

The cell surface metalloprotease ADAM17 (a disintegrin and metalloprotease 17) orchestrates cell-cell interactions and has a key role in inflammation, development and in diseases such as Rheumatoid Arthritis and Cancer. ADAM17 is responsible for liberating the pro-inflammatory cytokine TNFα and most EGFR-ligands from their membrane anchor, thereby keeping a close reign on the TNFα- and EGFR signaling pathways. In a recent study, the inactive Rhomboid-like proteins iRhoms 1 and 2 emerged as crucial upstream regulators of ADAM17 during mouse development.1 In this study, mice lacking both iRhoms resembled mice lacking ADAM17, whereas the animals lacking only iRhom1 or iRhom2 appeared normal and indistinguishable from wild type littermates. Thus, either iRhom can, in principle, have compensatory or redundant functions in the absence of the other. But two organs are notable exceptions, the immune system and the brain. Interestingly, little or no iRhom1 is expressed in immune cells, mostly macrophages and the macrophage-like microglia in the brain, leaving the regulation of ADAM17 to iRhom2 in these cells.1 On the other hand, little or no iRhom2 is expressed in the rest of brain (i.e. neurons and glial cells), so iRhom1-/- mice have almost no mature and functional ADAM17 in the brain.1 (Fig. 1). The brain is responsible for cognition, memory, reasoning and many other aspects of daily life, and is one of the most enigmatic and complex structures in the body. Therefore the remarkable division of labor between iRhom1 and iRhom2 raises intriguing questions about their functions in different cell types in the brain. It is conceivable that iRhom2 has a role in neuroinflammation and neuroinflammatory diseases, whereas iRhom1 is perhaps more likely to play a role in physiological processes in the brain.
Figure 1.
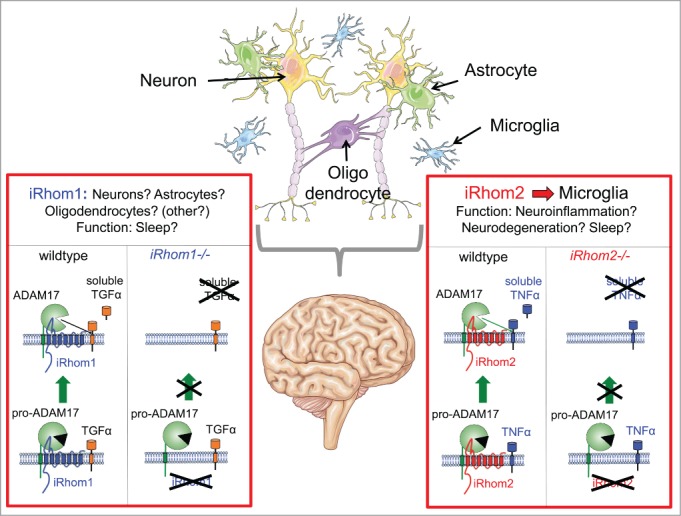
Differential regulation of ADAM17 by iRhom1 and iRhom2 in the brain. iRhom2 regulates ADAM17-dependent TNFα release in microglia, likely contributing to neuroinflammation, neurodegeneration and sleep. iRhom1 controls ADAM17 in the rest of the brain, although the exact cell type(s) remain(s) to be identified. Rhom1/ADAM17-dependent TGFα shedding and EGFR activation may induce sleep. Includes images from www.servier.com.
The normal activity of the brain can be severely impacted by neurodegenerative conditions such as Alzheimer disease and by traumatic brain injuries such as concussions. Both have an essential neuroinflammatory component. One important clue about possible roles of iRhom2 in the brain emerged recently from a study that found an association between the methylation status of the iRhom2 gene and Alzheimer disease.2 Inflammation is thought to play an important role in the later stages in the pathogenesis of AD, and the Aβ peptides that assemble to form the characteristic amyloid plaques in AD can activate microglia,3 lending additional support to the notion that iRhom2/ADAM17 might be critical mediators of this pathway. De-methylation of the iRhom2 gene may increase the expression of iRhom2, presumably leading to enhanced ADAM17-dependent release of TNFα (and other substrates), thereby exacerbating the inflammatory component of AD.2,3 Interestingly, concussions and other forms of traumatic brain injury can also lead to temporary or even permanent cognitive impairment. There is mounting evidence that this depends, at least in part, on activation of the TNFα pathway.4 Moving forward, it will be interesting to learn more about the functions of the iRhom2/ADAM17-dependent production of soluble TNFα in TBI, AD and other brain diseases that could be exacerbated by inflammation. If the iRhom2/ADAM17/TNFα pathway is indeed activated by brain injury or amyloid plaques, and if blocking components of this pathway can ameliorate or prevent the cognitive impairment associated with animal models of AD or TBI, then iRhom2 could become an attractive target to prevent cognitive decline caused by neuroinflammation.
The observation that iRhom1-/- mice have very little, if any mature ADAM17 in their brains, yet don’t display any evident abnormalities is also quite unexpected. In this case, clues to possible functions of iRhom1 are provided by its role in regulating sleep in Drosophila,5 and by the role of the EGFR and its ligand TGFα (an iRhom1/ADAM17 substrate) in regulating locomotor activity in rodents.6 Interestingly, inflammation and the iRhom2/ADAM17 substrate TNFα are also known to affect sleep in mice and humans, so the iRhoms could be involved in controlling different aspects of this restorative and protective process.7
Overall, the serendipitous discovery of the differential regulation of ADAM17 by iRhoms1 and 2 in the brain promises to open up a new frontier for exploration of the function of these molecules. The evolutionarily conserved role of the EGFR and its ligands in sleep and regulating locomotor activity raises questions about the role of iRhom1/ADAM17-dependent shedding of TGFα in this process. Moreover, the iRhom2/ADAM17-dependent processing of TNFα in microglia brings up questions about the role of this pathway in neurodegeneration in AD and TBI, and in the somnolence caused by infections and inflammation. Moving forward, it will also be fascinating to learn more about the substrate repertoire and functions of iRhom1/ADAM17 in the different cell types in the brain. In addition to the known substrates like EGFR-ligands and TNFα, we anticipate that many other membrane proteins with unique and interesting roles in the brain will emerge as substrates for iRhom1 or 2 and ADAM17. We hope that a better understanding of the differential regulation of ADAM17 by iRhom1 and 2 in the brain will help uncover new and selective therapeutic opportunities for modulation of sleep, neurodegeneration and possibly also other, yet to be discovered physiologically and pathologically relevant pathways in the brain.
References
- 1.Li X, et al.. Proc Natl Acad Sci U S A 2015; 112:6080–5; PMID:25918388; http://dx.doi.org/ 10.1073/pnas.1505649112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Jager PL, et al.. Nat Neurosci 2014; 17:1156–63; PMID:25129075; http://dx.doi.org/ 10.1038/nn.3786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heppner FL, et al.. Nat Rev Neurosci 2015; 16:358–72; PMID:25991443; http://dx.doi.org/ 10.1038/nrn3880 [DOI] [PubMed] [Google Scholar]
- 4.Sriram K, O'Callaghan JP. J Neuroimmune Pharmacol 2007; 2:140–53; PMID:18040839; http://dx.doi.org/ 10.1007/s11481-007-9070-6 [DOI] [PubMed] [Google Scholar]
- 5.Zettl M, et al.. Cell 2011; 145:79–91; PMID: 21439629; http://dx.doi.org/ 10.1016/j.cell.2011.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kramer A, et al.. Science 2001; 294:2511–5; PMID:11752569; http://dx.doi.org/ 10.1126/science.1067716 [DOI] [PubMed] [Google Scholar]
- 7.Clark IA, Vissel B. J Neuroinflammation 2014; 11:51; PMID:24655719; http://dx.doi.org/ 10.1186/1742-2094-11-51 [DOI] [PMC free article] [PubMed] [Google Scholar]