70 but whether the same regulation by DSS1 applies is still unclear.
Concluding remarks and implications for tumorigenesis
BRCA2 germline mutations documented in breast cancer databases are spread throughout its primary sequence, therefore, which function(s) or region(s) might be more relevant for tumor suppression remains an open question.
Gathering information from deleterious missense variants can be very informative as, contrary to truncating mutations, they can be ascribed to a specific function. However, because most of the missense variants are rare their clinical classification is challenging. Thus, considerable efforts are underway to collect clinical and functional information to determine their causality.71 As of today, BRCA2 missense variants classified as deleterious have only been described in the C-terminal region.72 One reason for this might be that several interactions accumulate in the C-terminal part of BRCA2, in particular, in its DNA-binding domain. These include, DSS1, DNA, midbody components such as Filamin A and NMCII, or other partners such as p53. For example, D2723H is a known deleterious mutation disrupting the DSS1 binding site, this mutation results in the exposure of a NES of BRCA2 and renders the protein cytoplasmic.19 Other deleterious mutations that lie in the DNA binding domain include W2626C or R3052W.73 However, it is striking that the principal role of BRCA2 in HR is dependent on its interaction with RAD51 through the BRC repeats and yet, no deleterious missense variants have been located in that region. One reason for this might be that the other BRC repeats can compensate for the mutated one. However, this explanation is at odds with the prediction from the structural analysis of variants affecting this region by which the BRC repeats would form a Velcro-strip like structure where the mutation of one BRC repeat would affect the interaction of the other BRC repeats with RAD51.23 Another possibility is that the mutations in this region are rare and there might not be enough family history information to calculate the odds of causality with accuracy. This is in fact a general problem for many BRCA2 missense variants, collectively known as VUS (Variants of Unknown clinical Significance).74
In spite of the established importance of the DNA-binding domain of BRCA2 for HR, some studies point out a partial restoration of HR activity of cells expressing a BRCA2 truncation that lacks this region.75,76 Hence, suggesting that other regions of the protein may substitute or complement for this function. Interestingly, in many instances, the N- and C-terminal regions of BRCA2 are linked to the same function. For example, there are at least 3 regions particularly important for HR activity: the BRC repeats, the N-terminal PALB2- binding site and the C-terminal region that binds DNA and RAD51. Likewise, the N- and C-terminal regions are both involved in interactions important for maintaining the structure of the midbody. For example, the variant T582P located in the N-terminus and R3005A located in the C-terminus impede interactions with midbody proteins that lead to aberrant cytokinesis.57 Whether the cytokinetic function of BRCA2 is associated to cancer is still inconclusive, as the missense mutations specifically leading to this phenotype without affecting HR have not been classified yet.
Although no BRCA2 missense variant has been reported as pathogenic in the N-terminal region so far, functional analysis of VUS blocking PALB2 interaction (ex. W31R, W31C) disrupt BRCA2 function in HR rendering cells sensitive to DNA damage.77 In addition, mounting evidence from co-segregation analysis indicates that deletion of exon 3, comprising the transactivation core, EMSY and PALB2 binding sites, is pathogenic.78
Another example of the connection between the N- and C-terminal functions is the fact that BRCA2 is phosphorylated at the N- and C-terminal ends by CDK1/CDK2, which in turn regulates RAD51 phosphorylation by PLK1 and control HR.66 Two VUS altering the phosphorylation in the N- and C-terminus by CDKs, T77A and P3292L, have been reported in breast cancer cases, however, their implications for cancer causality are still unknown. Nevertheless, these observations and the plasticity of BRCA2 reported using truncations of the protein75 rise the question of whether the N-terminal and C-terminal modules are functionally connected. In support of this idea, the recent electron microscopy reconstruction of BRCA2 suggests that the N- and C-terminus of BRCA2 engage to form a dimer.70 If confirmed, these observations would have important implications making even more relevant the functional evaluation of missense variants in the context of the full-length protein.
The active research on (i) the different functions and partners of BRCA2, its structure and regulation, (ii) the new gene editing techniques that allow the functional evaluation of variants in their physiological context and (iii) the advent of next generation sequencing technologies facilitating genetic testing promises an improved calling of cancer predisposing mutations. These findings should bring opportunities for preventing the development of BRCA2-associated tumors.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Catharina von Nicolai and Åsa Ehlén from the Carreira lab and Jose M. Jimenez-Gomez for insightful comments on the manuscript.
Funding
This work was supported by EC-Marie Curie Career Integration grant CIG293444, Fondation pour la Recherche Médicale grant AJE201101, La Ligue Contre le Cancer grant and the ATIP-AVENIR CNRS/INSERM Young Investigator grant 201201 to AC that also funded CB; JSM was supported by a postdoctoral Fellowship from Fondation de France.
References
- 1.Mavaddat N, Peock S, Frost D, Ellis S, Platte R, Fineberg E, Evans DG, Izatt L, Eeles RA, Adlard J, et al.. Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. J Natl Cancer Inst 2013; 105:812-22; PMID:23628597; http://dx.doi.org/ 10.1093/jnci/djt095 [DOI] [PubMed] [Google Scholar]
- 2.Connor F, Smith A, Wooster R, Stratton M, Dixon A, Campbell E, Tait TM, Freeman T, Ashworth A. Cloning, chromosomal mapping and expression pattern of the mouse Brca2 gene. Hum Mol Genet 1997; 6:291-300; PMID:9063750; http://dx.doi.org/ 10.1093/hmg/6.2.291 [DOI] [PubMed] [Google Scholar]
- 3.Wooster R, Bignell GR, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995; 378:789-92; PMID:8524414; http://dx.doi.org/ 10.1038/378789a0 [DOI] [PubMed] [Google Scholar]
- 4.Sharan SK, Morimatsu M, Albrecht U, Lim DS, Regel E, Dinh C, Sands A, Eichele G, Hasty P, Bradley A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 1997; 386:804-10; PMID:9126738; http://dx.doi.org/ 10.1038/386804a0 [DOI] [PubMed] [Google Scholar]
- 5.Yu VP, Koehler M, Steinlein C, Schmid M, Hanakahi LA, van Gool AJ, West SC, Venkitaraman AR. Gross chromosomal rearrangements and genetic exchange between nonhomologous chromosomes following BRCA2 inactivation. Genes Dev 2000; 14:1400-6; PMID:10837032 [PMC free article] [PubMed] [Google Scholar]
- 6.Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J Biol Chem 1997; 272:31941-4; PMID:9405383; http://dx.doi.org/ 10.1074/jbc.272.51.31941 [DOI] [PubMed] [Google Scholar]
- 7.Mizuta R, LaSalle JM, Cheng HL, Shinohara A, Ogawa H, Copeland N, Jenkins NA, Lalande M, Alt FW. RAB22 and RAB163/mouse BRCA2: proteins that specifically interact with the RAD51 protein. Proc Natl Acad Sci USA 1997; 94:6927-32; PMID:9192668; http://dx.doi.org/ 10.1073/pnas.94.13.6927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 2005; 434:598-604; PMID:15800615; http://dx.doi.org/ 10.1038/nature03404 [DOI] [PubMed] [Google Scholar]
- 9.Carreira A, Hilario J, Amitani I, Baskin RJ, Shivji MKK, Venkitaraman AR, Kowalczykowski SC. The BRC Repeats of BRCA2 Modulate the DNA-Binding Selectivity of RAD51. Cell 2009; 136:1032-43; PMID:19303847; http://dx.doi.org/ 10.1016/j.cell.2009.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carreira A, Kowalczykowski SC. Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc Natl Acad Sci USA 2011; 108:10448-53; PMID:21670257; http://dx.doi.org/ 10.1073/pnas.1106971108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jensen RB, Carreira A, Kowalczykowski SC. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010; 467:678-83; PMID:20729832; http://dx.doi.org/ 10.1038/nature09399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen P-L, Lee W-H, Pavletich NP. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science 2002; 297:1837-48; PMID:12228710; http://dx.doi.org/ 10.1126/science.297.5588.1837 [DOI] [PubMed] [Google Scholar]
- 13.San Filippo J, Chi P, Sehorn MG, Etchin J, Krejci L, Sung P. Recombination mediator and Rad51 targeting activities of a human BRCA2 polypeptide. J Biol Chem 2006; 281:11649-57; PMID:16513631; http://dx.doi.org/ 10.1074/jbc.M601249200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu J, Doty T, Gibson B, Heyer W-D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol 2010; 17:1260-2; PMID:20729859; http://dx.doi.org/ 10.1038/nsmb.1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thorslund T, McIlwraith MJ, Compton SA, Lekomtsev S, Petronczki M, Griffith JD, West SC. The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat Struct Mol Biol 2010; 17:1263-5; PMID:20729858; http://dx.doi.org/ 10.1038/nsmb.1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagaraju G, Scully R. Minding the gap: the underground functions of BRCA1 and BRCA2 at stalled replication forks. DNA Repair 2007; 6:1018-31; PMID:17379580; http://dx.doi.org/ 10.1016/j.dnarep.2007.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Q, Mazloum N, Mao N, Kojic M, Holloman WK. Dss1 regulates interaction of Brh2 with DNA. Biochemistry 2009; 48:11929-38; PMID:19919104; http://dx.doi.org/ 10.1021/bi901775j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Zou C, Bai Y, Wazer DE, Band V, Gao Q. DSS1 is required for the stability of BRCA2. Oncogene 2005; 25:1186-94; http://dx.doi.org/ 10.1038/sj.onc.1209153 [DOI] [PubMed] [Google Scholar]
- 19.Jeyasekharan AD, Liu Y, Hattori H, Pisupati V, Jonsdottir AB, Rajendra E, Lee M, Sundaramoorthy E, Schlachter S, Kaminski CF, et al.. A cancer-associated BRCA2 mutation reveals masked nuclear export signals controlling localization. Nat Struct Mol Biol 2013; 20:1191-8; PMID:24013206; http://dx.doi.org/ 10.1038/nsmb.2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao W, Vaithiyalingam S, San Filippo J, Maranon DG, Jimenez-Sainz J, Fontenay GV, Kwon Y, Leung SG, Lu L, Jensen RB, et al.. Promotion of BRCA2-Dependent Homologous Recombination by DSS1 via RPA Targeting and DNA Mimicry. Mol Cell 2015; 59:176-87; PMID:26145171; http://dx.doi.org/ 10.1016/j.molcel.2015.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gudmundsdottir K, Lord CJ, Witt E, Tutt ANJ, Ashworth A. DSS1 is required for RAD51 focus formation and genomic stability in mammalian cells. EMBO Rep 2004; 5:989-93; PMID:15359272; http://dx.doi.org/ 10.1038/sj.embor.7400255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kojic M, Yang H, Kostrub CF, Pavletich NP, Holloman WK. The BRCA2-interacting protein DSS1 is vital for DNA repair, recombination, and genome stability in Ustilago maydis. Mol Cell 2003; 12:1043-9; PMID:14580353; http://dx.doi.org/ 10.1016/S1097-2765(03)00367-8 [DOI] [PubMed] [Google Scholar]
- 23.Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, Venkitaraman AR. Insights into DNA recombination from the structure of a RAD51–BRCA2 complex. Nature 2002; 420:287-93; PMID:12442171; http://dx.doi.org/ 10.1038/nature01230 [DOI] [PubMed] [Google Scholar]
- 24.Chen CF, Chen PL, Zhong Q, Sharp ZD, Lee WH. Expression of BRC repeats in breast cancer cells disrupts the BRCA2-Rad51 complex and leads to radiation hypersensitivity and loss of G(2)/M checkpoint control. J Biol Chem 1999; 274:32931-5; PMID:10551859; http://dx.doi.org/ 10.1074/jbc.274.46.32931 [DOI] [PubMed] [Google Scholar]
- 25.Abe T, Branzei D. High levels of BRC4 induced by a Tet-On 3G system suppress DNA repair and impair cell proliferation in vertebrate cells. DNA Repair 2014; 22:153-64; PMID:25218467; http://dx.doi.org/ 10.1016/j.dnarep.2014.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Magwood AC, Mundia MM, Baker MD. High Levels of Wild-Type BRCA2 Suppress Homologous Recombination. J Mol Biol 2012; 421:38-53; PMID:22579622; http://dx.doi.org/ 10.1016/j.jmb.2012.05.007 [DOI] [PubMed] [Google Scholar]
- 27.Ayoub N, Rajendra E, Su X, Jeyasekharan AD, Mahen R, Venkitaraman AR. The carboxyl terminus of Brca2 links the disassembly of Rad51 complexes to mitotic entry. Curr Biol 2009; 19:1075-85; PMID:19540122; http://dx.doi.org/ 10.1016/j.cub.2009.05.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell 2006; 22:719-29; PMID:16793542; http://dx.doi.org/ 10.1016/j.molcel.2006.05.022 [DOI] [PubMed] [Google Scholar]
- 29.Oliver A, Oliver AW, Swift S, Swift S, Lord CJ, Ashworth A, Pearl LH, Pearl L. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep 2009; 10:990-6; PMID:19609323; http://dx.doi.org/ 10.1038/embor.2009.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buisson R, Dion-Côté A-M, Coulombe Y, Launay H, Cai H, Stasiak AZ, Stasiak A, Xia B, Masson J-Y. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol 2010; 17:1247-54; PMID:20871615; http://dx.doi.org/ 10.1038/nsmb.1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, Yu X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol 2009; 19:524-9; PMID:19268590; http://dx.doi.org/ 10.1016/j.cub.2009.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkäs K, Roberts J, Lee A, Subramanian D, De Leeneer K, Fostira F, et al.. Breast-cancer risk in families with mutations in PALB2. N Engl J Med 2014; 371:497-506; PMID:25099575; http://dx.doi.org/ 10.1056/NEJMoa1400382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer 2011; 11:467-80; PMID:21701511; http://dx.doi.org/ 10.1038/nrc3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, et al.. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002; 297:606-9; PMID:12065746; http://dx.doi.org/ 10.1126/science.1073834 [DOI] [PubMed] [Google Scholar]
- 35.Cipak L, Watanabe N, Bessho T. The role of BRCA2 in replication-coupled DNA interstrand cross-link repair in vitro. Nat Struct Mol Biol 2006; 13:729-33; PMID:16845393; http://dx.doi.org/ 10.1038/nsmb1120 [DOI] [PubMed] [Google Scholar]
- 36.Adamo A, Collis SJ, Adelman CA, Silva N, Horejsí Z, Ward JD, Martinez-Perez E, Boulton SJ, La Volpe A. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell 2010; 39:25-35; PMID:20598602; http://dx.doi.org/ 10.1016/j.molcel.2010.06.026 [DOI] [PubMed] [Google Scholar]
- 37.Chapman JR, Taylor MRG, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 2012; 47:497-510; PMID:22920291; http://dx.doi.org/ 10.1016/j.molcel.2012.07.029 [DOI] [PubMed] [Google Scholar]
- 38.Kim H, D'Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev 2012; 26:1393-408; PMID:22751496; http://dx.doi.org/ 10.1101/gad.195248.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patel KJ, Yu VP, Lee H, Corcoran A, Thistlethwaite FC, Evans MJ, Colledge WH, Friedman LS, Ponder BA, Venkitaraman AR. Involvement of Brca2 in DNA repair. Mol Cell 1998; 1:347-57; PMID:9660919; http://dx.doi.org/ 10.1016/S1097-2765(00)80035-0 [DOI] [PubMed] [Google Scholar]
- 40.Lomonosov M, Anand S, Sangrithi M, Davies R, Venkitaraman AR. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev 2003; 17:3017-22; PMID:14681210; http://dx.doi.org/ 10.1101/gad.279003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 2011; 145:529-42; PMID:21565612; http://dx.doi.org/ 10.1016/j.cell.2011.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schlacher K, Wu H, Jasin M. A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-BRCA1/2. Cancer Cell 2012; 22:106-16; PMID:22789542; http://dx.doi.org/ 10.1016/j.ccr.2012.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klein HL. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair 2008; 7:686-93; PMID:18243065; http://dx.doi.org/ 10.1016/j.dnarep.2007.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buisson R, Niraj J, Pauty J, Maity R, Zhao W, Coulombe Y, Sung P, Masson J-Y. Breast cancer proteins PALB2 and BRCA2 stimulate polymerase η in recombination-associated DNA synthesis at blocked replication forks. CellReports 2014; 6:553-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raghunandan M, Chaudhury I, Kelich SL, Hanenberg H, Sobeck A. FANCD2, FANCJ and BRCA2 cooperate to promote replication fork recovery independently of the Fanconi Anemia core complex. Cell Cycle 2015; 14:342-53; PMID:25659033; http://dx.doi.org/ 10.4161/15384101.2014.987614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Min J, Choi ES, Hwang K, Kim J, Sampath S, Venkitaraman AR, Lee H. The breast cancer susceptibility gene BRCA2 is required for the maintenance of telomere homeostasis. J Biol Chem 2012; 287:5091-101; PMID:22187435; http://dx.doi.org/ 10.1074/jbc.M111.278994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Badie S, Escandell JM, Bouwman P, Carlos AR, Thanasoula M, Gallardo MM, Suram A, Jaco I, Benítez J, Herbig U, et al.. BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat Struct Mol Biol 2010; 17:1461-9; PMID:21076401; http://dx.doi.org/ 10.1038/nsmb.1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aguilera A, García-Muse T. R Loops: From Transcription Byproducts to Threats to Genome Stability. Mol Cell 2012; 46:115-24; PMID:22541554; http://dx.doi.org/ 10.1016/j.molcel.2012.04.009 [DOI] [PubMed] [Google Scholar]
- 49.Skourti-Stathaki K, Proudfoot NJ, Gromak N. Human Senataxin Resolves RNA/DNA Hybrids Formed at Transcriptional Pause Sites to Promote Xrn2-Dependent Termination. Mol Cell 2011; 42:794-805; PMID:21700224; http://dx.doi.org/ 10.1016/j.molcel.2011.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhatia V, Barroso SI, García-Rubio ML, Tumini E, Herrera-Moyano E, Aguilera A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014; 511:362-5; PMID:24896180; http://dx.doi.org/ 10.1038/nature13374 [DOI] [PubMed] [Google Scholar]
- 51.Menzel T, Nähse-Kumpf V, Kousholt AN, Klein DK, Lund-Andersen C, Lees M, Johansen JV, Syljuåsen RG, Sørensen CS. A genetic screen identifies BRCA2 and PALB2 as key regulators of G2 checkpoint maintenance. EMBO Rep 2011; 12:705-12; PMID:21637299; http://dx.doi.org/ 10.1038/embor.2011.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuks F, Milner J, Kouzarides T. BRCA2 associates with acetyltransferase activity when bound to P/CAF. Oncogene 1998; 17:2531-4; PMID:9824164; http://dx.doi.org/ 10.1038/sj.onc.1202475 [DOI] [PubMed] [Google Scholar]
- 53.Lin H-R, Ting NSY, Qin J, Lee W-H. M phase-specific phosphorylation of BRCA2 by Polo-like kinase 1 correlates with the dissociation of the BRCA2-P/CAF complex. J Biol Chem 2003; 278:35979-87; PMID:12815053; http://dx.doi.org/ 10.1074/jbc.M210659200 [DOI] [PubMed] [Google Scholar]
- 54.Choi E, Park P-G, Lee H-O, Lee Y-K, Kang GH, Lee JW, Han W, Lee HC, Noh D-Y, Lekomtsev S, et al.. BRCA2 fine-tunes the spindle assembly checkpoint through reinforcement of BubR1 acetylation. Developmental Cell 2012; 22:295-308; PMID:22340495; http://dx.doi.org/ 10.1016/j.devcel.2012.01.009 [DOI] [PubMed] [Google Scholar]
- 55.Lekomtsev S, Guizetti J, Pozniakovsky A, Gerlich DW, Petronczki M. Evidence that the tumor-suppressor protein BRCA2 does not regulate cytokinesis in human cells. J Cell Sci 2010; 123:1395-400; PMID:20356927; http://dx.doi.org/ 10.1242/jcs.068015 [DOI] [PubMed] [Google Scholar]
- 56.Daniels MJ, Wang Y, Lee M, Venkitaraman AR. Abnormal cytokinesis in cells deficient in the breast cancer susceptibility protein BRCA2. Science 2004; 306:876-9; PMID:15375219; http://dx.doi.org/ 10.1126/science.1102574 [DOI] [PubMed] [Google Scholar]
- 57.Mondal G, Rowley M, Guidugli L, Wu J, Pankratz VS, Couch FJ. BRCA2 localization to the midbody by filamin A regulates cep55 signaling and completion of cytokinesis. Developmental Cell 2012; 23:137-52; PMID:22771033; http://dx.doi.org/ 10.1016/j.devcel.2012.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takaoka M, Saito H, Takenaka K, Miki Y, Nakanishi A. BRCA2 phosphorylated by PLK1 moves to the midbody to regulate cytokinesis mediated by nonmuscle myosin IIC. Cancer Res 2014; 74:1518-28; PMID:24448238; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-0504 [DOI] [PubMed] [Google Scholar]
- 59.Lee M, Daniels MJ, Garnett MJ, Venkitaraman AR. A mitotic function for the high-mobility group protein HMG20b regulated by its interaction with the BRC repeats of the BRCA2 tumor suppressor. Oncogene 2011; 30:3360-9; PMID:21399666; http://dx.doi.org/ 10.1038/onc.2011.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Evers B, Drost R, Schut E, de Bruin M, van der Burg E, Derksen PWB, Holstege H, Liu X, van Drunen E, Beverloo HB, et al.. Selective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin Cancer Res 2008; 14:3916-25; PMID:18559613; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-4953 [DOI] [PubMed] [Google Scholar]
- 61.Milner J, Ponder B, Hughes-Davies L, Seltmann M, Kouzarides T. Transcriptional activation functions in BRCA2. Nature 1997; 386:772-3; PMID:9126734; http://dx.doi.org/ 10.1038/386772a0 [DOI] [PubMed] [Google Scholar]
- 62.Hughes-Davies L, Huntsman D, Ruas M, Fuks F, Bye J, Chin S-F, Milner J, Brown LA, Hsu F, Gilks B, et al.. EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell 2003; 115:523-35; PMID:14651845; http://dx.doi.org/ 10.1016/S0092-8674(03)00930-9 [DOI] [PubMed] [Google Scholar]
- 63.Shin S, Verma IM. BRCA2 cooperates with histone acetyltransferases in androgen receptor-mediated transcription. Proc Natl Acad Sci USA 2003; 100:7201-6; PMID:12756300; http://dx.doi.org/ 10.1073/pnas.1132020100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rajagopalan S, Andreeva A, Rutherford TJ, Fersht AR. Mapping the physical and functional interactions between the tumor suppressors p53 and BRCA2. Proc Natl Acad Sci USA 2010; 107:8587-92; PMID:20421506; http://dx.doi.org/ 10.1073/pnas.1003689107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pefani D-E, Latusek R, Pires I, Grawenda AM, Yee KS, Hamilton G, van der Weyden L, Esashi F, Hammond EM, O'Neill E. RASSF1A-LATS1 signalling stabilizes replication forks by restricting CDK2-mediated phosphorylation of BRCA2. Nat Cell Biol 2014; 16:962-8; PMID:25218637; http://dx.doi.org/ 10.1038/ncb3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yata K, Bleuyard J-Y, Nakato R, Ralf C, Katou Y, Schwab RA, Niedzwiedz W, Shirahige K, Esashi F. BRCA2 coordinates the activities of cell-cycle kinases to promote genome stability. CellReports 2014; 7:1547-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spain BH, Larson CJ, Shihabuddin LS, Gage FH, Verma IM. Truncated BRCA2 is cytoplasmic: implications for cancer-linked mutations. Proc Natl Acad Sci USA 1999; 96:13920-5; PMID:10570174; http://dx.doi.org/ 10.1073/pnas.96.24.13920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Han X, Saito H, Miki Y, Nakanishi A. A CRM1-mediated nuclear export signal governs cytoplasmic localization of BRCA2 and is essential for centrosomal localization of BRCA2. Oncogene 2008; 27:2969-77; PMID:18059333; http://dx.doi.org/ 10.1038/sj.onc.1210968 [DOI] [PubMed] [Google Scholar]
- 69.Zhou Q, Kojic M, Cao Z, Lisby M, Mazloum NA, Holloman WK. Dss1 interaction with Brh2 as a regulatory mechanism for recombinational repair. Mol Cell Biol 2007; 27:2512-26; PMID:17261595; http://dx.doi.org/ 10.1128/MCB.01907-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shahid T, Soroka J, Kong EH, Malivert L, McIlwraith MJ, Pape T, West SC, Zhang X. Structure and mechanism of action of the BRCA2 breast cancer tumor suppressor. Nat Struct Mol Biol 2014; 21(11):962-8; PMID:24389540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Spurdle AB, Healey S, Devereau A, Hogervorst FBL, Monteiro ANA, Nathanson KL, Radice P, Stoppa-Lyonnet D, Tavtigian S, Wappenschmidt B, et al.. ENIGMA-Evidence-based network for the interpretation of germline mutant alleles: An international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum Mutat 2011; 33:2-7; PMID:21990146; http://dx.doi.org/ 10.1002/humu.21628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guidugli L, Carreira A, Caputo SM, Ehlen A, Galli A, Monteiro ANA, Neuhausen SL, Hansen TVO, Couch FJ, Vreeswijk MPG. Functional assays for analysis of variants of uncertain significance in BRCA2. Hum Mutat 2014; 35:151-64; PMID:24323938; http://dx.doi.org/ 10.1002/humu.22478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guidugli L, Pankratz VS, Singh N, Thompson J, Erding CA, Engel C, Schmutzler R, Domchek S, Nathanson K, Radice P, et al.. A classification model for BRCA2 DNA binding domain missense variants based on homology-directed repair activity. Cancer Res 2013; 73:265-75; PMID:23108138; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Plon SE, Eccles DM, Easton DF, Foulkes WD, Genuardi M, Greenblatt MS, Hogervorst FBL, Hoogerbrugge N, Spurdle AB, Tavtigian SV, et al.. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 2008; 29:1282-91; PMID:18951446; http://dx.doi.org/ 10.1002/humu.20880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Siaud N, Barbera MA, Egashira A, Lam I, Christ N, Schlacher K, Xia B, Jasin M. Plasticity of BRCA2 Function in Homologous Recombination: Genetic Interactions of the PALB2 and DNA Binding Domains. PLoS Genet 2011; 7:e1002409; PMID:22194698; http://dx.doi.org/ 10.1371/journal.pgen.1002409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008; 451:1111-5; PMID:18264088; http://dx.doi.org/ 10.1038/nature06548 [DOI] [PubMed] [Google Scholar]
- 77.Biswas K, Das R, Eggington JM, Qiao H, North SL, Stauffer S, Burkett SS, Martin BK, Southon E, Sizemore SC, et al.. Functional evaluation of BRCA2 variants mapping to the PALB2-binding and C-terminal DNA-binding domains using a mouse ES cell-based assay. Hum Mol Genet 2012; 21:3993-4006; PMID:22678057; http://dx.doi.org/ 10.1093/hmg/dds222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Muller D, Rouleau E, Schultz I, Caputo S, Lefol C, Bièche I, Caron O, Nogues C, Limacher JM, Demange L, et al.. An entire exon 3 germ-line rearrangement in the BRCA2 gene: pathogenic relevance of exon 3 deletion in breast cancer predisposition. 2011; 12:121; PMID:21939546 [DOI] [PMC free article] [PubMed] [Google Scholar]