

Abstract
Tumor cells with defective apoptosis pathways often respond to chemotherapy by entering irreversible cell cycle arrest with features of senescence. However, rare cells can bypass entry to senescence, or re-enter cell cycle from a senescent state. Deficiency in senescence induction and maintenance may contribute to treatment resistance and early relapse after therapy. Senescence involves epigenetic silencing of cell cycle genes and reduced rRNA transcription. We found that senescence-inducing treatments such as DNA damage and RNA polymerase I inhibition stimulate the binding between the nucleolar protein NML (nucleomethylin) and SirT1. The NML complex promotes rDNA heterochromatin formation and represses rRNA transcription. Depletion of NML reduced the levels of H3K9Me3 and H3K27Me3 heterochromatin markers on rDNA and E2F1 target promoters in senescent cells, increased rRNA transcription, and increased the frequency of cell cycle re-entry. Depletion of the nucleolar transcription repressor factor TIP5 also promoted escape from senescence. Furthermore, tumor tissue staining showed that breast tumors without detectable nucleolar NML expression had poor survival. The results suggest that efficient regulation of nucleolar rDNA transcription facilitates the maintenance of irreversible cell cycle arrest in senescent cells. Deficiency in nucleolar transcription repression may accelerate tumor relapse after chemotherapy.
Keywords: chemotherapy, heterochromatin, nucleolus, NML, p53, rDNA, senescence
Introduction
Replicative senescence was initially defined as a cell culture phenomenon in which extensive passage of primary fibroblasts results in telomere erosion, activation of DNA damage signaling, and irreversible cell cycle arrest.1 Senescent cells exhibit large flat cell morphology, increased p21/p16 levels, active metabolism and autophagy, and increased lysosomal SA-β-gal activity. Oncogene activation and chemotherapy also induce premature senescence that is similar to replicative senescence in cell morphology and the expression profile of molecular markers.2 SA-β-gal staining of human tissues suggests that senescence occurs in vivo in aged tissues, pre-malignant lesions, and in tumors treated with chemotherapy.3
Cellular senescence has complex and highly context-dependent effects for the host organism.1 Secretion of cytokines and matrix metalloproteases by senescent mesenchymal cells (the SASP phenotype) may promote inflammation, accelerated aging, degenerative diseases, and tumor growth.4 Removal of senescent cells in mice has been shown to reduce aging phenotypes.5 Chemotherapy-induced tumor cell senescence may prevent apoptosis and lead to drug resistance.6,7 However, in certain settings drug-induced senescence may mediate the anti-tumor effect of chemotherapy. Mouse models showed that tumor cells with defective apoptosis pathway respond to chemotherapy by entering senescence.8 The SASP phenotype promotes clearance of senescent tumor cells by the immune system.9 In such context, senescence is beneficial and may lead to a stable disease state or tumor clearance after therapy. However, cell culture showed that after drug treatment, rare tumor cells may bypass the initiation of senescence or re-enter cell cycle from a senescent state.10 The escape from senescence may allow a subset of treated tumor cells to evade immune clearance and cause relapse of tumors after therapy.
During induction of senescence in cultured cell, multiple small nucleoli characteristic of proliferating cells often fuse to form a single large nucleolus, indicating significant changes in nucleolar rDNA chromatin structure and function. rRNA synthesis consumes >50% of cellular transcriptional activity, and is tightly coupled to nutrient availability and growth signaling.11 The c-Myc oncogene is a potent activator of rRNA transcription. RNA polymerase I (Pol I) activity is regulated by mTOR, which promotes the recruitment of Pol I to rDNA promoters.12 The basal factor UBF of the Pol I initiation complex is also targeted by the Rb, ARF and p53 tumor suppressor proteins.13-15 Therefore, numerous growth and stress signals converge on regulating Pol I activity and rRNA transcription. Recently the RNA Pol I-specific inhibitor CX5461 has shown promise as an anti-tumor agent in animal models, in part by activating p53 through the nucleolar stress signaling mechanism that inhibits MDM2 function.16,17
Nearly 50% of rDNA repeats are present as heterochromatin in growing cells, which is important for maintaining genetic stability.18 The NoRC (nucleolar remodeling complex) is important for switching rDNA between silent and active state. NoRC is a chromatin remodeling complex that recruits DNMT and HDAC to the promoter to trigger heterochromatin formation and silencing.19 A recent study identified a novel repressor complex (eNoSC) that also regulates rRNA transcription.20 The eNoSC complex contains SirT1, SUV39H1, and a novel nucleolar protein NML (nucleomethylin).20 Knockdown of NML increases rRNA transcription and prevents the inhibition of rRNA synthesis by glucose starvation. NML represses rDNA by promoting H3K9 methylation and establishing heterochromatin across the rDNA. NML has an N terminal half that binds H3K9me2, and a C-terminal domain homologous to SAM-dependent methyltransferase.20 The factors present in the NML complex suggest that it can promote the spreading of heterochromatin marks across the rDNA.
The biochemical properties of NML suggest that it may contribute to the regulation of senescence. Senescence involves stable epigenetic silencing of proliferation genes and reduced rRNA transcription.3 Silenced E2F target genes form heterochromatin foci (SAHF) visible in some senescent human cells.21 Presumably, senescence requires establishing and maintaining positive feedback loops in the heterochromatinization of key proliferation genes. Heterochromatin proteins HP1, SUV39H1, and NML bind to methylated H3K9 and then promote further methylation of adjacent H3K9. Therefore they have the ability to establish self-sustaining silencing when given proper initiation signals. Recent study suggested that the RNA sensing ability of NML provides a novel positive feedback mechanism in regulating rDNA silencing.22
In this report we describe evidence that NML facilitates tumor cells to initiate and maintain irreversible cell cycle arrest during drug-induced senescence. Deficiency in NML expression increases the probability of tumor cell escape from drug-induced senescence, and correlates with poor survival in breast cancer patients. The results reveal a role of nucleolar transcription repression in maintaining cellular senescence, and implicate abnormal NML expression as a marker of poor survival in breast cancer.
Results
Depletion of NML reduces the efficiency of senescence response
To investigate the role of NML in regulating drug-induced senescence, NML was stably knocked down in A549 cells using 2 different shRNA (Fig. 1A). Both A549-NML shRNA cell lines showed increased pre-rRNA level as expected (Fig. 1B). The control shRNA and NML shRNA expressing cells showed similar growth rates under normal culture conditions (data not shown).
Figure 1.

NML knockdown promotes escape from drug-induced senescence. (A) A549 cells were stably infected with retrovirus expressing NML shRNAs. NML knockdown efficiency was confirmed by protein gel blot. (B) Pre-rRNA expression levels in stable NML knockdown cell lines were determined by RT-qPCR. Values are mean ± SD of triplicates. The results are reproducible in multiple experiments. (C) A549 stable NML knockdown cells were treated with 0.1 µM doxorubicin for 7 days, cultured in drug-free medium for 7 days, and stained for SA-β-gal activity. Circled area indicates SA-β-gal-negative cells that may have bypassed senescence during drug treatment or undergone reversal after drug removal. Colonies formed over a senescent cell monolayer were stained by crystal violet at 21 d Bar=20 µm. (D) Colony formation efficiency by A549 with NML knockdown after senescence induction with 0.1 µM doxorubicin for 7 d followed by drug washout for 21 d without re-seeding. (E) A549 cells were treated with 0.1 µM CX5461 for 7 d and stained for SA-β-gal. Bar=20 µm. (F) H1299 stably infected with NML shRNA retroviruses were treated with doxorubicin for 5 d and cultured in drug-free medium for 9 d Colonies were stained with crystal violet.
Prolonged treatment of A549 cells by low-dose doxorubicin caused senescence phenotypes such as cell cycle arrest and SA-β-gal positive staining. Importantly, the treated cells showed low probability of colony formation after drug removal for 21 d (Fig. 1C, top panels). Therefore, the majority of the cells (>99.9%) have established irreversible cell cycle arrest during the drug treatment, which is an important feature of senescence. NML knockdown significantly increased the frequency of colony formation after drug washout (Fig. 1D, lower panels). SA-β-gal staining showed that a fraction of NML knockdown cells remained negative for this senescence marker after doxorubicin treatment (Fig. 1C), suggesting that some of these cells may be responsible for colony formation. Two different NML shRNAs produced similar results (Fig. 1D). Recent study showed that the RNA Pol I inhibitor CX5461 is an efficient inducer of senescence.16 Knockdown of NML also reduced the efficiency of CX5461-induced senescence based on the SA-β-gal marker staining (Fig. 1E) and colony formation assay (data not shown). Therefore, NML facilitates the initiation of drug-induced senescence. The presence of SA-β-gal negative cells immediately after drug treatment, and the increased formation of colonies after drug washout suggest that a small subset of cells (<0.1%) did not fully commit to irreversible arrest during treatment and quickly resumed proliferation after drug removal.
To test whether NML knockdown led to the emergence of drug-resistant cells, the NML-deficient colonies formed after drug washout were pooled and re-treated with doxorubicin for 7 days, followed by drug removal and long-term culture. The colony formation efficiency after such treatment was similar to the original NML-deficient cells (data not shown). Therefore, the colonies retained the same level of drug sensitivity as the parental NML-deficient cells, suggesting that they had not undergone permanent mutations or epigenetic changes that confer classic drug resistance.
A549 cells express wild type p53, which is an important inducer of senescence. To test the role of NML in senescence response in other cell lines with different genetic background, we knocked down NML in H1299 (lung cancer, p53 null) and MCF7 (breast cancer, p53 wild type). The frequency of colony formation after doxorubicin treatment was also increased in both H1299 (Fig. 1F) and MCF7 cells with NML knockdown (data not shown), suggesting that NML regulates senescence initiation or maintenance in multiple cell types.
NML expression prevents reversal from senescence
Although senescence is often perceived as being synonymous with irreversible cell cycle arrest, previous study showed that certain senescent cells could revert to active cell cycle after inactivation of p53.23 To test whether NML expression is also needed to prevent reversal from senescence, A549 cells were pre-treated with doxorubicin for 7 d. The senescent cells were then transiently transfected with NML siRNA (Fig. 2A), followed by incubation in drug-free medium for 21 d. The result showed that transient NML knockdown in senescent cells caused an increase in colony formation (Fig. 2B), suggesting a small number of senescent cells reverted to active cell cycle. As expected, transient knockdown of p53 also caused significant colony formation in senescent A549 cells (Fig. 2B). These results suggest that in cells already committed to senescence, NML expression continues to play a role in maintaining cell cycle arrest similar to p53, contributing to the irreversibility of senescence phenotype.
Figure 2.

Depletion of NML in senescent cells promotes escape. (A) A549 cells were treated with 0.1 μM doxorubicin for 7 d and then transfected with NML siRNA for 2 d. NML knockdown in senescent cells was verified by western blot. (B) A549 cells were treated with 0.1 µM doxorubicin for 7 d to induce senescence, followed by transfection with NML or p53 siRNA. Colonies formed over the senescent monolayer were stained after 21 d (C) A549 cells were stably infected with retrovirus expressing TIP5 shRNA, the knockdown efficiency was confirmed by RT-qPCR. Values are mean ± SD of triplicates. (D) Pre-rRNA expression levels in A549 TIP5 knockdown cells were determined by RT-qPCR. Values are mean ± SD of 3 experiments. (E) A549 TIP5 knockdown cells were treated with 0.1 µM doxorubicin for 7 d Colony formation in drug-free medium was determined without re-seeding.
Nucleolar silencing facilitates induction of senescence
Since NML functions in the nucleolus, the above results suggest that efficient silencing of nucleolar transcription facilitates the initiation or maintenance of senescent cells. If this is the case, perturbing other nucleolar transcription repressors should also compromise senescence response. To test this hypothesis, the TIP5 subunit of nucleolar repressor complex NoRC was knocked down using 2 shRNAs (Fig. 2C). As expected, inactivation of NoRC caused increased pre-rRNA expression (Fig. 2D), and increased the frequency of colony formation after treatment with doxorubicin (Fig. 2E). TIP5 knockdown also increased the fraction of SA-β-gal negative cells after drug treatment (data not shown). Overall, these results suggest that precise regulation of nucleolar rRNA transcription level facilitates the induction and maintenance of senescence.
NML promotes nucleolar rDNA heterochromatin formation in senescent cells
Previous study showed that rRNA binds to NML and inhibits the formation of NML-SirT1 complex, possibly providing a positive feedback mechanism.22 Consistent with this finding, transient (2 hrs) inhibition of RNA Pol I using CX5461 stimulated endogenous NML-SirT1 binding (Fig. 3A). Senescent cells undergo significant changes in nucleolar morphology, which is likely to involve changes in the functional status of NML. To test this possibility, A549 cells treated with doxorubicin and CX5461 for 4–7 d were analyzed for pre-rRNA level as a surrogate for rRNA transcription. The level of pre-rRNA in the senescent cells decreased by ˜50% (Fig. 3B). NML-SirT1 binding was increased in cells chronically treated with doxorubicin or CX5461 (Fig. 3C), suggesting NML was in an activated state in senescent cells. NML knockdown correlates with increased pre-rRNA level in senescent cells (Fig. 3D), suggesting that it is required for efficient regulation of rRNA transcription during senescence.
Figure 3.

NML knockdown prevents heterochromatin formation on rDNA. (A) A549 cells were treated with 0.3 µM CX5461 for 2 hrs and 24 hrs. Endogenous NML-SirT1 binding was analyzed by SirT1 IP and NML protein gel blot. (B) Pre-rRNA level in senescent cells induced by 0.1 µM doxorubicin or 0.3 µM CX5461 for 7 d was determined by RT-qPCR. Values are mean ± SD of triplicates. (C) A549 cells were incubated with indicated drugs for 4 d The endogenous SirT1-NML complex was detected by IP-western blot. (D) Pre-rRNA level in A549 NML knockdown cells treated with 0.1 µM doxorubicin for 24 and 72 hrs was analyzed by RT-qPCR. Values are mean ± SD of triplicates. (E, F) A549 NML knockdown cells were treated with 0.3 µM CX5461 for 1 and 7 d H3K9Me3 and H3K27Me3 levels on rDNA promoter were detected by ChIP and qPCR. (G) A549 expressing 2 different NML shRNA were treated with CX5461 for 7 d and analyzed for histone methylation at the rDNA promoter by ChIP.
Consistent with the down regulation in pre-rRNA level, induction of senescence was accompanied by increased repressive chromatin marks H3K9me3 and H3K27me3 on the rDNA promoter. Knockdown of NML significantly reduced both markers on rDNA, correlating with elevated pre-rRNA expression (Fig.. 3E-F). Two different NML shRNA produced the same effects (Fig. 3G). The results suggest that initiation of senescence involves increased heterochromatin formation at the rDNA loci, which in part requires the function of NML.
Loss of NML delays induction of senescence and irreversible arrest
To further test the mechanism by which NML enhances senescence response, A549 cells were treated with doxorubicin for different time points and analyzed for cell cycle markers by western blot. The results revealed that NML knockdown reduced the magnitude of p21 induction by doxorubicin (Fig. 4A). The down regulation of cyclin B level was incomplete in NML knockdown cells after 3 d of doxorubicin treatment, although they were reduced to the same level as control cells after 7 d of treatment (Fig. 4A). Similar results were also observed using CX5461 as inducer of senescence (Fig. 4A). NML knockdown did not prevent p53 stabilization by doxorubicin, suggesting that the reduced p21 level may be due to deficiency in p53 activity or p53-independent mechanisms. These results suggest that a small sub-population of NML-deficient cells have a delayed cell cycle arrest response after DNA damage, which may prevent establishment of robust epigenetic modifications needed for irreversible arrest, resulting in rapid resumption of proliferation after drug removal.
Figure 4.
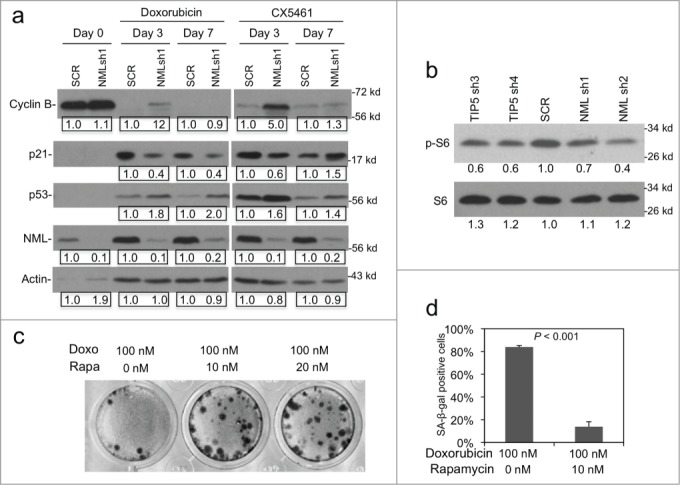
NML knockdown compromises cell cycle regulation. (A) A549 NML knockdown cells were treated with 0.1 µM doxorubicin or 0.3 µM CX5461 for 3 and 7 d. Expression levels of cell cycle markers were analyzed by protein gel blot. Numbers in boxes show pair-wise quantitation of control shRNA and NML shRNA. (B) The level of phosphorylated ribosomal protein S6 (Ser240/Ser244) was determined in A549 NML and TIP5 knockdown cells by western blot. Numbers represent relative quantitation of bands. (C) A549 cells were co-treated with doxorubicin and rapamycin for 7 d and cultured in drug-free medium for 21 d to detect colony formation over the senescent monolayer. (D) A549 cells were co-treated with doxorubicin and rapamycin for 7 d and the percentage of SA-β-gal positive cells was determined.
Recent studies suggested that mTOR activity is required for the induction of senescence.24 We found that the phosphorylation level of ribosomal protein S6 (a substrate of the mTOR target S6 kinase) was moderately reduced in A549 cells after knockdown of NML or TIP5 (Fig. 4B), suggesting that abnormal increase in rRNA transcription may down regulate mTOR activity. mTOR is a key driver of biosynthesis and its activity is stimulated by cellular ATP, glucose and amino acid levels. NML knockdown causes increased rRNA synthesis that consumes cellular resources, these changes may trigger negative feedback signals to lower mTOR activity in order to maintain homeostasis. To determine whether mTOR activity affects the initiation of senescence in A549 cells, the effect of mTOR inhibitor rapamycin was tested. The result showed that cells simultaneously treated with doxorubicin/rapamycin formed colonies with higher frequency compared to doxorubicin alone (Fig. 4C), and had weaker SA-β-gal staining (Fig. 4D). These results suggest that deficiency in nucleolar transcription silencing dampens p21 response to DNA damage and also reduces mTOR activity, both changes may contribute to the partial deficiency in senescence induction and maintenance.
Partial activation of senescence-related genes in NML-deficient cells
Senescence involves activation and repression of numerous genes in cell cycle, growth signaling, autophagy, and secretion pathways. As an unbiased survey of NML knockdown on senescence-related gene expression, qRT-PCR was performed for a panel of 84 genes implicated in senescence. A549 control and NML knockdown cells were compared before and after CX5461 treatment for 5 d. The results showed that among the 84 genes analyzed, 18 were induced >2 fold and 6 were repressed >5 fold after CX5461 treatment for 5 d in control A549 cells (Fig. 5A). Most of the top activated genes were expressed at lower levels in NML knockdown cells after treatment (Fig. 5B, lower left shaded area). Furthermore, most of the strongly repressed genes were expressed at higher levels in NML-deficient cells (Fig. 5B upper right shaded area). Overall, 6 out of 18 genes that were activated in senescent cells were expressed at 1.5 to 3-fold lower levels after NML knockdown, whereas 5 out of 6 genes repressed in senescent cells were expressed at 1.3 to 1.6-fold higher levels in NML-deficient cells. Analysis of cells after 7 d of CX5461 treatment confirmed most of the gene expression changes observed in the 5 day treatment (data not shown). Therefore, NML knockdown caused partial deficiencies in the activation and repression of multiple genes involved in senescence induction.
Figure 5.
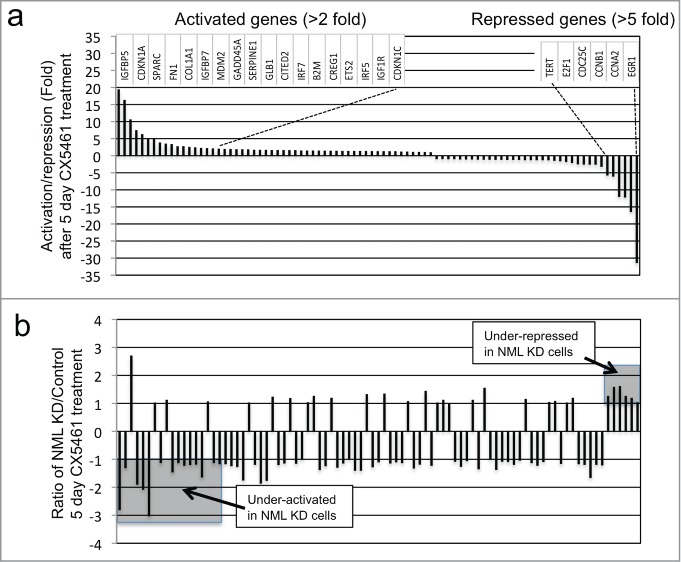
NML depletion alters the regulation of senescence-related genes. (A) A549-control shRNA cells were treated with 0.3 µM CX5461 for 5 d and the mRNA levels of 84 senescence-related genes were analyzed by RT-PCR. Fold changes were determined by comparing treated to untreated cells. The genes were sorted from strongest induction (left) to strongest repression (right). The names of the top 18 activated genes and top 6 repressed genes are shown. (B) Comparison of senescence gene expression levels after 5 d of CX5461 treatment. The mRNA levels for control and NML knockdown cells after 5 d of CX5461 treatment were compared and the ratios were shown in the same gene order as (A). A ratio of +1 or -1 indicates the gene was induced to the same degree in control and knockdown cells. A ratio of >+1 indicates stronger induction in NML knockdown cells compared to control cells, a ratio of <-1 indicates weaker induction in NML knockdown cells compared to control cells. The results are representative of 3 experiments.
Deficiency in E2F target gene heterochromatin formation
The results described above showed that the majority of NML-deficient cells (>99.9%) were still capable of initiating and maintaining irreversible cell cycle arrest after drug treatment despite partial deficiencies in senescent gene expression. However, a minute fraction of the cells have increased frequency of resuming clonal growth. We hypothesized that the multi-gene expression changes after NML knockdown leads to less robust epigenetic modifications on certain promoters. While the compromised gene regulation is still sufficient for irreversible arrest, the probability of reactivation by sporadic events after drug removal is increased.
Of particular interest was the partial defect in E2F1 mRNA down regulation in NML-deficient cells, which resulted in 1.5-fold higher level of E2F1 after 5 d of treatment (Fig. 5B). To test whether NML deficiency affects heterochromatin formation at E2F1 target genes, ChIP analyses were performed using H3K9me3 and H3K27me3 antibodies. Analysis of several E2F1 target promoters (PCNA, Cyclin A, Cyclin E) showed that histone methylation at these promoters were increased in senescent cells, with H3K27me3 level increased more significantly compared to H3K9me3. Knockdown of NML was associated with reduced H3K9me3 and H3K27me3 levels at these promoters, in both proliferating cells and senescent cells (Fig. 6A-B). Similar analysis showed that TIP5 knockdown also reduced the induction of H3K9me3 and H3K27me3 at the E2F1 target promoters (Fig. 6C-D). Therefore, deficiency in nucleolar transcription repression not only affects heterochromatin formation on rDNA repeats, but prevents the modification of nucleoplasmic promoters. The results suggest that the rapid emergence of a small number of proliferating cells after senescence induction was probably due to a population-wide deficit in heterochromatin modifications at E2F1 targets, which increased the probability of sporadic re-activation.
Figure 6.
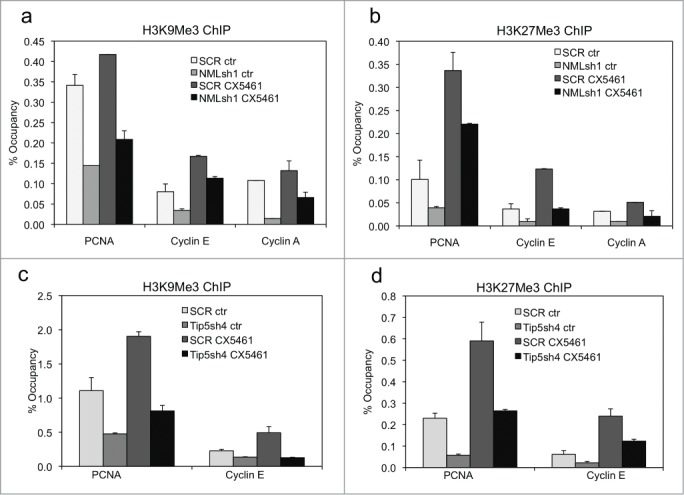
Depletion of NML reduces heterochromatin formation at E2F target genes. (A, B) A549 NML knockdown cells were treated with 0.3 µM CX5461 for 7 d. H3K9Me3 and H3K27Me3 levels at indicated E2F1 target promoters were detected by ChIP and qPCR. Values are mean ± SD of triplicates. (C, D) A549 TIP5 knockdown cells were treated with 0.3 µM CX5461 for 7 d. H3K9Me3 and H3K27Me3 levels at indicated E2F1 target promoters were detected by ChIP and qPCR. Values are mean ± SD of triplicates. All results are reproducible in multiple experiments.
Down regulation of NML in breast cancer correlates with poor survival
Given the important role of the nucleolus in cell growth and proliferation, NML down regulation may occur during tumor development. An Oncomine query revealed that NML (aka RRP8) is heterozygously deleted in a subset of ovarian, bladder, brain, and esophagus tumors (Fig. 7A). Certain microarray data sets in Oncomine showed that NML mRNA level is reduced in breast tumors (Fig. 7B). Analysis using a gene expression database with 3450 systemically treated breast cancer patients showed that increased NML mRNA expression was associated with better relapse-free and overall survival (Fig. 7C).25 Higher expression of TIP5 mRNA was also associated with better relapse-free survival (Fig. 7D). Therefore, reduced NML expression may facilitate tumor formation and contribute to poor survival in breast cancer.
Figure 7.
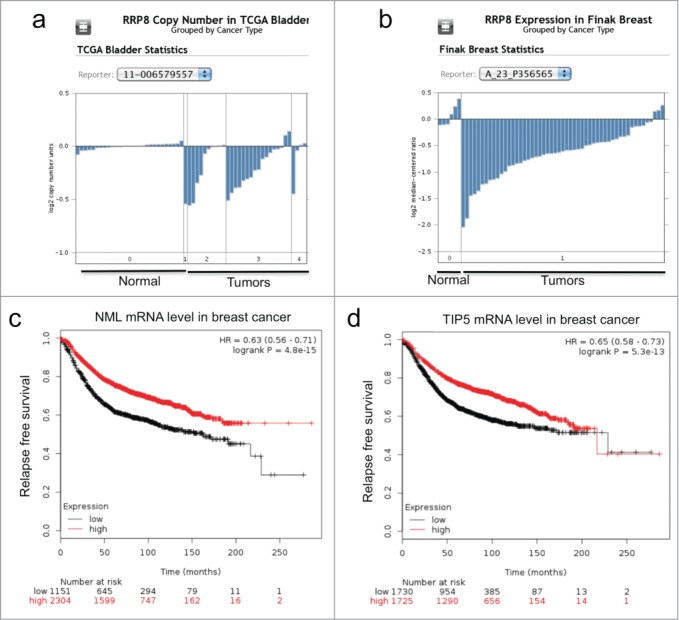
NML expression is downregulated in a subset of tumors. (A) Reduced NML gene (RRP8) copy number in a bladder cancer cohort (Oncomine). A subset of tumor samples showed heterozygous deletion at the RRP8 locus. (B) Reduced NML mRNA level in an invasive breast cancer cohort compared to normal tissue (Oncomine). (C, D) Kaplan-Meyer plots of 3450 patients with breast tumors following systemic treatment showing that low NML (RRP8, probe set 203170_at) and TIP5 (TIP5, probe set 201355_s_at) mRNA levels are associated with shorter relapse free survival.
To further test the significance of NML expression, a breast TMA (n=121) was analyzed by IHC staining. The result showed that 54% (n=65) had weak to strong nucleolar staining (NML-positive. Examples in Fig. 8A-C), 46% (n=56) of the samples had no detectable staining (NML-negative, Example in Fig. 8D). NML-negative staining was correlated with early death overall (Fig. 8E), strongly correlated with early death in ER-negative populations (n=31) (Fig. 8G). The ER-positive population also suggested a similar trend but the result did not reach statistical significance (Fig. 8F). Due to inherently better prognosis in the ER-positive population, a larger sample size will be needed to determine whether NML expression correlates with survival in this group. These results suggest that absence or low NML expression in breast tumors is associated with poor survival and early recurrence for the high-risk ER-negative population.
Figure 8.

Absence of NML staining is associated with poor survival in ER-negative breast cancer. (A, B, C, D) NML expression in breast tumors was detected by IHC staining. Arrows indicate nucleolar NML staining in normal breast control and a subset of breast tumors. Bar=20 µm. (E, F, G) Kaplan-Meier plots of breast tumor survival stratified based on nucleolar NML positive and negative staining and ER status.
Discussion
The nucleolus was traditionally regarded as simply a ribosome factory. Recent studies suggest that it is also an important sensor of cellular stress and DNA damage.26,27 Inhibition of nucleolar rRNA transcription by chemotherapy agents or serum starvation leads to activation of p53, mediated by excess ribosomal proteins that bind to MDM2 and prevent p53 degradation.28 The nucleolus has also been suggested to act as a depot for factors that act outside the nucleolus, such as the ARF tumor suppressor that function as an inhibitor of MDM2. Certain nucleolar proteins such as NPM1 has dual roles in the nucleolus and nucleoplasm; its abnormal distribution after nucleolar perturbation may also affect the regulation of nucleoplasmic genes.29 Knockdown of the NoRC nucleolar repressor subunit TIP5 has recently been shown to promote transformation of NIH3T3 cells.30 Our data in this report suggests that nucleolar silencing may have an important role in maintaining cellular senescence.
During classical replicative senescence of human fibroblast cultures, cell cycle arrest is caused by critical telomere shortening that activates DNA damage signaling and p53 accumulation. In the absence of telomerase reactivation or ectopic expression, inactivation of p53 or pRb can only restore cell proliferation for limited number of cycles.23,31 Premature senescence of tumor cells induced by oncogenic stress or DNA damage does not involve telomere shortening and is not restricted by the structural defect as replicative senescence. Nonetheless, drug-induced senescent cell culture can remain arrested for weeks or longer in the absence of further drug treatment, suggesting that a stable epigenetic silencing state has been established on the proliferation genes, which prevents cell cycle re-entry.
Our results suggest that efficient regulation of nucleolar rRNA transcription is necessary for irreversible senescence. Maintenance of senescence after drug removal presumably requires self-sustaining heterochromatinization of proliferation genes in cooperation with sustained activation of growth-inhibitory factors such as IGFBP5 and p21. Heterochromatin proteins HP1, SUV39H1, and NML all have the ability to bind methylated H3K9 and promote methylation of adjacent nucleosomes. Therefore they are well-suited for self-reinforcing heterochromatin formation and spreading. Recent work showed that NML-SirT1 binding is competitively inhibited by rRNA.22 NML provides positive feedback to amplify signals that stimulate or inhibit rRNA transcription. Signals that inhibit RNA Pol I will stimulate NML-SirT1 binding, resulting in further increase of heterochromatin marks on rDNA repeats. This property may allow NML to play a key role in maintaining heterochromatin on the rDNA by forming a positive feedback loop. Loss of NML expression reduces the level of heterochromatin marks on rDNA, increasing the probability of stochastic re-activation by external disturbances or internal fluctuations.
Depletion of NML or TIP5 also reduced the levels of heterochromatin marks on E2F1 target gene promoters unrelated to the rDNA locus, suggesting that nucleolar silencing has an important role in regulating nucleoplasmic gene expression. Recent studies identified the ribosomal protein-MDM2-p53 pathway in connecting nucleolar dysfunction to cell cycle arrest. However, NML deficiency also promotes senescence escape in cells without p53, suggesting the presence of p53-independent mechanisms. We speculate that there are multiple inside-out signaling pathways that enable the nucleolus to influence nucleoplasmic genes. The pRb tumor suppressor also undergoes nucleolar translocation, which may be regulated by nucleolar heterochromatin level.32 The RelA subunit of NFkB, which is also required for inducing senescence, is also regulated by nucleolar localization.33,34 The distribution of other factors such as NPM1 that function in both nucleoplasm and nucleolus may also be affected by nucleolar heterochromatin formation. rRNA may serve as a messenger to regulate heterochromatin proteins in the nucleoplasm, since HP1 also interacts with RNA.35 Many genomic loci are closely associated with and are co-purified with the nucleolus.36,37 The nucleolus is known to promote the silencing of X chromosome through spatial proximity.38 Therefore, the nucleolar heterochromatin domain may directly facilitate the silencing of non-nucleolar genes. A hyperactive nucleolus in senescent cells may increase the probability of reactivating silenced cell cycle genes.
The biological roles of cellular senescence are complex and context-dependent. It is a predominant mode of response to chemotherapy agents such as doxorubicin in vivo.6,39 Our results suggest that loss of NML expression in breast cancer correlates with poor patient survival in ER-negative cases. Whether NML status also correlates with survival in ER-positive cases remains to be determined with a larger cohort. Although the effect of NML appears to be independent of cell type in vitro (e.g., NML knockdown affects ER-positive MCF7 cells), the in vivo impact may depend on what is the main driver of survival in each tumor type. Surgery and adjuvant chemotherapy with DNA damaging drugs such as doxorubicin often achieve long-term disease-free survival for breast cancer patients. However, a small number of tumor cells are known to exist in a state of dormancy and may cause relapse years later.40 It is possible that dormant tumor cells are stabilized by senescence, and escape from senescence causes recurrence. Loss of NML expression may accelerate the escape of senescent tumor cells, causing early relapse and poor survival. The results of our study suggest that treatments that promote nucleolar transcription silencing may be beneficial against tumors that are resistant to apoptosis but retain senescence response. The RNA Pol I inhibitor CX5461 has shown therapeutic potential in mouse leukemia model.17 It will be important to investigate the role of RNA Pol I inhibition in solid tumors that have low apoptotic potential and rely on senescence as a treatment response mechanism.
Materials and Methods
Cell lines and reagents
H1299 (human non-small cell lung carcinoma), A549 (human lung adenocarcinoma), MCF7 (human breast adenocarcinoma) were maintained in Dulbecco modified Eagle medium (DMEM) with 10% fetal bovine serum. A549 cells with stable knockdown of NML and TIP5 were generated by infection with pSuperior-retro-NML and pSuperior-retro-TIP5 virus followed by puromycin selection (pSuperior-retro-puro vector, OligoEngine). CX5461 was obtained from Selleckchem. Rapamycin was obtained from LC laboratories. Rabbit polyclonal antibody for NML was raised against His6-NML-1–200. Anti-SirT1 monoclonal antibody 10E4 was purchased from Millipore. p21 and p53 antibodies were purchased from BD PharMingen. S6 and phosphor-S6 were from Cell Signaling Technology. Senescence gene RT-PCR profiling was performed using the Human Cellular Senescence PCR Array (Qiagen, PAHS-050Z). Data processing was performed using the manufacturer website.
Immunoprecipitation
Cells were lysed in lysis buffer (50 mM Tris-HCl pH 8.0, 5 mM EDTA, 150 mM NaCl, 0.5% NP-40, 1 mM phenylmethylsulfonyl fluoride, protease inhibitor cocktail) and centrifuged for 10 min at 14,000 x g to remove the insoluble debris. The supernatant was used for immunoprecipitation and protein gel blotting. For detecting endogenous SirT1-NML complex, the insoluble debris were re-suspended in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate), and mixed with the supernatant. Cell lysate (200–1000 µg of protein) was immunoprecipitated with 10E4 against SirT1 and protein A agarose beads for 18 hrs at 4˚C. After washing 4 times with SNNTE (50 mM Tris-HCl pH 7.4, 5 mM EDTA, 500 mM NaCl, 0.1% NP40, and 5% sucrose), the beads were boiled in Laemmli sample buffer for 5 min and subjected to SDS-PAGE and western blot to detect SirT1-NML interaction using affinity purified rabbit-anti-NML antibody.
Chromatin immunoprecipitation and quantitative-PCR
ChIP assay was performed using standard procedure. Proteins were cross-linked to genomic DNA with 1% formaldehyde for 10 min at 23˚C. The cross-link was stopped by 0.125 M glycine for 5 min at 23˚C. The cells were washed 3 times with ice-cold phosphate-buffered saline (PBS) and lysed in 0.45 ml of RIPA buffer with proteinase inhibitor cocktails on ice for 10 min. Cell pellet was sonicated 8 cycles using Bioruptor XL (8 min/cycle, 30 sec on and 30 sec off) and then spun down at 14,000 g for 10 min to remove debris. The lysates were pre-cleared by incubating with a salmon sperm DNA/protein A agarose slurry for 30 min at 4˚C with rotation. The cleared lysates were diluted 1:6 in ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH8.0, and 167 mM NaCl). The samples were incubated with 1 μg antibody for 18 hrs at 4˚C with rotation. Protein A agarose slurry (50 µl) were added and incubated for 2 hrs at 4˚C. The beads were washed once each with low salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH8.0, 150 mM NaCl), high salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH8.0, 500 mM NaCl), LiCl buffer (250 mM LiCl, 1% NP40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris-HCl pH8.0), and TE buffer (10 mM Tris-HCl pH8.0, 1 mM EDTA). The beads were eluted twice with a total of 200 μl of elution buffer (1% SDS, 100 mM NaHCO3) at 25˚C for 15 min with shaking (1000 rpm). NaCl was added to 200 mM and the samples were de-crosslinked at 65˚C for 18 hrs. Proteinase K (0.1 mg/ml), EDTA (10 mM) and Tris-HCl pH6.8 (40 mM) were added and incubated at 42˚C for 1 hr. DNA was extracted using GenEluteTM PCR clean-up kit (Sigma). The Samples were subjected to SYBR Green real-time PCR analysis using the following primers. rDNA promoter H0F: 5'- GGTATATCTTTCGCTCCGAG; H0R: 5'-GACGACAGGTCGCCAGAGGA. Cyclin AF: GGCCAACACCCATAAGAGAA; Cyclin AR: TGAACGCAGGCTGTTTACTG. Cyclin EF: AAGGACAGAGGAGCTGTGGA; Cyclin ER: TTCGCATAAGGGTCAAATCC. PCNAF: TGGTGAAACCCCGTCTCTAC; PCNAR: CTCAGCCTCCCGAGTATCTG.
SA-β-galactosidase staining
Cells (2.5x104/well) were seeded to 24 well plates for 18 hrs and treated with doxorubicin or CX5461 for indicated duration. The cells were washed twice with PBS and SA-β-gal was detected using Senescence histochemical staining kit (Sigma) according to manufacturer protocol. Cells were incubated at 37˚C for 18 hrs and photographed.
Colony formation assay
Cells (6x104/well) were seeded to 6 well plates for 18 hrs and incubated with doxorubicin or CX5461 for 5–7 d The cells were washed twice with drug-free medium and cultured in drug-free medium for 2–6 weeks with refeeding every 5 d until colonies are visible. Colonies were stained by crystal violet.
RNA interference (RNAi)
Post-senescence cells induced by 0.1 µM doxorubicin for 7 d were transfected with 50 nM control siRNA (AATTCTCCGAACGTGTCACGT), NML siRNA pool, or p53 siRNA pool (Dharmacon) using RNAiMAX (Invitrogen) according to instructions from the supplier. After 48 hrs of transfection, cells were cultured in normal DMEM medium until visible colonies formed.
Immunohistochemistry
The breast tumor microarray (Breast 4 TMA) was obtained from the Moffitt Cancer Center Tissue Core. After de-paraffinization with xylene and a graded ethanol series, TMA slides were incubated with 0.38 mg/ml of protease1 (Ventana Medical System, Inc.. Arizona, USA) for 4 min at 37°C for antigen retrieval. The sections were blocked with 0.3% of H2O2 for 5 min and 1% BSA in PBS for 30 min. The sections were incubated with rabbit anti-NML antibody (1:100; Sigma) for 1 hr at room temperature. The immunohistochemical staining was developed using IHC staining kit (Dako, CA, USA). The sections were then counterstained. NML staining was scored manually by 2 individuals. Due to the distinct and highly focused nucleolar staining pattern, the samples could be reliably categorized as being positive or negative for nucleolar NML, whereas semi-quantitative ranking by automated image analysis was found to be unreliable.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
Leixiang Yang, Tanjing Song, Lihong Chen designed and performed the experiments. Hatem Soliman provided important scientific input. Leixiang Yang and Jiandong Chen designed the experiments and wrote the manuscript.
Funding
This work was supported by grants CA141244 and CA109636 from the National Institutes of Health, 4BB14, 4KB12 from the Florida Department of Health, and a Miles For Moffitt award from the Moffitt Foundation to J. Chen. This work was also supported in part by the Tissue Core and Imaging Core at the H. Lee Moffitt Cancer Center & Research Institute, an NCI designated Comprehensive Cancer Center (P30-CA076292).
References
- 1.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 2005; 120:513-22; PMID:15734683; http://dx.doi.org/ 10.1016/j.cell.2005.02.003 [DOI] [PubMed] [Google Scholar]
- 2.d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nature Rev Cancer 2008; 8:512-22; http://dx.doi.org/ 10.1038/nrc2440 [DOI] [PubMed] [Google Scholar]
- 3.Schmitt CA. Cellular senescence and cancer treatment. Biochimica Et Biophysica Acta 2007; 1775:5-20; PMID:17027159 [DOI] [PubMed] [Google Scholar]
- 4.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol 2011; 192:547-56; PMID:21321098; http://dx.doi.org/ 10.1083/jcb.201009094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011; 479:232-6; PMID:22048312; http://dx.doi.org/ 10.1038/nature10600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson JG, Pant V, Li Q, Chang LL, Quintas-Cardama A, Garza D, Tavana O, Yang P, Manshouri T, Li Y, et al. . p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 2012; 21:793-806; PMID:22698404; http://dx.doi.org/ 10.1016/j.ccr.2012.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu PC, Wang Q, Grobman L, Chu E, Wu DY. Accelerated cellular senescence in solid tumor therapy. Exp Oncol 2012; 34:298-305; PMID:23070015 [PubMed] [Google Scholar]
- 8.Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, Lowe SW. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002; 109:335-46; PMID:12015983; http://dx.doi.org/ 10.1016/S0092-8674(02)00734-1 [DOI] [PubMed] [Google Scholar]
- 9.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445:656-60; PMID:17251933; http://dx.doi.org/ 10.1038/nature05529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberson RS, Kussick SJ, Vallieres E, Chen SY, Wu DY. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res 2005; 65:2795-803; PMID:15805280; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-1270 [DOI] [PubMed] [Google Scholar]
- 11.Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene 2006; 25:6384-91; PMID:17041624; http://dx.doi.org/ 10.1038/sj.onc.1209883 [DOI] [PubMed] [Google Scholar]
- 12.McStay B, Grummt I. The epigenetics of rRNA genes: from molecular to chromosome biology. Ann Rev Cell Dev Biol 2008; 24:131-57; http://dx.doi.org/ 10.1146/annurev.cellbio.24.110707.175259 [DOI] [PubMed] [Google Scholar]
- 13.Zhai W, Comai L. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol Cell Biol 2000; 20:5930-8; PMID:10913176; http://dx.doi.org/ 10.1128/MCB.20.16.5930-5938.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voit R, Schafer K, Grummt I. Mechanism of repression of RNA polymerase I transcription by the retinoblastoma protein. Mol Cell Biol 1997; 17:4230-7; PMID:9234680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ayrault O, Andrique L, Fauvin D, Eymin B, Gazzeri S, Seite P. Human tumor suppressor p14ARF negatively regulates rRNA transcription and inhibits UBF1 transcription factor phosphorylation. Oncogene 2006; 25:7577-86; PMID:16924243; http://dx.doi.org/ 10.1038/sj.onc.1209743 [DOI] [PubMed] [Google Scholar]
- 16.Drygin D, Lin A, Bliesath J, Ho CB, O'Brien SE, Proffitt C, Omori M, Haddach M, Schwaebe MK, Siddiqui-Jain A, et al. . Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res 2011; 71:1418-30; PMID:21159662; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-1728 [DOI] [PubMed] [Google Scholar]
- 17.Bywater MJ, Poortinga G, Sanij E, Hein N, Peck A, Cullinane C, Wall M, Cluse L, Drygin D, Anderes K, et al. . Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 2012; 22:51-65; PMID:22789538; http://dx.doi.org/ 10.1016/j.ccr.2012.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grummt I, Pikaard CS. Epigenetic silencing of RNA polymerase I transcription. Nat Rev Mol Cell Biol 2003; 4:641-9; PMID:12923526; http://dx.doi.org/ 10.1038/nrm1171 [DOI] [PubMed] [Google Scholar]
- 19.Santoro R, Li J, Grummt I. The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat Genet 2002; 32:393-6; PMID:12368916; http://dx.doi.org/ 10.1038/ng1010 [DOI] [PubMed] [Google Scholar]
- 20.Murayama A, Ohmori K, Fujimura A, Minami H, Yasuzawa-Tanaka K, Kuroda T, Oie S, Daitoku H, Okuwaki M, Nagata K, et al. . Epigenetic control of rDNA loci in response to intracellular energy status. Cell 2008; 133:627-39; PMID:18485871; http://dx.doi.org/ 10.1016/j.cell.2008.03.030 [DOI] [PubMed] [Google Scholar]
- 21.Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003; 113:703-16; PMID:12809602; http://dx.doi.org/ 10.1016/S0092-8674(03)00401-X [DOI] [PubMed] [Google Scholar]
- 22.Yang L, Song T, Chen L, Kabra N, Zheng H, Koomen J, Seto E, Chen J. Regulation of SirT1-Nucleomethylin Binding by rRNA Coordinates Ribosome Biogenesis with Nutrient Availability. Mol Cell Biol 2013; 33:3835-48; PMID:23897426; http://dx.doi.org/ 10.1128/MCB.00476-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003; 22:4212-22; PMID:12912919; http://dx.doi.org/ 10.1093/emboj/cdg417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle 2009; 8:1888-95; PMID:19471117; http://dx.doi.org/ 10.4161/cc.8.12.8606 [DOI] [PubMed] [Google Scholar]
- 25.Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treatment 2010; 123:725-31; http://dx.doi.org/ 10.1007/s10549-009-0674-9 [DOI] [PubMed] [Google Scholar]
- 26.Boisvert FM, van Koningsbruggen S, Navascues J, Lamond AI. The multifunctional nucleolus. Nat Rev Mol Cell Biol 2007; 8:574-85; PMID:17519961; http://dx.doi.org/ 10.1038/nrm2184 [DOI] [PubMed] [Google Scholar]
- 27.Rubbi CP, Milner J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J 2003; 22:6068-77; PMID:14609953; http://dx.doi.org/ 10.1093/emboj/cdg579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell 2009; 16:369-77; PMID:19878869; http://dx.doi.org/ 10.1016/j.ccr.2009.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colombo E, Alcalay M, Pelicci PG. Nucleophosmin and its complex network: a possible therapeutic target in hematological diseases. Oncogene 2011; 30:2595-609; PMID:21278791; http://dx.doi.org/ 10.1038/onc.2010.646 [DOI] [PubMed] [Google Scholar]
- 30.Guetg C, Lienemann P, Sirri V, Grummt I, Hernandez-Verdun D, Hottiger MO, Fussenegger M, Santoro R. The NoRC complex mediates the heterochromatin formation and stability of silent rRNA genes and centromeric repeats. EMBO J 2010; 29:2135-46; PMID:20168299; http://dx.doi.org/ 10.1038/emboj.2010.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wright WE, Shay JW. Cellular senescence as a tumor-protection mechanism: the essential role of counting. Curr Opin Genet Dev 2001; 11:98-103; PMID:11163158; http://dx.doi.org/ 10.1016/S0959-437X(00)00163-5 [DOI] [PubMed] [Google Scholar]
- 32.Angus SP, Solomon DA, Kuschel L, Hennigan RF, Knudsen ES. Retinoblastoma tumor suppressor: analyses of dynamic behavior in living cells reveal multiple modes of regulation. Mol Cell Biol 2003; 23:8172-88; PMID:14585976; http://dx.doi.org/ 10.1128/MCB.23.22.8172-8188.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stark LA, Dunlop MG. Nucleolar sequestration of RelA (p65) regulates NF-kappaB-driven transcription and apoptosis. Mol Cell Biol 2005; 25:5985-6004; PMID:15988014; http://dx.doi.org/ 10.1128/MCB.25.14.5985-6004.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vaughan S, Jat PS. Deciphering the role of nuclear factor-kappaB in cellular senescence. Aging 2011; 3:913-9; PMID:21990145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keller C, Adaixo R, Stunnenberg R, Woolcock KJ, Hiller S, Buhler M. HP1(Swi6) Mediates the Recognition and Destruction of Heterochromatic RNA Transcripts. Mol Cell 2012; 47:215-27; PMID:22683269; http://dx.doi.org/ 10.1016/j.molcel.2012.05.009 [DOI] [PubMed] [Google Scholar]
- 36.Nemeth A, Conesa A, Santoyo-Lopez J, Medina I, Montaner D, Peterfia B, Solovei I, Cremer T, Dopazo J, Längst G. Initial genomics of the human nucleolus. PLoS Genet 2010; 6:e1000889; PMID:20361057; http://dx.doi.org/ 10.1371/journal.pgen.1000889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Koningsbruggen S, Gierlinski M, Schofield P, Martin D, Barton GJ, Ariyurek Y, den Dunnen JT, Lamond AI. High-resolution whole-genome sequencing reveals that specific chromatin domains from most human chromosomes associate with nucleoli. Mol Biol Cell 2010; 21:3735-48; PMID:20826608; http://dx.doi.org/ 10.1091/mbc.E10-06-0508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang LF, Huynh KD, Lee JT. Perinucleolar targeting of the inactive X during S phase: evidence for a role in the maintenance of silencing. Cell 2007; 129:693-706; PMID:17512404; http://dx.doi.org/ 10.1016/j.cell.2007.03.036 [DOI] [PubMed] [Google Scholar]
- 39. te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res 2002; 62:1876-83; PMID:11912168 [PubMed] [Google Scholar]
- 40.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 2007; 7:834-46; PMID:17957189; http://dx.doi.org/ 10.1038/nrc2256 [DOI] [PMC free article] [PubMed] [Google Scholar]