

Abstract
Mitochondria are organelles that orchestrate a plethora of fundamental cellular functions that have been associated with various steps of tumor progression. However, we currently lack a mechanistic understanding of how mitochondrial dynamics, which reflects the organelles' exquisite heterogeneity in shape and spatial distribution, affects tumorigenesis. In a recent study, we uncovered a surprising new role of mitochondrial dynamics in response to PI3K therapy. We found that re-activation of Akt/mTOR signaling in tumor cells exposed to small molecule PI3K antagonists currently in the clinic triggered the transport of energetically active, elongated mitochondria to the cortical cytoskeleton of tumor cells. In turn, these repositioned mitochondria supported increased lamellipodia dynamics, faster turnover of focal adhesion complexes, heightened velocity and distance of random cell migration and increased tumor cell invasion. In this Extra View, we discuss the mechanistic basis of this paradoxical response to PI3K antagonists and propose possible strategies to disable mitochondrial adaptation.
Keywords: drug resistance, focal adhesion, Hsp90, mitochondria, metastasis, PI3K
Abbreviations
- Seventeen-AAG
17- allylaminogeldanamycin
- Akt
v-akt murine thymoma viral oncogene homolog 1
- mTOR
mammalian target of rapamycin
- ATP
adenosine triphosphate
- B-Raf
v-raf murine sarcoma viral oncogene homolog B
- Drp1
dynamin 1-like
- ERK
extracellular signal regulated kinase
- Erv1
growth factor, augmenter of liver regeneration
- FAK
focal adhesion kinase
- Hsp90
Heat shock protein of 90 kDa
- IL-6
interleukin-6
- IL-1β
interleukin-1β
- mtHsp90
mitochondrial Hsp90
- MYC
v-myc avian myelocytomatosis viral oncogene homolog
- OxPhos
oxidative phosphorylation
- PI3K
Phosphoinositide 3-kinase
- PI3Ki
PI3K inhibitor
- PTEN
phosphatase and tensin homolog
- PyK2
protein tyrosine kinase 2 β
- Rac
ras-related C3 botulinum toxin substrate
- Rho
ras homolog
- RTK
receptor tyrosine kinase
- TGFβ
transforming growth factor β
- TNFα
tumor necrosis factor α
- TRAP-1
tumor necrosis factor receptor associated protein-1
- ULK1
uncoordinated like kinase-1
- Wnt
wingless-type MMTV integration site
Introduction
Phosphoinositide 3-kinases (PI3K) are master regulators of cellular metabolism that transduce extracellular growth factor signals via receptor tyrosine kinases (RTKs) or G-protein coupled receptors. PI3K activation leads to phosphorylation of phosphatidylinositol lipids at the plasma membrane, which in turn recruit and/or activate downstream effectors, including the serine/threonine kinases protein kinase B (PKB/Akt) and mammalian target of rapamycin (mTOR). In normal cells, the phosphatase and tensin homolog (PTEN) restrains the pathway's activation. Hyperactivation of the PI3K pathway is one of the most common alterations in cancer, functions as a pivotal disease driver and can involve a variety of mechanisms, including copy number alterations, activating mutations in PI3K/Akt/mTOR, and deletion of PTEN.1 Together with the fact that PI3K and downstream kinases are amenable to pharmacological intervention, this pathway provides one of the most attractive targets for therapeutic intervention and personalized medicine approaches in cancer.2 However, and despite high expectations, the vast majority of small molecule PI3K antagonists evaluated in the clinic so far have shown significant toxicity and limited efficacy as monotherapy.3 The activity of these agents is likely limited by the emergence of treatment resistance mechanisms, including activation of compensatory signaling pathways (RTKs, ERK, MYC, Notch/Wnt); and a paradoxical reactivation of Akt/mTOR, the very same pathway that these agents are designed to inhibit.4-7
We recently demonstrated that Akt2-directed repurposing of mitochondrial functions provides a novel adaptive mechanism of tumor resistance to PI3K therapy.8 Importantly, disabling the addiction of tumors to PI3K-induced mitochondrial adaptation produced potent and synergistic anti-cancer activity in preclinical studies.8 In a follow-up study, we have now uncovered a surprising new role of mitochondrial dynamics in response to PI3K therapy.9 Accordingly, re-activation of Akt signaling in tumor cells exposed to PI3K therapy triggers the transport of energetically active mitochondria to the cortical cytoskeleton of tumor cells, where they support increased lamellipodia dynamics, faster turnover of focal adhesion complexes and increased tumor cell migration and invasion.9 Here we discuss the mechanistic basis of this paradoxical response to PI3K antagonists that engenders more aggressive disease traits, and propose possible strategies to disable adaptive mitochondrial rewiring for cancer therapeutics.
An Undesired Pro-Metastatic Effect of PI3K Therapy
PI3K targeted therapy induces extensive bioenergetics and transcriptional reprogramming in tumor cells that culminates with global changes in the secretory profile and activation of growth factor receptor kinases of treated tumors.8 Surprisingly, we found that PI3K inhibitors (PI3Ki) up-regulated 2 main gene networks of protection from apoptosis and increased cell motility.9 Indeed, tumor cells treated with various small molecule PI3Ki currently used in clinical trials (PX866, GDC0941, AZD6482, BKM120) showed increased motility and invasion (Fig. 1). At the cellular level, PI3Ki resulted in enhanced membrane cell dynamics and loss of directional migration in response to chemotactic gradients.
Figure 1.
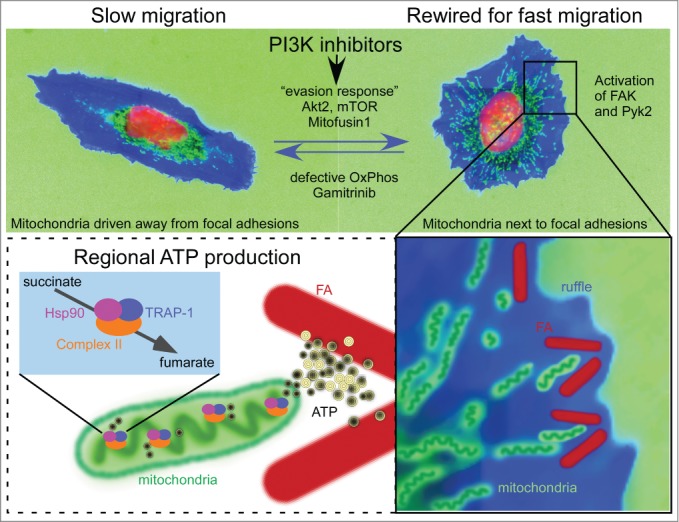
PI3K repositions mitochondria to increase tumor cell invasion. In this schematic representation, tumor cells are drawn based on representative cellular morphology and actual mitochondrial localization. Mitochondria are green, cytoskeleton is blue and the nucleus is red. Top, Treatment of tumor cells with PI3K inhibitors used in the clinic initiates a compensatory adaptive response centered on reactivation of Akt2 and mTOR. As a result, mitochondria travel to the cortical cytoskeleton, a process that requires elongation (Mitofusin1) and active mitochondrial respiration (OxPhos). Bottom, Rewired cells juxtapose mitochondria to focal adhesions (FA), where they provide a regional source of energy and accelerate focal adhesion dynamics. The mitochondrial Hsp90 chaperones overcome metabolic stress and maintain OxPhos by folding complex II. A mitochondrially-targeted Hsp90 inhibitor, Gamitrinib, prevents PI3Ki-induced pro-invasive responses.
Most signaling studies analyze the activity of PI3Ki in a 24 h window, leaving unaddressed the possibility that additional adaptive responses may eventually compensate for PI3K inhibition. To investigate this possibility, we followed up the status of the pathway for up to 3 d. As expected, and in agreement with the literature, a single dose of PI3Ki led to a transient inhibition of Akt. However, reactivation of Akt1 and Akt2, but also the mTOR pathway, followed at around 36 h after treatment.8,9 In agreement with the well-documented control of migration and metastasis by both Akt and mTOR,10,11 we found that reactivation of Akt2 or mTOR were essential for the paradoxical increase in tumor cell invasion associated with PI3K therapy. At the mechanistic level, mTOR has been shown to regulate actin cytoskeleton dynamics through the small GTPases Rho and Rac,12 as well as activation of protein kinase Cα.13 Recent studies implicated the mTOR substrate uncoordinated like kinase 1 (ULK1) in inhibition of tumor cell invasion.14 Both the well-known function of ULK1 in autophagy,15 as well as its novel role in preventing activation of focal adhesion kinase (FAK), are inhibited by mTOR phosphorylation. This constitutes a novel mechanism by which mTOR promotes tumor cell motility, via inactivation of ULK1 and release of FAK inhibition.14 Such mechanism might be at play in adaptive tumor responses to PI3Ki, as the increase in tumor cell invasion observed in these settings is associated with activation of FAK and the related focal adhesion kinase, Pyk2.9 In light of these results, one could speculate that FAK inhibitors might prevent the pro-metastatic adaptive responses to PI3K inhibitors. As proof of concept to test this model, we depleted tumor cells of FAK and Pyk2, and this prevented the increase in cell invasion after PI3K treatment.
Moving forward, mechanistic studies should be aimed at dissecting whether mTOR inactivation of ULK1 contributes to the increased invasion seen in PI3Ki-treated tumor cells. This is relevant in light of the recent development of a specific ULK1 inhibitor that shows promising anti-cancer activity.16 A second related question that warrants further study is whether autophagy-dependent or –independent functions of ULK1 are at play in the tumor adaptive response to PI3Ki.
Therapy-Induced Metastasis in Pre-Clinical Models
Although perhaps unexpected for PI3K therapy, it had already been noted that several other anti-cancer drugs produced a paradoxical pro-metastatic effect in pre-clinical settings (reviewed in17). Chemotherapeutic drugs were among the first to be associated with increased incidence of pulmonary metastasis in mice. For instance, several of the DNA-targeting agents (cyclophosphamide, bleomycin, doxorubicin, cytarabine, 5-azacytidine, aphidicoline), RNA targeting drugs (actinomycin D, mithramycin), and the anti-folate compound methotrexate were all associated with pro-metastatic effects.17 Certain molecular targeted therapies seem to engender similar responses; for instance antiangiogenic agents, geldanamycin-based Hsp90 inhibitors18 and B-Raf inhibitors19 were shown to paradoxically increase metastatic competency. It is interesting that a common feature of such disparate molecular therapies is activation of the PI3K/Akt signaling axis. Another example is molecular therapy against Akt, which has also been shown to exacerbate metastasis in mice models, aided by nuclear accumulation of β-catenin.20 Our recent findings place PI3K inhibitors in this class of drugs that may have unwanted pro-metastatic effects.9
The relevance of these findings to patient settings is still unclear, as we do not currently have reliable mouse models to predict a potential paradoxical effect of sustained treatment on disease progression.17 Thus, the appearance of a more invasive, potentially pro-metastatic phenotype associated with drug treatment may not necessarily predict disease progression in patients.17 On the other hand, as elegantly illustrated in the case of B-Raf inhibitors,19 integrating clinical data with pre-clinical observations confirmed a clear undesired pro-metastatic effect of these agents in the clinic. Therefore, before rushing these compounds to the clinic, it may be desirable to pursue retrospective analysis of patients treated with these agents, and see whether treatment failure was associated with the development new distal metastasis.
Rewiring of Mitochondrial Function and Mitochondrial Dynamics in Tumor Cell Invasion
Mitochondria are organelles that orchestrate a plethora of fundamental cellular functions, including bioenergetics, calcium homeostasis, reduction-oxidation status balance, reactive oxygen species generation and programmed cell death.21 Not surprisingly, mitochondria have been implicated in multiple steps of tumor progression22 and the exquisite heterogeneity in shape and spatial distribution of these organelles in cancer is quickly becoming a field of intense investigation.23 A complex, intertwined machinery of mitochondrial dynamics influences organelle division, fusion and subcellular transport.24 How these processes may couple to tumor progression has not been widely investigated. A key feature of our observation of increased tumor cell motility and invasion in response to PI3K therapy, was a clear requirement of mitochondrial elongation and intracellular trafficking to the cell periphery9 (Fig. 1).
This is not the first time that a link between mitochondrial localization and cell migration is noted. Previous reports observed re-distribution of mitochondria toward the leading edge of cells during persistent migration.25 Also, a proteomics analysis of invadopodia revealed enrichment of mitochondrial proteins,26 suggesting that mitochondria were present at sites of active cell invasion. Interestingly, alteration of mitochondrial “shaping” proteins (the regulators of fusion and fission) has been reported in breast and thyroid tumors.27,28 In these studies, the mitochondrial fission factor Drp1 played a central role in the acquisition of metastatic competency. Mechanistically, our recent findings connected pathways of mitochondrial elongation via the fusion protein Mitofusin1 to organelle trafficking to the cortical cytoskeleton.9 Also novel were the findings that cortical mitochondria become in close proximity to focal adhesions (approx. 0.5–1 μm) and that such mitochondrial proximity accelerates the turnover of focal adhesions (Fig. 1). Although not conclusively demonstrated, we speculate that mitochondria may be needed as a “regional” source of energy in proximity of focal adhesions. A second energy-driven consequence of mitochondria accumulation at the leading edge of the cell would be regional stabilization of detyrosinated microtubules by ATP.29 Alternatively, mitochondria might be needed for calcium signaling,30 or for the production of fatty acids and eicosanoids,31,32 important for membrane dynamics.
Mitochondrial Dynamics, Growth Factors and Cellular Stress
As the PI3K pathway is activated downstream of growth factors, it is conceivable that mitochondrial relocalization might not only be restricted to adaptation to therapy, but also involved in physiologic growth signals. A growing body of evidence is consistent with this model, as mitochondrial shape is regulated by the non-canonical Wnt ligand Wnt5a33; pro-inflammatory cytokines TNFα, IL-6, and IL-1β34; transforming growth factor-β (TGF-β)35; erv1-like growth factor36; and serum.37 On the other hand, the regulation of mitochondrial trafficking in response to growth factor stimulation has not been widely studied. In this context, we showed that stimulation of tumor cells with chemoattractant-rich media induced repositioning of mitochondria to the cortical cytoskeleton.9 Further studies are needed to characterize the individual growth signals that might reprogram mitochondrial trafficking.
A second related question that stems from our observations is whether mitochondrial dynamics can be modulated by environmental stress stimuli (e.g., hypoxia, nutrient deprivation, oxidative stress). Indeed, a connection between hypoxia and mitochondrial dynamics, mostly fission via Drp1, has been noted.38 In addition, increased mitochondrial fission by hypoxia was shown to enhance breast cancer invasion.39 Whether a conserved mitochondrial trafficking mechanism reminiscent of the one described in hypoxic neurons40,41 is at play in actively invasive tumor cells, awaits confirmation.
Other stress conditions of the tumor microenvironment have been associated with mitochondrial dynamics. For instance, mitochondrial elongation is a common response to nutrient deprivation (combinations of serum, glucose, glutamine or other aminoacids starvation) in cancer cells.42 It is interesting that not all starvation stimuli are created equal; amino acid starvation, in particular glutamine, are among the most potent inducers of mitochondrial fusion. Glucose starvation, on the other hand, did not induce changes in mitochondrial shape. Whether mTOR repression in amino acid-starved cells might explain mitochondrial fusion, is presently unknown. However, our results suggest that in nutrient-rich conditions, tumor cell invasion is supported by mitochondrial fusion coupled to mTOR-dependent trafficking of mitochondria to the leading edge of the tumor cell.9
Disabling Mitochondrial-Adaptive Drug Resistance
Although most tumors rewire their metabolism toward glycolysis (the Warburg effect),43 mitochondrial bioenergetics and biosynthesis are vital for many tumor traits.22 An emerging role of oxidative phosphorylation as a cancer driver is supported by fresh experimental evidences linking mitochondrial respiration to oncogene-dependent transformation,44 metabolic reprogramming,45 protein translation,46 tumor cell repopulation,47 cell motility48 and metastasis.14 Our group has been interested in these mechanisms, and we have recently uncovered a key role of adaptive mitochondrial reprogramming in drug resistance8 and metastatic competency.9 Mechanistic aspects of how tumors may regulate oxidative phosphorylation have also come into better focus, with a key role of protein folding quality control maintained by mitochondria-localized Heat Shock Protein-90 (mtHsp90) chaperones,49 as well as organelle proteases,50 in ensuring the stability and function of respiratory protein complexes.
Together, these mechanistic observations suggest that mitochondrially-targeted drugs may represent a promising new dimension to interfere with adaptive tumor reprogramming, and ultimately eliminate drug-resistant cancer cells.51 This concept has been successfully pursued with the clinical development of modulators of apoptosis, a process regulated at the outer mitochondrial membrane. Nevertheless, the concept of targeting mitochondrial metabolism or organelle-driven adaptation in cancer is still underdeveloped. Against this backdrop, our laboratory designed Gamitrinib52 to target abundant pools of Hsp90 and its structurally related chaperone, TRAP-1 in mitochondria, selectively of tumor cells.53 There is a compelling rationale to think that these organelle-localized chaperones may provide promising targets for cancer therapy. First, their activity likely improves protein folding quality control in mitochondria, an ideal mechanism to buffer the risk of proteotoxic stress typical of highly bioenergetically active (tumor) cells.54 Second, a proteomics screen of mitochondrial molecules that require Hsp90 for folding identified key regulators of virtually every organelle function,49 suggesting that disabling this pathway may irreversibly compromise global organelle homeostasis. Third, mitochondrial Hsp90 chaperones have been shown to directly sustain tumor cell invasion and metastasis by dampening the activation of autophagy and the unfolded protein response.14,55
Gamitrinib relies on a combinatorial structure where the Hsp90 ATPase inhibitory module of 17-allylaminogeldanamycin (17-AAG), a first-generation Hsp90 inhibitor, is linked to the mitochondria-targeting moiety of triphenylphosphonium.52 This enables fast and efficient mitochondrial accumulation of Gamitrinib, with virtually no inhibition of cytosolic Hsp90. Conversely, none of the first or second-generation Hsp90 antagonists currently in the clinic had the ability to accumulate in mitochondria.52 Functionally, targeting mitochondrial Hsp90s resulted in catastrophic and irreversible effects on mitochondrial functions,52 disabled Complex II-dependent oxidative phosphorylation,49 and induced acute mitochondrial permeability transition and apoptosis,56 delivering potent anticancer activity in localized and disseminated xenograft and genetic tumor models.57
In our studies, we found that oxidative phosphorylation was required for mitochondrial trafficking to the cortical cytoskeleton. In respiration-deficient tumor cells, focal adhesion complexes were devoid of mitochondria and invasion was impaired,9 reinforcing the rationale of targeting mitochondrial Hsp90s as novel anti-metastatic therapy. Accordingly, Gamitrinib prevented Pi3Ki-induced mitochondrial infiltration of the cortical cytoskeleton (Fig. 1), interfered with mitochondrial fusion and potently suppressed tumor cell invasion.9 Consistent with its mechanism of action of targeting mitochondrial integrity, Gamitrinib potently synergized with PI3K therapy in a high-throughput drug combination screen, converting a transient, cytostatic effect of these agents into potent, cytotoxic anticancer activity in preclinical tumor models.8
Closing Remarks
Our recent studies identified an additional adaptive mechanism deployed by tumor cells to evade PI3K therapy. This is characterized by mitochondrial fusion and relocation to the cell periphery providing a regional energy source to fuel increased tumor cell motility and invasion. As dysregulation of mitochondrial shape, number and subcellular topography takes center stage in tumor adaptation, reaching a better understanding of how exactly mitochondrial dynamics contribute to cancer traits has become an urgent priority. Although proper mitochondrial dynamics is key to mitochondrial function, its impact on metabolic reprogramming and tumor progression needs further investigation. Future studies aimed at defining the molecular requirements of mitochondrial trafficking and organelle dynamics in response to PI3Ki are presently underway. In particular, it will be of interest to test whether tumor cells hijack a well-established mitochondrial trafficking machinery that has been shown in neurons and other cell types, including lymphocytes, to reposition mitochondria at subcellular sites of high energy demands, thus powering up mechanisms of invasion and metastasis.
From a more translational and disease-relevant standpoint, our results caution against the use of single-agent PI3Ki therapy, and argue that the combination of these agents with approaches to disable mitochondrial reprogramming, including Gamitrinib, FAK inhibitors, Akt and mTOR inhibitors may provide rational and more effective anticancer regimens in the clinic.
Funding
This work was supported by the National Institutes of Health (NIH) grants P01 CA140043, R01 CA78810 and CA190027 (DCA.), F32CA177018 (M.C.C.), a Challenge Award from the Prostate Cancer Foundation (DCA. and M.C.C.), and the Office of the Assistant Secretary of Defense for Health Affairs through the Prostate Cancer Research Program under Award No. W81XWH-13-1-0193 (DCA).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Yuan TL, Cantley LC, PI3K pathway alterations in cancer: variations on a theme. Oncogene 2008; 27(41):5497-510; PMID:18794884; http://dx.doi.org/ 10.1038/onc.2008.245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell 2003; 4(4):257-62; PMID:14585353; http://dx.doi.org/ 10.1016/S1535-6108(03)00248-4 [DOI] [PubMed] [Google Scholar]
- 3.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov 2009; 8(8):627-44; PMID:19644473; http://dx.doi.org/ 10.1038/nrd2926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 2014; 13(2):140-56; PMID:24481312; http://dx.doi.org/ 10.1038/nrd4204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chakrabarty A, Sánchez V, Kuba MG, Rinehart C, Arteaga CL. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad Sci U S A 2012; 109(8):2718-23; PMID:21368164; http://dx.doi.org/ 10.1073/pnas.1018001108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011; 19(1):58-71; PMID:21215704; http://dx.doi.org/ 10.1016/j.ccr.2010.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Serra V, Scaltriti M, Prudkin L, Eichhorn PJ, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M, Rodriguez S, et al.. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 2011; 30(22):2547-57; PMID:21278786; http://dx.doi.org/ 10.1038/onc.2010.626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghosh JC, Siegelin MD, Vaira V, Faversani A, Tavecchio M, Chae YC, Lisanti S, Rampini P, Giroda M, Caino MC, et al.. Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J Natl Cancer Inst 2015; 107(3): PMID:25650317; http://dx.doi.org/ 10.1093/jnci/dju502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caino MC, Ghosh JC, Chae YC, Vaira V, Rivadeneira DB, Faversani A, Rampini P, Kossenkov AV, Aird KM, Zhang R, et al.. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc Natl Acad Sci U S A 2015; 112(28):8638-43; PMID:26124089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007; 129(7):1261-74; PMID:17604717; http://dx.doi.org/ 10.1016/j.cell.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou H, Huang S. Role of mTOR signaling in tumor cell motility, invasion and metastasis. Curr Protein Pept Sci 2011; 12(1):30-42; PMID:21190521; http://dx.doi.org/ 10.2174/138920311795659407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 2004; 6(11):1122-8; PMID:15467718; http://dx.doi.org/ 10.1038/ncb1183 [DOI] [PubMed] [Google Scholar]
- 13.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 2004; 14(14):1296-302; PMID:15268862; http://dx.doi.org/ 10.1016/j.cub.2004.06.054 [DOI] [PubMed] [Google Scholar]
- 14.Caino MC, Chae YC, Vaira V, Ferrero S, Nosotti M, Martin NM, Weeraratna A, O'Connell M, Jernigan D, Fatatis A, et al.. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J Clin Invest 2013; 123(7):2907-20; PMID:23921130; http://dx.doi.org/ 10.1172/JCI67841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong PM, Puente C, Ganley IG, Jiang X. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 2013; 9(2):124-37; PMID:23295650; http://dx.doi.org/ 10.4161/auto.23323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra-Panickar D, Yang CC, Sheffler DJ, et al.. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell 2015; 59(2):285-97; PMID:26118643; http://dx.doi.org/ 10.1016/j.molcel.2015.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol 2011; 8(4):210-21; PMID:21364524; http://dx.doi.org/ 10.1038/nrclinonc.2011.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price JT, Quinn JM, Sims NA, Vieusseux J, Waldeck K, Docherty SE, Myers D, Nakamura A, Waltham MC, Gillespie MT, et al.. The heat shock protein 90 inhibitor, 17-allylamino-17-demethoxygeldanamycin, enhances osteoclast formation and potentiates bone metastasis of a human breast cancer cell line. Cancer Res 2005; 65(11):4929-38; PMID:15930315; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-4458 [DOI] [PubMed] [Google Scholar]
- 19.Paraiso KH, Das Thakur M, Fang B, Koomen JM, Fedorenko IV, John JK, Tsao H, Flaherty KT, Sondak VK, Messina JL, et al.. Ligand-independent EPHA2 signaling drives the adoption of a targeted therapy-mediated metastatic melanoma phenotype. Cancer Discov 2015; 5(3):264-73; PMID:25542447; http://dx.doi.org/ 10.1158/2159-8290.CD-14-0293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tenbaum SP, Ordóñez-Morán P, Puig I, Chicote I, Arqués O, Landolfi S, Fernández Y, Herance JR, Gispert JD, Mendizabal L, et al.. β-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat Med 2012; 18(6):892-901; PMID:22610277; http://dx.doi.org/ 10.1038/nm.2772 [DOI] [PubMed] [Google Scholar]
- 21.Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles' heel? Nat Rev Cancer 2014; 14(11):709-21; PMID:25342630; http://dx.doi.org/ 10.1038/nrc3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wallace DC. Mitochondria and cancer. Nat Rev Cancer 2012; 12(10):685-98; PMID:23001348; http://dx.doi.org/ 10.1038/nrc3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alirol E, Martinou JC. Mitochondria and cancer: is there a morphological connection? Oncogene 2006; 25(34):4706-16; PMID:16892084; http://dx.doi.org/ 10.1038/sj.onc.1209600 [DOI] [PubMed] [Google Scholar]
- 24.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science 2012; 337(6098):1062-5; PMID:22936770; http://dx.doi.org/ 10.1126/science.1219855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desai SP, Bhatia SN, Toner M, Irimia D. Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys J 2013; 104(9):2077-88; PMID:23663851; http://dx.doi.org/ 10.1016/j.bpj.2013.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Attanasio F, Caldieri G, Giacchetti G, van Horssen R, Wieringa B, Buccione R. Novel invadopodia components revealed by differential proteomic analysis. Eur J Cell Biol 2011; 90(2-3):115-27; PMID:20609496; http://dx.doi.org/ 10.1016/j.ejcb.2010.05.004 [DOI] [PubMed] [Google Scholar]
- 27.Ferreira-da-Silva A, Valacca C, Rios E, Pópulo H, Soares P, Sobrinho-Simões M, Scorrano L, Máximo V, Campello S. Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS One 2015; 10(3):e0122308; PMID:25822260; http://dx.doi.org/ 10.1371/journal.pone.0122308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW, Tu Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013; 32(40):4814-24; PMID:23128392; http://dx.doi.org/ 10.1038/onc.2012.494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Infante AS, Stein MS, Zhai Y, Borisy GG, Gundersen GG. Detyrosinated (Glu) microtubules are stabilized by an ATP-sensitive plus-end cap. J Cell Sci 2000; 113 (Pt 22):3907-19; PMID:11058078 [DOI] [PubMed] [Google Scholar]
- 30.Chen Y, Sheng ZH. Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J Cell Biol 2013; 202(2):351-64; PMID:23857772; http://dx.doi.org/ 10.1083/jcb.201302040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang X, Kovalenko OV, Tang W, Claas C, Stipp CS, Hemler ME. Palmitoylation supports assembly and function of integrin-tetraspanin complexes. J Cell Biol 2004; 167(6):1231-40; PMID:15611341; http://dx.doi.org/ 10.1083/jcb.200404100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer 2010; 10(3):181-93; PMID:20168319; http://dx.doi.org/ 10.1038/nrc2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Godoy JA, Arrázola MS, Ordenes D, Silva-Alvarez C, Braidy N, Inestrosa NC. Wnt-5a ligand modulates mitochondrial fission-fusion in rat hippocampal neurons. J Biol Chem 2014; 289(52):36179-93; PMID:25336659; http://dx.doi.org/ 10.1074/jbc.M114.557009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hahn WS, Kuzmicic J, Burrill JS, Donoghue MA, Foncea R, Jensen MD, Lavandero S, Arriaga EA, Bernlohr DA. Proinflammatory cytokines differentially regulate adipocyte mitochondrial metabolism, oxidative stress, and dynamics. Am J Physiol Endocrinol Metab 2014; 306(9):E1033-45; PMID:24595304; http://dx.doi.org/ 10.1152/ajpendo.00422.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryu SW, Yoon J, Yim N, Choi K, Choi C. Downregulation of OPA3 is responsible for transforming growth factor-β-induced mitochondrial elongation and F-actin rearrangement in retinal pigment epithelial ARPE-19 cells. PLoS One 2013; 8(5):e63495; PMID:23658835; http://dx.doi.org/ 10.1371/journal.pone.0063495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Todd LR, Damin MN, Gomathinayagam R, Horn SR, Means AR, Sankar U. Growth factor erv1-like modulates Drp1 to preserve mitochondrial dynamics and function in mouse embryonic stem cells. Mol Biol Cell 2010; 21(7):1225-36; PMID:20147447; http://dx.doi.org/ 10.1091/mbc.E09-11-0937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beck H, Flynn K, Lindenberg KS, Schwarz H, Bradke F, Di Giovanni S, Knöll B. Serum response factor (SRF)-cofilin-actin signaling axis modulates mitochondrial dynamics. Proc Natl Acad Sci U S A 2012; 109(38):E2523-32; PMID:22927399; http://dx.doi.org/ 10.1073/pnas.1208141109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, Dillin A, Mercola M, Ronai ZA. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell 2011; 44(4):532-44; PMID:22099302; http://dx.doi.org/ 10.1016/j.molcel.2011.08.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han XJ, Yang ZJ, Jiang LP, Wei YF, Liao MF, Qian Y, Li Y, Huang X, Wang JB, Xin HB, et al.. Mitochondrial dynamics regulates hypoxia-induced migration and antineoplastic activity of cisplatin in breast cancer cells. Int J Oncol 2015; 46(2):691-700; PMID:25434519 [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Lim S, Hoffman D, Aspenstrom P, Federoff HJ, Rempe DA. HUMMR, a hypoxia- and HIF-1alpha-inducible protein, alters mitochondrial distribution and transport. J Cell Biol 2009; 185(6):1065-81; PMID:19528298; http://dx.doi.org/ 10.1083/jcb.200811033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zanelli SA, Trimmer PA, Solenski NJ. Nitric oxide impairs mitochondrial movement in cortical neurons during hypoxia. J Neurochem 2006; 97(3):724-36; PMID:16606371; http://dx.doi.org/ 10.1111/j.1471-4159.2006.03767.x [DOI] [PubMed] [Google Scholar]
- 42.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A 2011; 108(25):10190-5; PMID:21646527; http://dx.doi.org/ 10.1073/pnas.1107402108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsu PP, Sabatini DM. Cancer cell metabolism: warburg and beyond. Cell 2008; 134(5):703-7; PMID:18775299; http://dx.doi.org/ 10.1016/j.cell.2008.08.021 [DOI] [PubMed] [Google Scholar]
- 44.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 2010; 107(19):8788-93; PMID:20421486; http://dx.doi.org/ 10.1073/pnas.1003428107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al.. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009; 458(7239):762-5; PMID:19219026; http://dx.doi.org/ 10.1038/nature07823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morita M, Gravel SP, Chénard V, Sikström K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, et al.. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab 2013; 18(5):698-711; PMID:24206664; http://dx.doi.org/ 10.1016/j.cmet.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 47.Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, et al.. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014; 514(7524):628-32; PMID:25119024; http://dx.doi.org/ 10.1038/nature13611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Körbel C, Laschke MW, Gimotty PA, Philipp SE, et al.. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013; 23(6):811-25; PMID:23764003; http://dx.doi.org/ 10.1016/j.ccr.2013.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chae YC, Angelin A, Lisanti S, Kossenkov AV, Speicher KD, Wang H, Powers JF, Tischler AS, Pacak K, Fliedner S, et al.. Landscape of the mitochondrial Hsp90 metabolome in tumours. Nat Commun 2013; 4:2139; PMID:23842546; http://dx.doi.org/ 10.1038/ncomms3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cole A, Wang Z, Coyaud E, Voisin V, Gronda M, Jitkova Y, Mattson R, Hurren R, Babovic S, Maclean N, et al.. Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2015; 27(6):864-76; PMID:26058080; http://dx.doi.org/ 10.1016/j.ccell.2015.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov 2010; 9(6):447-64; PMID:20467424; http://dx.doi.org/ 10.1038/nrd3137 [DOI] [PubMed] [Google Scholar]
- 52.Kang BH, Plescia J, Song HY, Meli M, Colombo G, Beebe K, Scroggins B, Neckers L, Altieri DC, et al.. Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J Clin Invest 2009; 119(3):454-64; PMID:19229106; http://dx.doi.org/ 10.1172/JCI37613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 2007; 131(2):257-70; PMID:17956728; http://dx.doi.org/ 10.1016/j.cell.2007.08.028 [DOI] [PubMed] [Google Scholar]
- 54.Chae YC, Caino MC, Lisanti S, Ghosh JC, Dohi T, Danial NN, Villanueva J, Ferrero S, Vaira V, Santambrogio L, et al.. Control of tumor bioenergetics and survival stress signaling by mitochondrial HSP90s. Cancer Cell 2012; 22(3):331-44; PMID:22975376; http://dx.doi.org/ 10.1016/j.ccr.2012.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Joo JH, Dorsey FC, Joshi A, Hennessy-Walters KM, Rose KL, McCastlain K, Zhang J, Iyengar R, Jung CH, Suen DF, et al.. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol Cell 2011; 43(4):572-85; PMID:21855797; http://dx.doi.org/ 10.1016/j.molcel.2011.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Siegelin MD, Dohi T, Raskett CM, Orlowski GM, Powers CM, Gilbert CA, Ross AH, Plescia J, Altieri DC. Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells. J Clin Invest 2011; 121(4):1349-60; PMID:21364280; http://dx.doi.org/ 10.1172/JCI44855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kang BH, Siegelin MD, Plescia J, Raskett CM, Garlick DS, Dohi T, Lian JB, Stein GS, Languino LR, Altieri DC. Preclinical characterization of mitochondria-targeted small molecule hsp90 inhibitors, gamitrinibs, in advanced prostate cancer. Clin Cancer Res 2010; 16(19):4779-88; PMID:20876793; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-1818 [DOI] [PMC free article] [PubMed] [Google Scholar]