

Abstract
The clinical success of ALK targeted therapy is limited by resistance. To identify rational co-targeting strategies to enhance clinical outcomes, we explored the molecular basis of ALK oncogene dependence in ALK gene rearrangement positive (ALK+) lung adenocarcinoma. We discovered that the RAS-RAF-MEK-ERK pathway is the critical downstream pathway necessary for ALK+ tumor cell survival. Upfront co-targeting of ALK plus MEK enhanced response and forestalled resistance in preclinical ALK+ tumor models, providing rationale for a new approach the treatment of ALK+ patients.
Keywords: ALK, lung cancer, MAPK, MEK, polytherapy, RAS, resistance
Major advances in the treatment of lung cancer have occurred through the use of molecular targeted therapy acting against certain tumor-promoting genetic alterations present in specific subtypes of the disease.1 However, targeted therapy resistance is a significant problem. Unraveling the basis of resistance is critical to understand the fundamental processes of aberrant cell signaling in cancer cells and to design therapeutic strategies to suppress resistance and improve patient survival.1,2
We recently investigated the biological basis of resistance to targeted therapies against the anaplastic lymphoma kinase (ALK) in ALK gene rearrangement-positive lung adenocarcinoma (ALK+).3 ALK+ disease accounts for ∼5–7% cases of lung adenocarcinoma, and this subset of lung cancer patients is treated with ALK inhibitors such as crizotinib and ceritinib (among other agents currently in clinical trials).3 The initial ALK inhibitor response is typically incomplete in these patients, with acquired resistance occurring with ˜12 months on therapy.4 Furthermore, up to 40% of ALK+ patients fail to respond to initial ALK inhibitor treatment, exhibiting innate resistance.4
Generally, efforts to thwart resistance have focused on treating acquired resistance after it has emerged.2 An alternative approach to enhance initial response and combat acquired resistance is to use rational upfront polytherapies that target the main oncoprotein (such as oncogenic ALK) and an important downstream effector of that oncoprotein. For example, upfront (but not second-line) inhibition of BRAFV600E plus its main effector, MEK1/2, shows activity superior to RAF or MEK inhibitor monotherapy in BRAFV600E-mutant melanoma patients.5,6
The most appropriate upfront polytherapy strategy is less clear in cancers with an oncogenic receptor kinase, such as ALK, that engages multiple downstream pathways.7 We addressed this knowledge gap in ALK+ lung adenocarcinoma to identify a rational upfront polytherapy strategy to enhance patient survival.7 Using models of lung adenocarcinoma harboring the major oncogenic ALK fusion EML4-ALK (echinoderm microtubule associated protein like 4-ALK), we found that the RAS-MAPK pathway, but not other established ALK effectors such as PI-3K (phosphoinositide-3 kinase) or JAK/STAT, is essential for tumor cell survival. EML4-ALK activated RAS-MAPK (mitogen activated protein kinase) signaling via all 3 major RAS GTPase isoforms (H-, N-, K-RAS), acting through the HELP domain of EML4. MAPK pathway reactivation via either KRASWT (wild type) copy number gain or decreased expression of the MAPK phosphatase DUSP6 was sufficient to promote ALK inhibitor resistance in vitro. Furthermore, these molecular events were each associated with ALK inhibitor resistance in ALK+ patients. Dual ALK-MEK inhibition in the upfront setting enhanced both the magnitude and duration of response in ALK+ lung adenocarcinoma preclinical models, in vitro and in vivo. Altogether, our findings revealed RAS-MAPK dependence as a hallmark feature of ALK+ lung adenocarcinoma and indicate that upfront ALK-MEK targeted polytherapy may suppress or prevent resistance and improve patient outcomes.
The findings in our study prompt new areas for investigation. The data raise the possibility that combining an ALK inhibitor such as crizotinib or ceritinib with a MEK such as trametinib could induce complete tumor response in ALK+ patients (Fig. 1). This hypothesis warrants testing in clinical trials. Furthermore, KRASWT copy number and DUSP6 downregulation should be further investigated as novel biomarkers of ALK inhibitor response in ALK+ patients, and potentially other tumor subsets with similar MAPK pathway dependence such as those with oncogenic mutations in RAS-RAF-MEK-ERK or EGFR.
Figure 1.
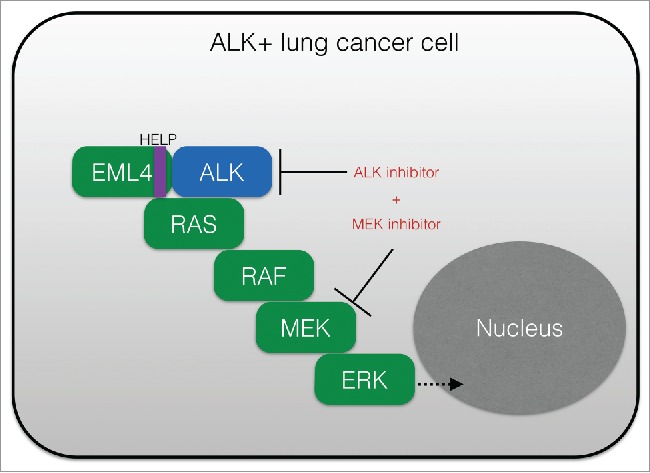
Upfront polytherapy targeting ALK and MEK in ALK+ lung adenocarcinoma. Shown is a schematic of the critical role of RAS-RAF-MEK-ERK (RAS-MAPK) signaling in ALK+ (EML4-ALK positive) tumor cells, and the rational co-targeting strategy to inhibit both ALK and MEK to enhance response. MAPK, mitogen activated protein kinase; ALK, anaplastic lymphoma kinase; HELP, HELP domain in EML4 (echinoderm microtubule associated protein like 4).
Moreover, our findings further suggest that the signaling properties of certain oncogenic fusion kinases (here, EML4-ALK) may be regulated by determinants within the fusion partner of the relevant kinase (here, EML4) (Fig. 1). Beyond a role in promoting dimerization of the kinase, our data indicate that EML4 (specifically its HELP domain) is crucial for proper intracellular localization of EML4-ALK and activation of RAS and downstream RAF-MEK-ERK signaling. Thus, a new layer of regulation of EML4-ALK oncogene function is revealed that could have relevance for the function of other ALK fusion proteins and of additional kinase fusion proteins more generally. The cell biological regulation of EML4-ALK/RAS engagement and signaling, and potentially of other kinase fusion proteins, is an interesting area for future investigation. Coordinated basic and translational studies will further define the role and regulation of RAS-MAPK signaling in ALK+ cancer cells, and those with other oncogenic fusion kinases, with important implications for improving oncogenic fusion-positive patient outcomes.
References
- 1.Pazarentzos E, Bivona TG. Oncogene 2015; PMID:25703329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garraway LA, Janne PA. Cancer Discov 2012; 2:214-26; PMID:22585993; http://dx.doi.org/ 10.1158/2159-8290.CD-12-0012 [DOI] [PubMed] [Google Scholar]
- 3.Hrustanovic G, et al.. Nat Med 2015; 21:1038-47; PMID:26301689; http://dx.doi.org/ 10.1038/nm.3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doebele RC, et al.. Clin Cancer Res 2012; 18:1472-82; PMID:22235099; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson DB, et al.. J Clin Oncol 2014; 32:3697-704; PMID:25287827; http://dx.doi.org/ 10.1200/JCO.2014.57.3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flaherty KT, et al.. N Engl J Med 2012; 367:1694-703; PMID:23020132; http://dx.doi.org/ 10.1056/NEJMoa1210093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hrustanovic G, et al.. Nat Med 2015; 21(9):1038-47; PMID:26301689 [DOI] [PMC free article] [PubMed] [Google Scholar]