

Abstract
P73, the homolog of p53, exists in 2 major forms: either as a pro-apoptotic TAp73 or an amino-terminally truncated DNp73, the latter lacking the first transactivation domain. While TAp73s tumor suppressive functions have been established, DNp73 is an anti-apoptotic protein conferring chemoresistance and is associated with poor survival. However, both forms are variably overexpressed in many human cancers. In this context, we have recently demonstrated that TAp73 is stabilized by hypoxia, a tumor-relevant condition that is associated with cell survival, via HIF-1α-mediated suppression of Siah1 E3 ligase that degrades TAp73. Consequently, hypoxic signals lead to TAp73-mediated activation of several angiogenic genes and blood vessel formation, thereby supporting tumorigenesis. We show here that, similar to TAp73, DNp73 is stabilized by hypoxia in a HIF-1α-dependent manner, which otherwise is degraded by Siah1. Moreover, DNp73 is capable of inducing the expression of Vegf-A, the prototypic angiogenic gene, and loss of DNp73 expression results in reduction in tumor vasculature and size. These data therefore indicate a common mode of regulation for both p73 forms by hypoxia, resulting in the promotion of angiogenesis and tumor growth, highlighting common functionality of these antagonistic proteins under specific physiological contexts.
Keywords: angiogenesis, DNp73, HIF-1α, hypoxia, Siah1, tumors, Vegf-A
Introduction
Homology among proteins often reflects common functionality, though structural variations due to truncations or addition of other domains could not only lead to loss of such common functions, but also to the acquisition of novel or even opposing ones. One group of proteins with such characteristics is the p53 family of tumor suppressors, which include the p63 and p73 proteins.1,2 Among them, while p53 is a classical tumor suppressor, p73 exists as either a tumor suppressor (the TAp73 form), or as the anti-apoptotic DNp73 form, the latter due to truncation of the amino-terminal transactivation domain.3 Expectedly, TAp73 has been shown to exhibit tumor suppressive properties in both cellular and animal model systems, whereas DNp73 expression is often associated with resistance to apoptosis and therapy, due to its ability to inhibit both p53 and TAp73.4,5 Consistently, cell death-inducing stress signals often lead to the stabilization and activation of TAp73, with the concomitant destabilization of DNp73,6-8 thus allowing apoptosis to ensue. However, clinical data are not entirely consistent with TAp73 being a classical tumor suppressor. Both TAp73 and DNp73 are often overexpressed in a variety of human cancers, without being mutated.4,9-12 Two possible scenarios could explain these clinical observations: firstly, DNp73 could negate the tumor suppressive properties of TAp73 when both are co-overexpressed, thereby providing a survival advantage. However, this reasoning does not explain all the cases where only TAp73 may be overexpressed.9,10 Alternatively, both TAp73 and DNp73 may have other common functions consistent with the promotion of carcinogenesis. We and others have previously demonstrated that TAp73 can indeed support cellular survival in combination with activator-protein (AP)-1 factors such as c-Jun, that are often activated in cancers,13,14 and via the transcriptional activation of genes involved in G1-S and G2-M progression,15 suggesting that contextual settings could dictate TAp73s functions. Additionally, growth promoting signals such as serum stimulation has been shown to activate TAp73.16 We have therefore been investigating if other physiologically relevant stimuli can promote p73s pro-proliferative properties. In this context, we have recently shown that hypoxia, a condition that is prevalent in cancers and contributes to chemo-resistance and cellular survival,17-19 leads to the stabilization of the TAp73 form, which allows the activation of the angiogenic transcriptional program to promote angiogenesis, and hence supports tumorigenesis.20 Given that the DNp73 form shares high sequence and structural homology to TAp73 and its role in cellular survival, we have examined if DNp73 is also subjected to comparable regulation by hypoxia, and its influence on angiogenesis. The results suggest a common mode of activation with similar consequential effects on tumor angiogenesis and growth.
Results
DNp73, like TAp73, is stabilized by hypoxia
We first determined if DNp73 could be stabilized by hypoxia. Since endogenous DNp73 expression is not detectable by conventional methods, we transfected the plasmids of both the α and β forms of TAp73 and DNp73 in p53 null H1299 cells followed by overnight exposure to hypoxia – ascertained by the upregulation of vegf-A and glut-1, key hypoxic target genes – which led to a significant increase in steady-state of both these forms (Fig. 1A), that occurred in a time-dependent manner (Fig. 1B). Similar effects were observed by treatment with deferoxamine (DFX) or dimethyloxaloylglycine (DMOG), prolyl hydroxylase inhibitors that mimic hypoxic conditions (Fig. 1C). Elevation of DNp73β following hypoxia correlated with increased half-life (T1/2) from ∼6 h to >9 h as measured by cycloheximide pulse-chase (Fig. 1D), similar to TAp73 as reported earlier.20 Moreover, the hypoxia-mediated upregulation of DNp73β was gradually reversed upon re-oxygenation (Fig. 1E), highlighting the oxygen-mediated regulation of DNp73 expression. These data therefore demonstrate that hypoxia stabilizes DNp73, in a similar fashion to TAp73.
Figure 1.
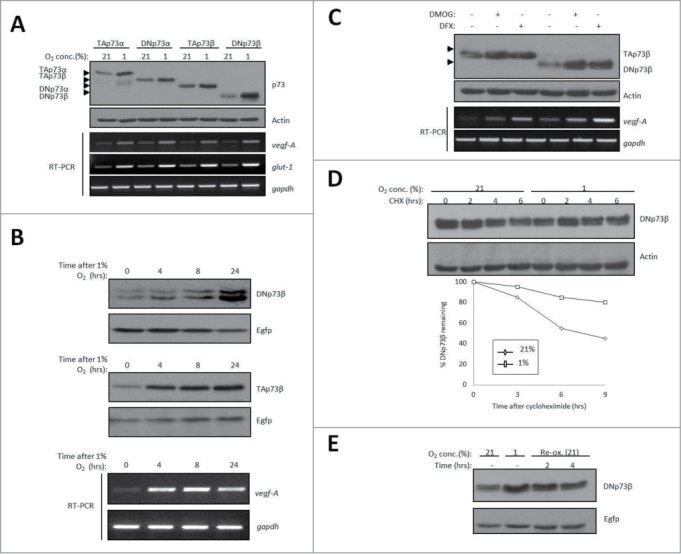
Hypoxia. Induces the stabilization of DNp73. (A-C) Levels of transfected DNp73α and DNp73β, as well as TAp73α and TAp73β in H1299 cells were assessed by immunoblot upon exposure to 1% O2 overnight (A) or for the indicated time points (B), or in the presence of DMOG (1 mM) and DFX (250 μM) (C,). Egfp levels are shown for equal transfection efficiency. Endogenous vegf-A and glut-1 expression relative to gapdh was measured in the same experiment by real-time qPCR analysis (lower panels). Arrowheads indicate position of the respective p73 forms. Part of (A) is reproduced from Dulloo et al., 2015.20 (D) Half-life of DNp73β in H1299 cells was measured in the presence of 1% O2 upon addition of 50 μg/ml cycloheximide (CHX) for the indicated time-periods (upper panel) and quantified (lower panel).(E) Levels of transfected DNp73β in H1299 cells were assessed upon exposure to 1% O2 for 24 hours, and upon re-oxygenation (Re-ox.) at 21% O2 for 2 hours and 4 hours.
DNp73 stabilization is dependent on HIF-1α and SIAH1
As TAp73 was regulated by the HIF-1α/Siah1 axis,20 and given the high similarity of both these major p73 forms, we next examined if this will also be the case with DNp73. However, to rule out the involvement of cullin-based Von Hippel-Lindau (VHL) E3 ligase complex, which is a critical regulator of HIF-1α expression,21 we utilized cells inducibly expressing the dominant-negative C111S mutant Nedd8-conjugating enzyme Ubc12 (dnUbc12), capable of inhibiting all cullin-based ligases,22 as shown by the increase of endogenous p27 levels (Fig. 2A), in which DNp73 was transfected. Like TAp73, hypoxia-mediated upregulation and re-oxygenation-mediated reversal of DNp73β steady-state levels were not affected by dnUbc12 expression, ruling out a role for VHL in DNp73 regulation by hypoxia. We next tested the role of HIF-1α in DNp73 stabilization. Silencing of hif-1α led to a decrease in the levels of DNp73 even in normoxic conditions, and the increase upon hypoxia was compromised, similar to TAp73 (Fig. 2B). In addition, co-expression of wild-type Siah1 (Siah1-WT), but not the catalytic mutant (Siah1-ΔRING), with DNp73β led to the marked reduction of the latter's steady-state levels (Fig. 2C). Moreover, silencing of siah1 upregulated the basal level of DNp73 and rescued the decrease in its level observed in the presence of hif-lα siRNA (Fig. 2D). Thus, these data indicate that DNp73 is stabilized by HIF-1α, which acts upstream of Siah1 to negate its destabilizing activity on DNp73 under hypoxic conditions.
Figure 2.
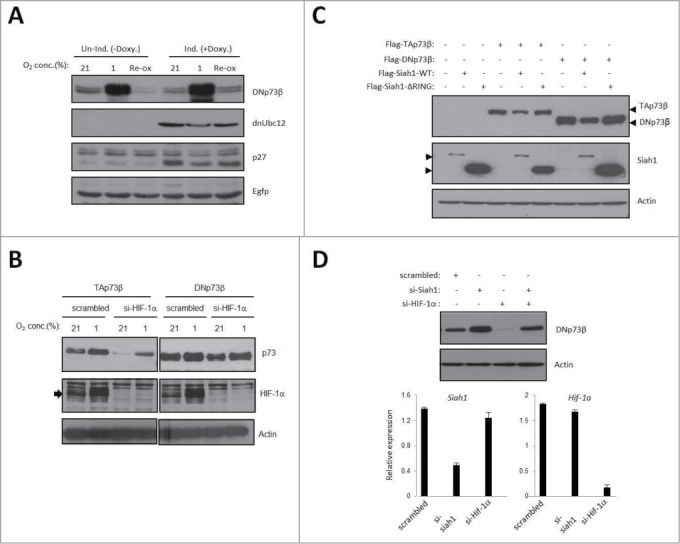
HIF-1α and Siah1 regulate DNp73 levels. (A) Effects of 1% O2 and re-oxygenation (Re-ox.) with 21% O2 on transfected DNp73β in HEK293 cells inducibly expressing the dominant negative form of Ubc12 (dnUbc12) were assessed, as described.20. (B) Levels of TAp73β and DNp73β upon hypoxia were determined after silencing the expression of hif-1α in H1299 cells. Arrowhead indicates HIF-1α. (C) Effects of co-expression of either wild-type or ΔRING mutant Siah1 on TAp73β and DNp73β levels in H1299 cells are shown. Arrowheads indicate position of the respective p73 forms. Arrowheads indicate position of the respective Siah proteins. (D) Effect of siRNA-mediated siah1 silencing alone or after co-silencing with hif-1α on transfected DNp73β levels in H1299 cells. Efficiency of knockdown for both siRNAs is shown by real-time qPCR in the lower panel.
DNp73 regulates Vegf-A expression and angiogenesis
Consequential effect of DNp73 stabilization by hypoxia was evaluated by the analysis of vegf-A expression, the prototypic angiogenic gene regulated by hypoxia. Real-time qPCR analysis of endogenous vegf-A upon transfection of various p73 plasmids revealed that similar to TAp73β and HIF1-α, DNp73β was capable of inducing vegf-A expression (Fig. 3A). However, DNp73β was incapable of transactivating Mdm2 efficiently, unlike TAp73β, highlighting that the amino-terminal transactivation domain of TAp73 might not be involved in vegf-A regulation. Similar results were obtained using the vegf-A promoter linked to a luciferase reporter gene (Fig. 3B). To evaluate if DNp73 binds to the same site as TAp73 on vegf-A promoter,20 we performed chromatin immunoprecipitation using anti-p73 antibody in cells transfected with either TAp73β or DNp73β. As shown in Figure. 3C, there was specific binding of both TAp73β and DNp73β onto the same site on the vegf-A promoter, which does not contain a HIF1-α binding site,20 whereas TAp73β was much more proficient in Mdm2 promoter binding than DNp73. Furthermore, endogenous vegf-A expression in mouse embryonic fibroblasts lacking DNp73 was reduced both at the basal level and particularly upon exposure to hypoxia (Fig. 3D), without any effect on glut-1 expression, altogether highlighting DNp73s role in the specific regulation of vegf-A expression, likely in a manner independent of HIF1-α promoter binding site.
Figure 3.
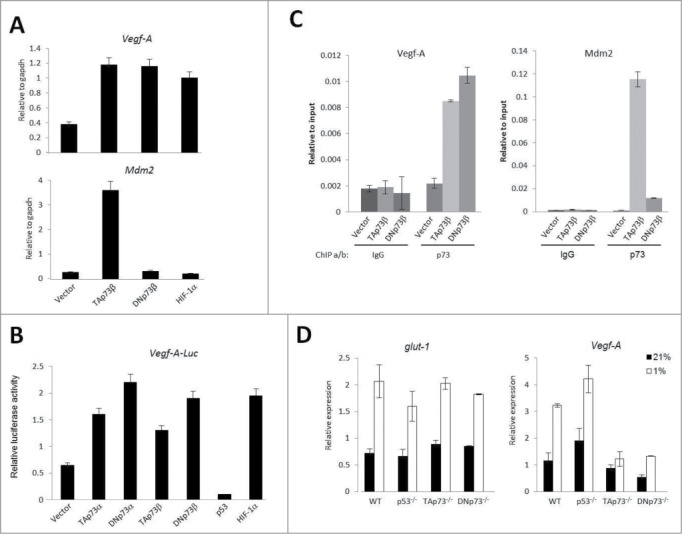
Vegf-A expression is induced by DNp73β. (A) Vegf-A and Mdm2 levels were determined by real-time qPCR analysis after transfection of indicated plasmids in H1299 cells. (B) Effect of indicated plasmids on Vegf-A promoter luciferase construct (Vegf-A-Luc) was assessed by luciferase assay in H1299 cells. (C) Chromatin immunoprecipitation (ChIP) analyses with anti-p73 and anti-IgG antibodies on promoters of Vegf-A and Mdm2 were carried out using H1299 cells inducibly expressing either TAp73β or DNp73β after 8 hours induction. Real-time qPCR was used to quantify the fragments pulled-down with anti-p73 and IgG antibodies, relative to input. (D) Vegf-A and glut-1 levels were determined by real-time qPCR analysis in wild-type, p53−/−, TAp73−/− and DNp73−/− MEFs upon exposure to 1% O2 for 4 hours.
We finally examined the effects of DNp73 absence on in vivo tumor angiogenesis. Using E1a/Ras transformed DNp73−/− and DNp73+/+ MEFs, which were injected into nude mice, we observed a reduction in the tumor volume/size due to the absence of DNp73 (tumor size [mm3] - DNp73−/− vs. DNp73+/+ MEFs: 384.91 vs. 1031.37) (Fig. 4A). Histological analysis of these tumors for CD34 staining to detect blood vessels revealed a correspondingly significant reduction in blood vessel density DNp73 (Fig. 4B). Collectively, these data indicate that similar to TAp73, DNp73 is also an important regulator of vegf-A expression and efficient blood vessel formation in tumors.
Figure 4.
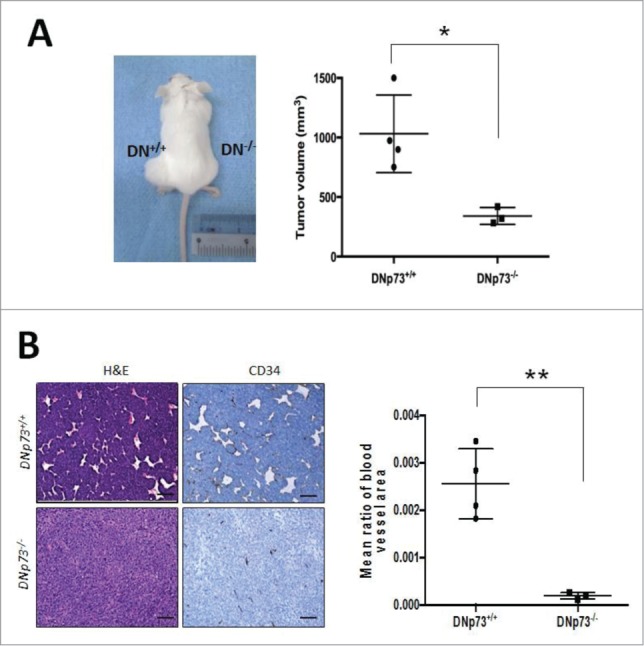
DNp73 supports tumor formation and angiogenesis. (A and B) SCID mice were injected subcutaneously on either flanks with E1a/Ras transformed DNp73+/+ and DNp73−/− cells and tumors were analyzed for tumor growth and blood vessel formation. Graph shows the size of tumors derived from each mouse [each dot represents a mouse/tumor; n = 4 (WT), n = 3 (KO)] with a representative mouse shown (A). Tumors were stained for H&E and CD34 (B). Scale bars: 100μm. Image is representative of all tumors analyzed. *p<0 .05 **p<0 .01.
Discussion
The data presented here, together with that in our recent publication,20 alludes to a scenario in which both TAp73 and DNp73 are stabilized by hypoxia through a mechanism involving HIF-1α-mediated Siah1 suppression, leading to the subsequent activation of vegf-A and an array of angiogenic genes, thereby resulting in enhanced tumor blood vessel formation. These data thus highlight a common pro-survival role for both these p73 forms, which in other contexts such as in response to DNA damage,23-25 are differentially regulated to perform opposite functions on cell survival. It is interesting to note that although TAp73 is often induced by multiple stress signals leading to its stabilization, DNp73 is often degraded by similar stimuli,23-25 with the net effect of controlling cellular proliferation and inducing death. Therefore, it has always been expected that this inverse correlation is maintained for the tumor suppressive effects of TAp73 to be felt. However, human tumors often exhibit elevated TAp73, DNp73 or both, insinuating that other possible scenarios can occur. Our data indicate that tumor-associated hypoxia is a condition that is exception to the reciprocal regulation of TAp73 and DNp73; positively regulating both p73 forms, thereby supporting survival.
It is noteworthy that upregulation of both these members by hypoxia lead to transactivation of the canonical angiogenic gene, vegf-A. Although DNp73 lacks the amino-terminal transactivation domain,1,3 it has been shown to retain some transactivation potential due to the presence of a putative second transactivation domain, which includes the N-terminal 13 unique amino acid residues and the PXXP motif, and was shown to weakly activate targets such as p21, 14–3–3 and Gadd45.26 Additionally, it has been reported that the C-terminal end of p73 within amino acid residues 381 to 399, common in both TAp73 and DNp73, can function as a putative transactivation domain required for preferential regulation of genes involved in cell cycle progression.27 We have also recently shown that both TAp73 and DNp73 are capable of inducing the transactivation of caspase-2S, an anti-apoptotic member of the caspase family,28 alluding to a distinct transactivation domain common to both these forms that is required for the regulation of specific target genes. An interesting aspect that emerges from these findings is that the dominant-negative effect of DNp73 on TAp73 might be lost in growth-promoting conditions such as hypoxia. Whether stimuli-specific protein modifications and/or recruitment of interactors enable this phenomenon is currently under investigation.
A recent publication using cells deficient for both TAp73 and DNp73 also alluded to the requirement for p73 in vasculogenesis, especially in the context of development.29 Moreover, knockdown of DNp73 relative to that of TAp73, was noted to have a more profound effect, supporting the data that both forms contribute to the angiogenic process, perhaps to varying degrees. However, loss of either TAp73 or DNp73 alone appears to be sufficient to retard tumor growth and angiogenesis,20,30 suggesting that while both proteins have a role in promoting angiogenesis, they may not be able to compensate for the loss of the other. This alludes to the possibility of TAp73 and DNp73 having both overlapping as well as distinct targets that are critical in regulating the angiogenic process, highlighting probable non-redundant functions of TAp73 and DNp73 in tumorigenesis, as well as non-overlapping functions that could be spatially and temporally controlled. Nonetheless, common growth promoting signals appear to similarly regulate both the major p73 forms, to enable the concerted activation of target genes to promote survival, such as in cancers. Thus, understanding the similarity and dichotomy in the regulation of both these forms under specific physiological contexts will enable better strategies to be developed for their modulation in pathological contexts.
In conclusion, work from us and others demonstrate a common role for both the TAp73 and DNp73 forms in promoting tumor growth and angiogenesis, suggesting that these proteins could serve as targets as well as a biomarker for anti-angiogenic therapy.
Materials and Methods
Cells and reagents
Cells lines used were as follows: human cell lines - p53 null H1299 lung adenocarcinoma cells; H1299 cells stably expressing TAp73β or DNp73β; HEK293 human embryonic kidney cells that stably express the mutant dnUbc12-HA under tet-on system. Mouse fibroblasts – DNp73+/+ and DNp73−/− (gift from Dr. Tak Mak) stably expressing E1a and Ras constructs, p53−/− and wild-type MEFs. Two μg/ml of doxycycline (Sigma-Aldrich) was used for induction. All cells were tested for mycoplasma contamination. Chemicals used: dimethyloxaloylglycine, deferoxamine (Sigma-Aldrich).
Hypoxia was induced by incubating cells in a modular incubator chamber (Billups-Rothenberg) containing 1% O2 at 37°C for 24 hours, unless otherwise stated. Re-oxygenation was carried out by placing the hypoxic cells back at 21% O2, for indicated time points. For half-life determination under hypoxia, medium containing 50 μg/ml cycloheximide was de-gased 2 hours before adding to hypoxic cells.
Plasmids, siRNAs and transfection
Plasmids used in this study: pCDNA3.1, Flag-TAp73β, TAp73β, Flag-DNp73β, DNp73β, p53, β-gal, Egfp. Vegf-A-luc was a gift from Dr. Fatima Mechta-Grigoriou. Wild-type Siah1 and ΔRING mutant Siah1 Flag-tagged plasmids were gifts from Dr. Shu-Ichi Matsuzawa. Two×105 cells were transfected with the various plasmids (0.2–0.5 μg of DNA) using Lipofectamine Plus-Reagent (Life Technologies). Cells were generally collected 24 hours after transfection for luciferase analyses and immunoblotting. siRNA transfections were performed using Transmessenger transfection kit (Qiagen) and cells were harvested 72 hours post transfection. Target sequence of siRNAs used: human hif-1α: 5′ CTAACTGGACACAGTGTGT 3′; human Siah1: 5′GATAGGAACACGCAAGCAA 3′.
Luciferase assay
Cells were transiently transfected with 0.2 μg of the relevant plasmids along with the reporter construct (Vegf-A-luc) and 0.1 μg of β-galactosidase construct to normalize for transfection efficiency. Luciferase assays were performed in triplicates as described.13
Real-time qPCR analyses
Total RNA was prepared from cells using TRIZOL Reagent (Life Technologies) and converted into single-strand cDNA using Superscript II Reverse Transcriptase (Life Technologies). Real-time quantitative RT-PCR (qPCR) was carried out using Rotor-Gene 6000 (Corbett Life Science) and Maxima® SYBR Green (Fermentas) according to manufacturers' protocol. Primers for RT-PCR used in this study are: mouse/human- GLUT1-Fwd: 5′ TGGATGTCCTATCTGAGCATCGTG 3′; GLUT1-Rev: 5′ CTTTGAAGTAGGTGAAGATGAAGA 3′; VEGFA-Fwd: 5′ ATGCCAAGTGGTCCCAGGCTGCAC 3′; VEGFA-Rev: 5′ TGTGCTGTAGGAAGCTCATCTCTC 3′. human- MDM2-Fwd: 5′ GAATCATCGGACTCAGGTACATC 3′; MDM2-Rev: 5′ TCTGTCTCACTAATTGCTCTCCT 3′.
Immunoblotting
Immunoblot analysis was performed essentially as described.6 Typically, 50 μg of protein lysate were used for immunoblot. Detection was done using enhanced Chemiluminescent reagent (Amersham) or Super signal West Dura (Pierce). Antibodies used in this study are: anti-p73 (ER15; 1:1000; Calbiochem), anti-p73 (GC15; 1:1000; Calbiochem), anti-FLAG (M2; 1:2000; Stratagene), anti-p27 (C-19; 1:500; Santa Cruz Biotechnology), anti-Actin (AC-74, 1:5000; Sigma-Aldrich), anti-Egfp (B-2; 1:2000; Santa Cruz Biotechnology), anti-HIF-1α (1:250; BD Biosciences).
Chromatin immunoprecipitation
Inducible cells were fixed with 1% formaldehyde for 10 mins at room temperature, and the reaction was stopped by addition of glycine to final concentration of 125 mM for 15 mins. Cells were then processed as described.20 For all ChIP experiments, real-time qPCR analyses were performed. Relative occupancy values were calculated after normalization with ChIP input DNA. All ChIP experiments were repeated at least 3 times. The primers used for real-time qPCR to quantify the ChIP-enriched DNA are: MDM2-Fwd: 5′ GATCGCAGGTGC-CTGTCGGGTCACTA 3′; MDM2-Rev: 5′ GGTCTACCCTCCAATCGCCACTGAACACA 3′; -1162-VEGFA-Fwd: 5′ CTACAGACGTTCCTTAGTGCTG 3′; -800-VEGFA-Rev: 5′ CTCATCTGGC-CTGCA GACATCA 3′
Mouse xenograft studies
E1a/RAS transformed MEFs (2×106 per 0.1 ml of PBS) were injected sub-cutaneously into both the left and right flanks of 5-weeks old immunodeficient SCID female mice. Tumors were allowed to grow for 5 weeks and tumor volume was measured prior to harvest (max tumor size of 1500 mm−3). Tumors were formalin-fixed and used for immunohistochemistry studies.
Numbers of mice for each group are indicated in figure legends. Tumor volume was calculated with the equation V (in mm−3) = a × b2 × 0.5, where a is the longest diameter and b is the shortest diameter. All animal experiments were carried with the approval of the Singhealth Institutional Animal Care and User Committee (IACUC).
Immunohistochemistry
Paraffin sections were processed using Bond Epitope Retrieval Buffer, Bond Leica Automated Staining and Bond Refine Kit (Leica Microsystems). Antibody used for mouse tissues: anti-CD34 (MEC14.7; Santa Cruz Biotechnology), at a dilution of 1:100. All slides were counterstained with haematoxylin. Species- and subtype-matched antibodies were used as negative controls.
Quantification of blood vessels size was done using ImageJ software. Values were calculated based on the mean size of 5 randomly chosen CD34-stained blood vessels for each tumor in each genotype and based on pixel numbers.
Statistical Analysis
Results were expressed as mean ± standard deviation (s.d.) of 3 or more biological replicates and representative data are shown from 3 independent experiments, unless otherwise indicated. Student's t-test was used to determine if the observed difference was statistically significant. All analyzes were done using GraphPad Prism. * p<0 .05; ** p<0 .01.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Drs Fatima Mechta-Grigoriou, Shu-Ichi Matsuzawa and Tak Wah Mak for the vegf-A promoter–luciferase, siah1 WT/ΔRING mutant plasmids, and the DNp73−/− and DNp73+/+ MEFs, respectively.
Author Contributions
ID designed the experiments, performed the research work and analyzed the data. BHP performed the research work and analyzed the data. KS designed the experiments, analyzed the data, supervised the project and wrote the manuscript with the co-authors input. All authors reviewed the manuscript.
Funding
This study was supported by grants from the National Medical Research Council to K.S.
References
- 1.Melino G, De Laurenzi V, Vousden KH. p73: Friend or foe in tumorigenesis. Nat Rev Cancer 2002; 2:605-15; PMID:12154353 [DOI] [PubMed] [Google Scholar]
- 2.Deyoung MP, Ellisen LW. p63 and p73 in human cancer: defining the network. Oncogene 2007; 26:5169-83; PMID:17334395; http://dx.doi.org/ 10.1038/sj.onc.1210337 [DOI] [PubMed] [Google Scholar]
- 3.Ozaki T, Nakagawara A. p73, a sophisticated p53 family member in the cancer world. Cancer Sci 2005; 96:729-37; PMID:16271066; http://dx.doi.org/ 10.1111/j.1349-7006.2005.00116.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stiewe T, Pützer BM. Role of p73 in malignancy: tumor suppressor or oncogene? Cell Death Differ 2002; 9:237-45; PMID:11859406; http://dx.doi.org/ 10.1038/sj.cdd.4400995 [DOI] [PubMed] [Google Scholar]
- 5.Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC, Khan F, Itie-Youten A, Wakeham A, Tsao MS, et al.. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008; 22:2677-91; PMID:18805989; http://dx.doi.org/ 10.1101/gad.1695308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toh WH, Siddique MM, Boominathan L, Lin KW, Sabapathy K. c-Jun regulates the stability and activity of the p53 homologue, p73. J Biol Chem 2004; 279:44713-22; PMID:15302867; http://dx.doi.org/ 10.1074/jbc.M407672200 [DOI] [PubMed] [Google Scholar]
- 7.Rossi M, De Laurenzi V, Munarriz E, Green DR, Liu YC, Vousden KH, Cesareni G, Melino G. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J 2005; 24:836-48; PMID:15678106; http://dx.doi.org/ 10.1038/sj.emboj.7600444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bisso A, Collavin L, Del Sal G. p73 as a pharmaceutical target for cancer therapy. Curr. Pharm Des 2011; 17:578-90; PMID:21391908; http://dx.doi.org/ 10.2174/138161211795222667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang MJ, Park BJ, Byun DS, Park JI, Kim HJ, Park JH, Chi SG. Loss of imprinting and elevated expression of wild-type p73 in human gastric adenocarcinoma. Clin. Cancer Res 2000; 6:1767-71; PMID:10815895 [PubMed] [Google Scholar]
- 10.Zaika AI, Kovalev S, Marchenko ND, Moll UM. Overexpression of the wild type p73 gene in breast cancer tissues and cell lines. Cancer Res 1999; 59:3257-63; PMID:10397274 [PubMed] [Google Scholar]
- 11.Concin N, Becker K, Slade N, Erster S, Mueller-Holzner E, Ulmer H, Daxenbichler G, Zeimet A, Zeillinger R, Marth C, et al.. Transdominant DeltaTAp73 isoforms are frequently up-regulated in ovarian cancer. Evidence for their role as epigenetic p53 inhibitors in vivo. Cancer Res 2004; 64:2449-60; PMID:15059898; http://dx.doi.org/ 10.1158/0008-5472.CAN-03-1060 [DOI] [PubMed] [Google Scholar]
- 12.Domínguez G, García JM, Peña C, Silva J, García V, Martínez L, Maximiano C, Gómez ME, Rivera JA, García-Andrade C, et al.. DeltaTAp73 upregulation correlates with poor prognosis in human tumors: putative in vivo network involving p73 isoforms, p53, and E2F-1. J Clin Oncol 2006; 24:805-15; PMID:16380414; http://dx.doi.org/ 10.1200/JCO.2005.02.2350 [DOI] [PubMed] [Google Scholar]
- 13.Vikhanskaya F, Toh WH, Dulloo I, Wu Q, Boominathan L, Ng HH, Vousden KH, Sabapathy K. p73 supports cellular growth through c-Jun-dependent AP-1 transactivation. Nat. Cell Biol 2007; 9:698-705; PMID:17496887 [DOI] [PubMed] [Google Scholar]
- 14.Subramanian D, Bunjobpol W, Sabapathy K. Interplay between TAp73 and selected Activator Protein-1 family members promotes AP-1 target gene activation and cellular growth. J Biol Chem 2015; 290(30):18636-49; pii: jbc.M115.636548; PMID:26018080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lefkimmiatis K, Caratozzolo MF, Merlo P, D'Erchia AM, Navarro B, Levrero M, Sbisa' E, Tullo A. p73 and p63 sustain cellular growth by transcriptional activation of cell cycle progression genes. Cancer Res 2009; 69:8563-71; PMID:19861536; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-0259 [DOI] [PubMed] [Google Scholar]
- 16.Weiss RH, Howard LL. p73 is a growth-regulated protein in vascular smooth muscle cells and is present at high levels in human atherosclerotic plaque. Cell Signal 2001; 13: 727-33; PMID:11602183; http://dx.doi.org/ 10.1016/S0898-6568(01)00195-4 [DOI] [PubMed] [Google Scholar]
- 17.Harris AL. Hypoxia-a key regulatory factor in tumour growth. Nat Rev Cancer 2002; 2:38-47; PMID:11902584; http://dx.doi.org/ 10.1038/nrc704 [DOI] [PubMed] [Google Scholar]
- 18.Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010; 29:625-34; PMID:19946328; http://dx.doi.org/ 10.1038/onc.2009.441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer 2008; 8:967-75; PMID:19946328; http://dx.doi.org/ 10.1038/nrc2540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dulloo I, Phang BH, Othman R, Tan SY, Vijayaraghavan A, Goh LK, Martin-Lopez M, Marques MM, Li CW, Wang DY, et al.. Hypoxia-inducible TAp73 regulates the angiogenic transcriptome and supports tumorigenesis. Nat Cell Biol 2015; 17:511-23; PMID:25774835; http://dx.doi.org/ 10.1038/ncb3130 [DOI] [PubMed] [Google Scholar]
- 21.Kaelin WG, Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev. Cancer 2008; 8:865-73; PMID:18923434 [DOI] [PubMed] [Google Scholar]
- 22.Chew EH, Poobalasingam T, Hawkey CJ, Hagen T. Characterization of cullin-based E3 ubiquitin ligases in intact mammalian cells–evidence for cullin dimerization. Cell Signal 2007; 19:1071-80; PMID:17254749; http://dx.doi.org/ 10.1016/j.cellsig.2006.12.002 [DOI] [PubMed] [Google Scholar]
- 23.Lin KW, Nam SY, Toh WH, Dulloo I, Sabapathy K. Multiple stress signals induce p73beta accumulation. Neoplasia 2004; 6:546-57; PMID:15548364; http://dx.doi.org/ 10.1593/neo.04205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oberst A, Rossi M, Salomoni P, Pandolfi PP, Oren M, Melino G, Bernassola F. Regulation of the p73 protein stability and degradation. Biochem Biophys Res Commun 2005; 331:707-12; PMID:15865926; http://dx.doi.org/ 10.1016/j.bbrc.2005.03.158 [DOI] [PubMed] [Google Scholar]
- 25.Dulloo D, Gopalan G, Melino G, Sabapathy K. The anti-apoptotic DeltaNp73 is degraded in a c-Jun-dependent manner upon genotoxic stress through the antizyme-mediated pathway. Proc Nat Acad Sci 2010; 107:4902-7; PMID:20185758; http://dx.doi.org/ 10.1073/pnas.0906782107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu G, Nozell S, Xiao H, Chen X. DeltaNp73beta is active in transactivation and growth suppression. Mol Cell Biol 2004; 24:487-501; PMID:14701724; http://dx.doi.org/ 10.1128/MCB.24.2.487-501.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nyman U, Vlachos P, Cascante A, Hermanson O, Zhivotovsky B, Joseph B. Protein kinase C-dependent phosphorylation regulates the cell cycle-inhibitory function of the p73 carboxy terminus transactivation domain. Mol Cell Biol 2009; 29:1814-25; PMID:19158275; http://dx.doi.org/ 10.1128/MCB.00585-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toh WH, Logette E, Corcos L, Sabapathy K. TAp73beta and DNp73beta activate the expression of the pro-survival caspase-2S. Nucleic Acids Res 2008; 36:4498-509; PMID:18611950; http://dx.doi.org/ 10.1093/nar/gkn414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandez-Alonso R, Martin-Lopez M, Gonzalez-Cano L, Garcia S, Castrillo F, Diez-Prieto I, Fernandez-Corona A, Lorenzo-Marcos ME, Li X, Claesson-Welsh L, et al.. p73 is required for endothelial cell differentiation, migration and the formation of vascular networks regulating VEGF and TGFβ signaling. Cell Death Differ 2015; 22(8):1287-99; PMID:25571973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Du W, Jiang P, Mancuso A, Stonestrom A, Brewer MD, Minn AJ, Mak TW, Wu M, Yang X. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat. Cell. Biol 2013; 15:991-1000; PMID:23811687 [DOI] [PMC free article] [PubMed] [Google Scholar]