

Abstract
Purpose: Over 90% of pancreatic adenocarcinoma PC express oncogenic mutant KRAS that constitutively activates the Raf-MEK-MAPK pathway conferring resistance to both radiation and chemotherapy. MEK inhibitors have shown promising anti-tumor responses in recent preclinical and clinical studies, and are currently being tested in combination with radiation in clinical trials. Here, we have evaluated the radiosensitizing potential of a novel MEK1/2 inhibitor GSK1120212 (GSK212,or trametinib) and evaluated whether MEK1/2 inhibition alters DNA repair mechanisms in multiple PC cell lines.
Methods: Radiosensitization and DNA double-strand break (DSB) repair were evaluated by clonogenic assays, comet assay, nuclear foci formation (γH2AX, DNA-PK, 53BP1, BRCA1, and RAD51), and by functional GFP-reporter assays for homologous recombination (HR) and non-homologous end-joining (NHEJ). Expression and activation of DNA repair proteins were measured by immunoblotting.
Results: GSK212 blocked ERK1/2 activity and radiosensitized multiple KRAS mutant PC cell lines. Prolonged pre-treatment with GSK212 for 24-48 hours was required to observe significant radiosensitization. GSK212 treatment resulted in delayed resolution of DNA damage by comet assays and persistent γH2AX nuclear foci. GSK212 treatment also resulted in altered BRCA1, RAD51, DNA-PK, and 53BP1 nuclear foci appearance and resolution after radiation. Using functional reporters, GSK212 caused repression of both HR and NHEJ repair activity. Moreover, GSK212 suppressed the expression and activation of a number of DSB repair pathway intermediates including BRCA1, DNA-PK, RAD51, RRM2, and Chk-1.
Conclusion: GSK212 confers radiosensitization to KRAS-driven PC cells by suppressing major DNA-DSB repair pathways. These data provide support for the combination of MEK1/2 inhibition and radiation in the treatment of PC.
Keywords: DNA repair, GSK1120212, KRAS, MEK inhibitor, Mekinist, pancreatic cancer, radiation, trametinib
Introduction
Pancreatic adenocarcinoma is the ninth most commonly diagnosed malignancy and the fourth leading cause of cancer-related death in the US.1 According to the National Cancer Institute, an estimated ∼46,000 new cases of pancreatic cancer were diagnosed and ∼40,000 deaths occurred in the US in 2014.2 At present, few therapeutic options are available for patients with this deadly disease, and there is an urgent need for the development of novel therapies for pancreatic carcinoma (PC). Approximately 80-85% of the patients are diagnosed with locally-advanced or metastatic disease precluding an option of surgical resection. Radiation alone or in combination with chemotherapy is an available treatment for locally advanced disease, and is sometimes used in the pre-surgical setting. It is remarkable that 90% or more of tumors harbor an oncogenic mutant KRAS, which other studies have shown confers resistance to radiation both in vitro and in vivo.3-5 Therefore, an in-depth understanding of how de-regulated KRAS contributes to the development of radioresistance in pancreatic as well as other malignancies expressing activated RAS oncogenes is critical for developing effective treatments.
Under physiologic conditions, growth factor-activated KRAS recruits and activates the RAF family of serine/threonine kinases, which in turn, activate the dual protein kinases MEK1 and MEK2 that subsequently activate MAP kinases ERK1 and ERK2. Then, activated ERK1 and ERK2 phosphorylate their target substrates promoting cell proliferation, survival, and differentiation.6,7 Aberrant activation of mutant KRAS results in the constitutive activation of this signaling pathway, which enhances the growth, survival and migration of pancreatic cancer cells.8,9 The oncogenic mutant KRAS also activates the PI3K-AKT/mTOR pathway that plays many critical roles in tumorigenesis.10,11 Importantly, we and others have demonstrated that KRAS-induced persistent activation of both MEK-ERK and PI3K-Akt pathways confers resistance to radiation and chemotherapy in pancreatic cancer.5,12-14 Although radiation therapy significantly improves local control in pancreatic cancer, radiation induces an activation of the MEK-ERK pathway, possibly promoting the proliferation and survival of tumor cells in vitro.12,15-17 Because there are apparent difficulties in targeting deregulated KRAS activity in pancreatic cancer, we and others have recently investigated the radiosensitizing potential of MEK inhibitors both in vitro in tumor cell lines and in vivo in mouse models.12,18-20 Although multiple MEK1/2 inhibitors have exhibited promising outcomes in preclinical studies, many early trials did not demonstrate benefit from MEK1/2 inhibition.21-23 This may be due to a narrow therapeutic window to spare non-tumor cells that rely on ERK1/2 signaling for their survival with older generation MEK inhibitors or lack of significant efficacy in unselected populations. However, more recent trials with more modern MEK1/2 inhibitors have shown substantial efficacy or improvements in clinical outcomes in BRAF/NRAS mutated melanoma, or KRAS mutated non-small cell lung cancer.24-27
A clinically useful MEK inhibitor is expected to produce prolonged MEK inhibition in tumor cells with minimal systemic toxicity resulting from targeting of non-tumor cells. To this end, GSK1120212 (GSK212 or trametinib), a potent and selective allosteric inhibitor of MEK1/2, has shown very promising antitumor activity in BRAF-driven melanoma.25-27 This ATP-non-competitive inhibitor produces sustained inhibition of ERK1/2 activity in human tumor cell lines expressing oncogenic BRAF and RAS in vitro and suppresses ERK1/2 activity in multiple tumor models when orally delivered in a single dose per day.28
While MEK1/2 inhibitors have shown activity pre-clinically as a radiosensitizer, a better characterization of the DNA repair pathways altered by MEK inhibition is needed. In the current study, GSK212 has been evaluated for its radiation sensitizing potential in vitro in human pancreatic cancer cells. DNA double-strand break (DSB) is recognized as the primary mechanism for radiation-induced tumor cell killing through the formation of chromosomal aberrations.29,30 Homologous recombination (HR) and non-homologous end-joining (NHEJ) are the 2 major pathways for repairing DSBs in mammalian cells and radioresistance is associated with upregulation of these pathways.30 Currently it is not clear whether MEK1/2 inhibitors sensitize cancer cells to radiation through alteration of DNA repair pathways. Here, we report that GSK212 significantly downregulates activity of both HR and NHEJ pathways through various DNA repair intermediates, thereby conferring radiosensitization to human pancreatic cancer cells.
Results
GSK212 produces dose- and time-dependent inhibition of ERK1/2 phosphorylation/activation and radiosensitization in multiple pancreatic cancer cell lines in vitro
To establish that the MEK1/2 inhibitor GSK1120212 (GSK212 or trametinib) sensitizes pancreatic cancer cells to ionizing radiation, we determined its minimum effective concentration(s) that completely inhibit(s) phosphorylation/activation of ERK1/2 by an early time point (1 hr). We found that 5–10 nM GSK212 completely inhibited ERK1/2 phosphorylation at 1hr in 2 representative KRAS mutant pancreatic cancer cell lines MIAPaCa-2 and AsPC-1 (Fig. 1A). Next, we measured the kinetics of ERK1/2 inhibition by 10 nM GSK212 in these 2 cell lines. We found that ERK1/2 phosphorylation was almost completely inhibited within 30 to 60 min at 10 nM and that complete inhibition was sustained for 24 hr in the continuous presence of the inhibitor (Fig. 1B). Similar results were obtained with additional pancreatic cancer cell lines (AsPC1 and BxPC3). These data clearly demonstrate that GSK212 at 10 nM concentration acts as an effective inhibitor of MEK1/2 with rapid and potent suppression of ERK1/2 activity in vitro in the pancreatic cancer cell lines examined.
Figure 1.
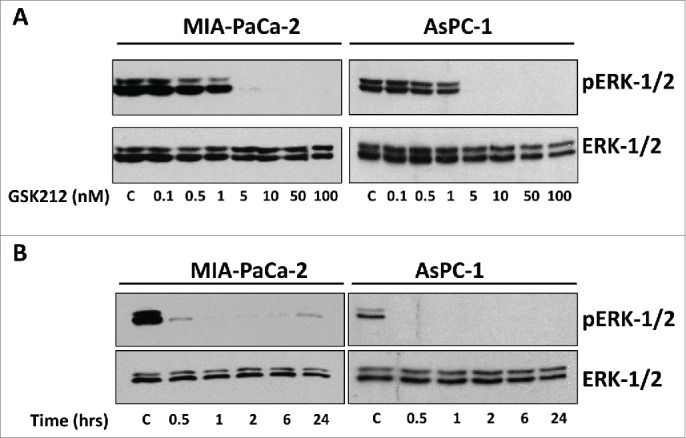
GSK212 is a potent MEK-1/2 inhibitor, demonstrating dose and time dependent inhibition of ERK-1/2. (A) Western blot images showing expression of total ERK-1/2 and phospho-ERK-1/2 in MIAPaCa-2 and AsPC1 cells treated for 1 hour with increasing doses of GSK212. ERK-1/2 activity is potently inhibited at 5–10 nM. (B) Western blot images showing rapid inhibition of ERK-1/2 activity within 30 min of treatment with 10 nM GSK212 that is sustained throughout 24 hours. C= control (DMSO). Multiple independent experiments were performed which showed similar results.
To determine the radiosensitizing potential of GSK212 in MIAPaCa-2 cells under conditions where ERK1/2 activity remained fully suppressed, radiation clonogenic survival assays were carried out. Our results found that treatment of MIAPaCa-2 cells for 1 hr with a low-dose of 10 nM GSK212 prior to delivering radiation (Fig. 2A) or 1 hr after irradiation (Fig. 2B) failed to radiosensitize cells in the continuous presence of GSK212 for 24 hr. However, when pretreated with 10 nM GSK212 for 24 hr (Fig. 2C) or 48 hr (Fig. 2D), MIAPaCa-2 cells became sensitized to radiation. These observations were confirmed in 2 other pancreatic cancer cells lines, AsPC-1 and BxPC-3 (Fig. S1A-B). As a control, GSK212 (10 nM) treatment for 48 hr prior to irradiation failed to radiosensitize a normal small intestinal epithelial cell line FHs74Int (Fig. S1C).
Figure 2.
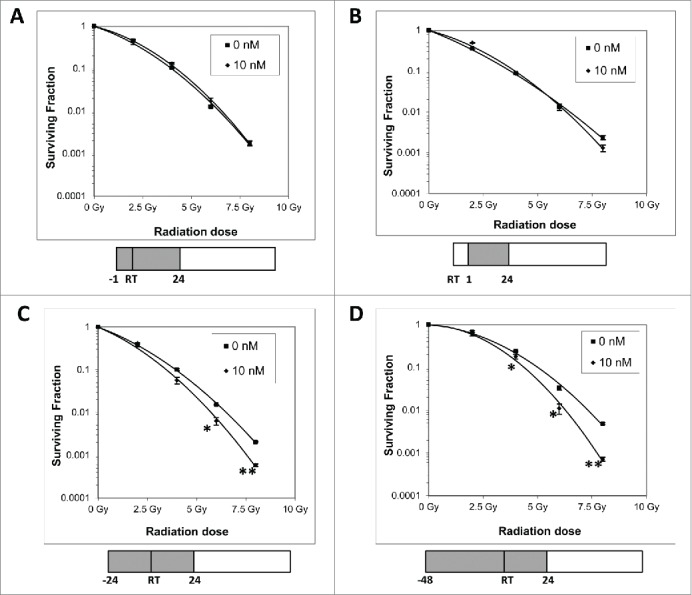
Prolonged MEK1/2 inhibition is required for effective radiosensitization. MIAPaCa-2 cells treated with 10 nM GSK212 or DMSO (“0 nM”) for 1 hour before radiation (A) or 1 hour after radiation (B) does not result in radiosensitization in clonogenic survival assays. However, pretreating cells with GSK212 for 24 (C) or 48 (D) hours prior to radiation results in significant radiosensitization. In all cases, GSK212 was left on for an additional 24 hours after radiation before removal from the medium. (*p <0.01; **p<0.001) For all assays, multiple independent experiments were performed which showed similar results.
Taken together, these data demonstrate that GSK212 at 5–10 nM concentrations completely inhibited the constitutive activation of ERK1/2 by 1 hr, and that sustained MEK inhibition for 24–48 hr is required for induction of significant radiosensitization compared with shorter time periods at which GSK212 produced very little or undetectable radiosensitization.
Effect of GSK212-mediated MEK1/2 inhibition on global DNA damage response pathways
The goal of radiation therapy is to eradicate tumor cells by producing a variety of lesions in DNA, including single-strand breaks and DSBs. To test if GSK212 alters the global DNA repair process in irradiated pancreatic cancer cells, neutral comet assays were conducted. The comet assay, also known as single cell gel electrophoresis (SCGE) assay, is a simple and sensitive technique for detecting DNA damage at the level of individual cells.31 We observed that pretreatment of MIAPaCa-2 cells with 10 nM GSK212 for 24 hr significantly delayed global DNA repair at 4 hr after exposure to 10 Gy radiation (Fig. 3A).
Figure 3.
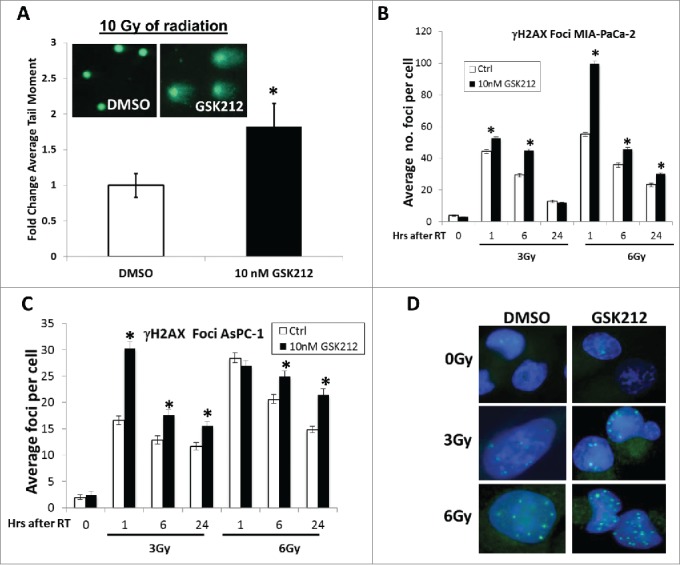
GSK212 treatment attenuates cellular DNA repair mechanisms. (A) Pretreatment of MIAPaCa-2 cells with 10 nM GSK212 for 24 hours prior to 10 Gy results in delayed repair compared to DMSO-treated cells, as measured 4 hours after radiation with a neutral comet assay (*p <0.05). Inset shows representative photos of comet tails taken with fluorescence microscopy at 4 hr after 10 Gy. (B-C) Pre-treatment of MIAPaCa-2 (B) or AsPC-1 (C) cells with 10 nM GSK212 for 24 hr followed by 3 or 6 Gy results in delayed resolution of nuclear γH2AX foci at 1, 6, and 24 hr after radiation compared to DMSO-treated (“Ctrl”) cells. (*p<0.001) (D) Representative photos of γH2AX foci at 24 hr after radiation for DMSO or 10 nM GSK212-treated MIAPaCa-2 cells. For all assays, multiple independent experiments were performed which showed similar results.
Efficient repair of DNA damage induces resistance to radiotherapy in cancer cells. The histone variant H2AX is a critical component of DNA repair process, which is rapidly phosphorylated by phophatidyl-inositol-3-kinase-like family Ser/Thr kinases including ATM, ATR and DNA-PK, on a unique C-terminal serine residue (Ser-139) to form γH2AX foci at nascent DSB sites.32 This is required for the mobilization of DNA repair proteins to the DSB sites.33 Thus, an accumulation of a large number of γH2AX molecules creates a focus at an individual DSB site, which allows the detection of DSBs with anti-γH2AX antibody.34 As DSBs are repaired, γH2AX foci are resolved. Here, the impact of GSK212 (10 nM) pretreatment for 24 hr on γH2AX foci resoluton was determined in MIAPaCa-2 and AsPC-1 cells at 1, 6, and 24 hr after 3 or 6 Gy radiation. We found that GSK212 pretreatment for 24 hr (followed by its continuous presence) delayed the resolution of radiation-induced γH2AX foci in multiple pancreatic cancer cell lines at 6 and 24 hr, suggesting that MEK1/2 activity is required for efficient repair of radiation-induced DSBs in pancreatic cancer cells (Fig. 3B-D).
Effect of GSK212-mediated MEK1/2 inhibition on DSB repair by HR
Since the development of radioresistance in human tumor cells can be caused by an up-regulation of DSB repair pathways, we examined the impact of GSK212 on both HR and NHEJ repair pathways in irradiated MIAPaCa-2 and AsPC-1 cells. BRCA1, encoded by the breast cancer associated gene 1 (BRCA1), plays an essential role in the HR repair pathway, and has controversial roles in the NHEJ and nucleotide excision repair pathways.30,35,36 In vitro studies reveal that BRCA1 deficiency is associated with increased radiosensitization resulting from defects in the HR repair pathway.37 Moreover, embryonic fibroblasts derived from BRCA1−/− mice are susceptible to radiation, implicating a central role of BRCA1 in DNA repair.30 These findings prompted us to examine the effects of GSK212 on radiation-induced BRCA1 foci formation in the nuclei of MIAPaCa-2 and AsPC-1 cells. Our data reveal that MEK1/2 inhibition in pancreatic cancer cells caused a substantial reduction in nuclear BRCA1 foci after irradiation (Figs. 4A-B). These data demonstrate that BRCA1 foci formation, which is required for facilitating HR repair in response to radiation-induced DSBs,38 was impaired by GSK212-mediated inhibition of MEK1/2.
Figure 4.

MEK1/2 inhibition results in attenuation of homologous recombination DNA double strand break repair. (A-B) Pre-treatment of MIAPaCa-2 (A) or AsPC-1 (B) cells with 10 nM GSK212 for 24 hr followed by 3 or 6 Gy results in defects in the formation of nuclear BRCA1 foci at 1, 6, and 24 hr after radiation compared to DMSO-treated (“Ctrl”) cells. (*p <0.001) (C) MIAPaCa-2-DR-GFP cells were treated with 5 μL of I-SceI adenovirus to induce DNA double-strand breaks in the target DNA recognition sequence, and subjected to flow cytometry either in the presence of DMSO (vehicle), or 10 nM GSK212 for either 24 hr (“late GSK212”) or 42 hr (“early GSK212”). Red indicates the presence of GSK212 in the medium of cells in the schematics accompanying each panel. Percent GFP-positive cells were quantified by flow cytometry. Note that early treatment of GSK212, but not late treatment, results in defects in functional HR repair as measured by a decrease in the %GFP positive cells compared to DMSO-treated cells (15.5% DMSO, 16.3% late GSK212, 6.6% early GSK212). (D) Mean of 3 independent experiments shown, which independently showed similar results.
To provide further evidence of dysregulation of the HR repair pathway, we studied the formation of RAD51 foci. RAD51 molecules accumulate at DSB sites and form nucleoprotein filaments which can be visualized by immunofluorescence microscopy as RAD51 foci.39 Because overexpression of RAD51 upregulates the HR repair process and increases radioresistance in eukaryotic cells,40 we examined if GSK212 compromises RAD51 foci formation in irradiated pancreatic cancer cells. We found that GSK212 also significantly decreased the number of RAD51 foci after irradiation of MIAPaCa-2 cells (Fig. S2).
To further demonstrate that GSK212-mediated MEK1/2 inhibition suppresses HR repair in irradiated pancreatic cancer cells, we used a previously validated MIAPaCa-2 stable cell line bearing a green fluorescent protein (GFP) reporter construct DR-GFP.41 This DR-GFP construct measures homology-directed repair of I-SceI endonuclease-induced DSBs within the genome-integrated DR-GFP reporter construct.42 MIAPaCa-2-DR-GFP cells were infected with adenovirus expressing I-SceI, to measure HR repair activity in the presence and absence of GSK212, by determining the percentage of GFP-positive cells 42 hr post-infection by flow cytometry. Our data confirm that GSK212 markedly decreased functional HR repair in cells when MEK1/2 inhibition occurred at the time of DNA damage induced by I-SceI, compared with MEK1/2 inhibition applied after induction of DNA damage (Fig. 4C, D).
Taken together, our data demonstrate that GSK212-mediated radiosensitization of pancreatic cancer cells is associated with the downregulation of DSB repair activity through the HR repair pathway.
Effect of GSK212-mediated MEK1/2 inhibition on DSB repair by NHEJ
NHEJ is another dominant repair pathway for radiation-induced DSBs in mammalian cells in which Ku70/Ku80 heterodimer recruits the catalytic subunit of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) to DSB sites. Subsequent dimerization of DNA-PKcs upregulates its catalytic activity through autophosphorylation and phosphorylation of other proteins involved in the NHEJ repair process.30,43,44 Since upregulation of DNA-PK enhances the DSB repair process rendering tumors radioresistant in preclinical 45 and clinical studies,46-48 we determined the effects of GSK212 on radiation-induced DNA-PK foci formation in MIAPaCa-2 and AsPC-1 cells. We noted that GSK212 significantly increased the presence of DNA-PK foci at various time points after radiation in both cell lines, including persistence of nuclear foci at 6 and 24 hr after radiation (Figs. 5A-B).
Figure 5.

MEK1/2 inhibition attenuates non-homologous end joining repair DNA double strand break repair. (A-B) Pre-treatment of MIAPaCa-2 (A) or AsPC-1 (B) cells with 10 nM GSK212 for 24 hr followed by 3 or 6 Gy results in an increase in nuclear DNA-PK foci at 1, 6, and 24 hr after radiation in DMSO-treated (“Ctrl”) cells. (*p <0.001) (C). MIAPaCa-2-Pem1-GFP cells were treated with 5 μL of I-SceI adenovirus to induce DNA double-strand breaks in the NHEJ reporter construct, and subjected to flow cytometry either in the presence of DMSO (control), or 10 nM GSK212 for either 24 hr (“late GSK212”) or 40 hr (“early GSK212”). Percent GFP-positive cells were quantified by flow cytometry. Note that early treatment of GSK212 (but not late treatment) results in defects in functional NHEJ repair as measured by a decrease in the %GFP positive cells compared to DMSO-treated cells (8.7% DMSO, 3.1% late GSK212, 2.7% early GSK212). (D) Mean of 4 independent experiments shown, which independently showed similar results.
Another common marker of NHEJ repair is the 53BP1 protein. DSBs induce a rapid co-localization of the p53-binding protein 53BP1 with γH2AX and other DNA repairing proteins including BRCA1 to form discrete nuclear foci.49,50 Because mice lacking 53BP1 are highly susceptible to radiation,51,52 we sought to determine the effects of GSK212 on radiation-induced 53BP1 foci formation in MIAPaCa-2 and AsPC-1 cells. Here, we found that the kinetics of radiation-induced 53BP1 foci resolution were also altered by GSK212 treatment in both cell lines compared to DMSO-treated cells (Fig. S2). These results paralleled those observed with DNA-PK, with more 53BP1 foci observed after radiation. Taken together, these data initially suggested to us that activation of NHEJ was compensating for loss of HR repair activity, or that kinetics of DNA repair through NHEJ were altered/delayed.
To more directly determine whether GSK212 induces radiosensitivity by altering NHEJ repair, a fluorescent reporter construct that allows sensitive and quantitative measurement of NHEJ repair was employed. Similar to the DR-GFP construct for HR activity, this construct (Pem-1-NHEJ) contains an engineered GFP gene containing recognition sites for I-SceI endonuclease for induction of DSBs, which does not express GFP in the absence of NHEJ repair of I-SceI-induced DSBs. Successful repair of DSBs by NHEJ repair results in GFP expression, and the number of GFP positive cells counted by flow cytometry provides a quantitative measure of NHEJ repair activity.53 We created stable MIAPaCa-2-Pem1-GFP cells (clone N4) and validated reporter activity with a DNA-PK inhibitor, NU-7441 (Fig. S4). Next, we tested MIAPaCa-2-Pem1-GFP cells with GSK212 to determine the effects of MEK1/2 inhibition on NHEJ repair. We found that GSK212 also significantly attenuated NHEJ repair both when MEK was inhibited at the time of DNA damage or after, suggesting that GSK212 also acts as an effective inhibitor of NHEJ repair in pancreatic cancer cells (Figs. 5C,D).
GSK212 downregulates the phosphorylation and expression of multiple DSB repairing proteins
To further elucidate the biochemical mechanisms underlying GSK212-mediated radiosensitization of pancreatic cells, we evaluated the alterations in expression/activation of a number of key proteins that are involved in the global DNA damage response and repair pathways. For example, BRCA1 is phosphorylated on specific serine residues in response to radiation, and is differentially regulated in S- and G2/M phases of the cell cycle. Ionizing radiation induces phosphorylation of Ser-988/Ser-1524 on BRCA1 during the S phase and of Ser-988/Ser-1423 during the G2/M phase, which is catalyzed by a number of Ser/Thr kinases that include ATM, ATR, cdk2, and Chk2.54 We found that GSK212 pretreatment for 24 h markedly decreased total BRCA1 expression (and therefore the presence of radiation-induced phosphorylation of BRCA1 on Ser-1524) in MIAPaCa-2 cells (Fig. 6A, B). Interestingly, RAD51 expression was also markedly decreased in the presence of GSK212 (Fig. 6A). With regard to NHEJ repair, ionizing radiation induces rapid phosphorylation of DNA-PKcs on Thr-2609 cluster and on Ser-2056, which is critical for the repair of DSBs.55 Our data also show that GSK212 pre-treatment decreased total DNA-PK expression (and therefore the presence of radiation-induced phosphorylation of DNA-PK on Ser-2056) in MIAPaCa-2 cells under the same conditions (Fig. 6A). Conversely, 53BP1 expression was upregulated in cells treated with GSK212.
Figure 6.
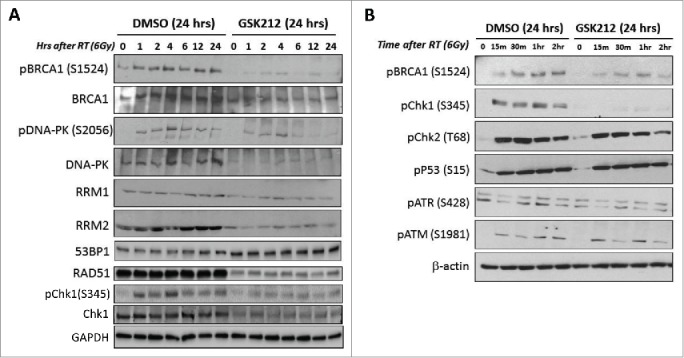
MEK1/2 inhibition results in inactivation or loss of multiple DNA repair pathway intermediates including BRCA1, DNA-PK, RRM2, RAD51, and Chk1. MIAPaCa-2 cells were pre-treated with DMSO (control) or GSK212 for 24 hr then radiated with 6 Gy. (A) At 0, 1, 2, 4, 6, 12, and 24 hours after 6 Gy, lysates were made and subjected to immunoblotting. Note the loss of radiation-induced activation of BRCA1 and DNA-PK, as well as loss of RRM2. (B) At 0.25, 0.5, 1, and 2 hr after 6 Gy, lysates were made and subjected to immunoblotting. Note the attenuation or loss of radiation-induced activation of BRCA1 and Chk1, that appears independent of ATM, ATR, p53, or Chk2.
Ribonucleotide reductase (RR) catalyzes the synthesis of deoxyribonucleoside triphosphates necessary for both DNA synthesis and DNA repair, and is a heterodimer consisting of 2 subunits, M1 and M2.56 RRM2 expression increases with the progression of cell cycle while the RRM1 expression remains at a steady-state level throughout the cell cycle.57 We found that RRM2 baseline and radiation-induced expression were markedly decreased by GSK212 pretreatment of MIAPaCa-2 cells when compared with RRM1 expression (Fig. 6A). We have also examined the effects of GSK212 pretreatment on total Chk1, as well as the early phosphorylation status of Chk1 (Ser-345), Chk2 (Thr-68), p53 (Ser-15), ATR (Ser-428), and ATM (Ser-1981), which are involved in the initial regulation of DNA damage response.30 Our data show that under the present experimental conditions, only total and phospho-Chk1 were significantly attenuated by GSK212 treatment of MIAPaCa-2 cells, with the Chk2 and p53 pathways largely unaffected (Fig. 6A, B). This reduction in Chk1 phosphorylation persisted throughout 24 hr (Fig. 6A). Earlier studies have shown that phosphorylation of Chk1 on Ser-317 and Ser-345 catalyzed by the upstream kinase ATR is critical for checkpoint activation in the S-phase and DNA damage response in the G2-phase.58,59 We therefore evaluated this kinase as well at the ATM kinase which activates Chk2 after radiation, but detected no differences in the activation state of either kinase (Fig. 6B).
Together, our data reveal that MEK1/2 inhibition led to the downregulation of multiple intermediates of the DSB repair pathways, including BRCA1, RAD51, DNA-PK, RRM2, and Chk-1 in irradiated cells, providing potential mechanisms for GSK212-mediated radiosensitization.
Discussion
The use of chemotherapy and radiotherapy remains a standard practice for the treatment of unresectable pancreatic cancer. In addition, radiation is often utilized in the post-operative management of resectable pancreatic cancer. Despite this, patient outcomes for pancreatic cancer remain dismal. Since KRAS acquires an activating oncogenic mutation in almost all pancreatic cancer, which activates downstream signaling pathways (RAF-MEK-MAPK and PI3K-Akt), conferring resistance to radiotherapy as well as chemotherapy, there is an urgent need for optimizing the current therapeutic regimens by incorporating novel agents that sensitize malignant cells to radiation and/or chemotherapy. To this end, a number of MEK inhibitors have shown promising outcome in preclinical studies and clinical trials in different disease sites.18,24,26,28 In pancreatic cancer preclinical studies, the use of MEK inhibitors seems to show some promise either alone or in combination with other agents, such as STAT3 inhibitors or CDK4/6 inhibitors.60-62 These combinations have been shown to be highly effective and synergistic in certain models, resulting in potent inhibition of tumor cell growth. A number of preclinical studies have demonstrated the benefit of MEK1/2 inhibition in combination with radiation with various types of MEK-1/2 inhibitors.12,18,19,63 This finding has led to the development of a number of phase I clinical trials in the United States testing the addition of MEK1/2 inhibition in combination with radiation for rectal, lung, and brain cancer (NCT01912625; NCT02015117; NCT01740648).
GSK212 (trametinib or Mekinist™) is a novel and potent MEK-1/2 inhibitor, and is FDA-approved for the treatment of BRAF V600E or V600K mutated melanoma.25,26 One of the advantages of using newer generation MEK1/2 inhibitors such as GKS212, is that they are highly selective for their targets by virtue of binding to a unique pocket that is adjacent to, but distinct from, the ATP-binding site. This offers the potential for less mutation rates compared to other small molecule inhibitors which bind to the ATP-binding site. Furthermore MAP kinases ERK1 and ERK2 are the only known substrates for the MEK isoforms highlighting their tight specificity.64,65 Thus, blocking MEK1/2 will likely result in fewer off-target un-intended effects.
The goal of the current study was to evaluate the radiosensitizing potential of a GSK212 in pancreatic cancer cells, which has shown promising efficacy in BRAF-mutated melanoma.25,26 Our key findings reported in this article are the following ones: (1) MEK-1/2 inhibition by GSK212 radiosensitizes multiple pancreatic cancer cell lines including MIAPaCa-2 and AsPC-1, which both have KRAS oncogenic mutations (G12C and G12D, respectively) 66,67 (2) Longer treatment time with GSK212 is required for effective radiosensitization (which has implications for clinical implementation); (3) GSK212-mediated radiosensitization is caused by functional inhibition of both HR and NHEJ repair pathways resulting in delayed DNA repair as evidenced by comet assay and persistent γH2AX expression; and (4) GSK212-mediated MEK1/2 inhibition decreases the expression of established proteins directly or indirectly involved in DSB repair or DNA damage response signaling, which include BRCA1, RAD51, DNA-PK, RRM2, and Chk-1. These data support the clinical investigation of GSK212 in combination with radiation for pancreatic cancer, which harbors high rates of KRAS activating mutations which promote radioresistance. Furthermore, our data suggest that p-BRCA1 (S1524), BRCA1, RAD51, p-DNA-PK (S2056), DNA-PK, RRM2, Chk1, and pChk1 (S345) might serve as useful biomarkers to predict the clinical efficacy of GSK212 in clinical trials.
Ionizing radiation kills tumor cells largely by inducing DSBs in DNA leading to genomic instability, apoptosis, or post-mitotic death. These DSBs are primarily repaired by HR and NHEJ.30 However, we and others have observed that radiation also induces MEK-ERK and PI3K-Akt pathway activation, which provides both proliferation and survival signals in pancreatic cancer cells rendering them radioresistant, as blocking either or both of these pathways induces radiosensitization.12 Up-regulation of DNA repair pathways is recognized as a primary acquired mechanism by which malignant cells become radioresistant. In accord with this notion, we found that a potent and specific MEK1/2 inhibitor GSK212 radiosensitizes pancreatic cancer cells by inhibiting the repair of radiation-induced DSBs, by functionally decreasing efficacy of both HR and NHEJ pathways, which is a novel finding.
Our data reveal that GSK212 significantly reduces the number of nuclear foci formed by both BRCA1 and RAD51, demonstrating that the HR repair pathway is impaired by MEK1/2 inhibition. These observations were confirmed by using HR-specific functional reporter constructs in pancreatic cancer cells. Our initial data on DNA-PK and 53BP1 foci suggested to us that the cancer cells up-regulated NHEJ activity in order to compensate for reductions in HR activity with GSK212 treatment. However, on further evaluation, we found that the NHEJ repair pathway is also inhibited by GSK212 as assessed by a specific and well-characterized functional NHEJ reporter.53 Further confirmation of the NHEJ functional reporter data was provided by immunoblotting, which showed that GSK212 treatment resulted in significant reductions in DNA-PK and pDNA-PK expression, a critical NHEJ repair protein.
With regard to homologous recombination, RAD51 is an important component of HR-mediated DNA repair, as RAD51 over-expression results in increased homologous recombination repair and resistance to radiation.40 RAD51 has been shown to be overexpressed in the majority of pancreatic cancer, providing further rationale for the approach of combining GSK212 with radiation.64 Our data also show that BRCA1-foci formation was significantly impaired when GSK212-treated pancreatic cancer cells are exposed to radiation. G2/M cell cycle arrest is one of the multiple functions BRCA1 performs in the cellular response to DNA damage through its interactions with different protein partners.68 It has been previously shown that suppression of ERK1/2 activity using MEK1/2-specific inhibitor U0126 induces inhibition of BRCA1-mediated G2/M cell cycle arrest in breast cancer cells, indicating that ERK1/2 activity is required for G2/M arrest.69 This study has further established that ERK1/2 inhibition through MEK1/2 inhibition abrogated the effects of BRCA1 on components of the G2/M checkpoint, including activation of Chk1 kinase. Our data reveal that GSK212-mediated ERK1/2 inhibition impairs the phosphorylation/activation of BRCA1 and Chk1 in pancreatic cancer cells, suggesting that GSK212-mediated ERK1/2 inhibition prevents proper radiation-induced G2/M checkpoint arrest to allow for proper DNA repair. Presumably, this could lead to lethal genomic events, as unrepaired DNA damage results in post-mitotic cell death. This notion is supported by an earlier study demonstrating that MEK2 activation is essential for progression through the G2/M checkpoint arrest in cells that are exposed to ionizing radiation.70
Likewise, both RRM2 and Chk1 have roles in DNA repair. RRM2 is upregulated after DNA damage, and has been shown to be important for mediating repair of a broad array of DNA damage induced by ultraviolet radiation, ionizing radiation, genotoxic stress, or genotoxic compounds such as camptothecin or doxorubicin.71,72 Interestingly, Chk1 activation appears important for DNA damage induced upregulation of RRM2.71 Furthermore, RRM2 expression is up-regulated by oncogenic KRAS and controls tumor cell proliferation, while downregulation of RRM2 can sensitize cells to genotoxic compounds like gemcitabine, an inhibitor of RRM1.73,74 Chk1 has also been shown to be important in DNA repair, as Chk1 inhibition results in pancreatic cancer cell radiosensitization, through loss of the G2 cell cycle checkpoint and inhibition of HR repair, as mentioned above.41 Together, these proteins have defined roles in DNA damage response signaling to promote DNA repair, and whose function is down-regulated by MEK-1/2 inhibitors.
In summary, our data demonstrate that sustained suppression of constitutive (resulting from activating KRAS mutation) and radiation-induced activation of ERK1/2 by the MEK1/2-specific inhibitor GSK212 sensitizes pancreatic cancer cells (but not normal small intestinal epithelial cells) to radiation by impairing proper DSB repair through altering the functionality of both HR and NHEJ pathways. Together with the in vivo data previously published for MEK inhibition in combination with radiation for pancreatic cancer,12 these data support the development of a clinical trial combining MEK-1/2 inhibition and radiation for the treatment of pancreatic cancer.
Materials and Methods
Antibodies, chemicals, and cell culture
The human pancreatic adenocarcinoma cell lines MIAPaCa-2, AsPC-1, BxPC3, and FHs74Int were obtained from American Type Culture Collection (Manassas, VA) and were maintained at 37°C in 5% CO2 in DMEM media (MIAPaCa-2, FHs74Int) or RPMI 1640 (AsPC-1 and BxPC3), and supplemented with 10% fetal bovine serum (Hyclone, Logan, UT) and penicillin/streptomycin (Life Technologies, Grand Island, NY). GSK1120212 (GSK212) (ChemieTek, Indianapolis, IN) was dissolved in DMSO and added to the cells for 48 hours unless stated differently. Total ERK-1/2, phospho-ERK-1/2 (Thr202/Tyr204), phospho-BRCA1(Ser1524), phospho-DNAPK(Ser2056), total BRCA1, total DNAPK, phospho-H2AX, RAD51, 53BP1, RRM1,RRM2, phospho-Chk1(Ser345), phospho-Chk2(T68), phospho-p53(Ser15), phospho-ATR(Ser428), phospho-ATM(Ser1981) and β actin antibodies were purchased from Cell Signaling Technology (Danvers, MA).
Immunoblotting
Immunoblotting was performed as described previously.75 Briefly, cell lysates were prepared using RIPA buffer (ThermoFisher, Waltham MA) supplemented with 1x protease (Complete, Roche, Indianapolis, IN) and phosphatase inhibitors (PhosSTOP, Roche, Indianapolis, IN, Roche) followed by protein quantification by the Dc protein assay kit (Bio-Rad, Hercules, CA). Equal amounts of protein were loaded and resolved by SDS/PAGE and transferred to nitrocellulose membranes. Primary antibodies were allowed to bind overnight at 4°C, and used at a dilution of 1:500-1,000. After washing in TBS-Tween, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies diluted 1:2,500-1:5,000 for 1 hour. Membranes were washed with TBS-Tween and incubated for 1 minute with enhanced chemiluminescence reagent (Immobilon, Millipore, Billerica, MA) prior to film exposure.
Radiation Clonogenic Assays
Cells were trypsinized to generate single cell suspensions and seeded onto 60 mm tissue culture plates in triplicate. Cells were incubated with DMSO or GSK212 at the indicated times and then irradiated with various doses (0–8 Gy). Twenty-four hours after radiation, medium was changed to remove DMSO or GSK212. Ten to 14 days after seeding, colonies were fixed with Methanol/Acetic Acid, stained with 0.5% crystal violet and the numbers of colonies or colony forming units (CFU) containing at least 50 cells were counted using a dissecting microscope (Leica Microsystems, Inc.. Buffalo Grove, IL) and surviving fractions calculated. Experiments were repeated multiple, independent times.
Experimental Radiation
Irradiation was performed essentially as described previously with 160kV, 25mA at a dose rate of approximately 113cGy/min using a RS-2000 biological irradiator (RadSource,GA).75
Neutral Comet Assay
Cells were treated with DMSO or GSK212 for different time points as stated. Neutral comet assay was performed according to the manufacturer's protocol (Trevigen, USA). Briefly, cells were suspended and mixed with low melting-point agarose at 37°C. The mixture was pipetted onto slides and placed in the dark at 4°C for 10 minutes. Slides were immersed in cold lysis solution overnight at 4°C. On the next day, slides were immersed in 50 ml of cold Neutral Electrophoresis Buffer for 30 minutes. Neutral electrophoresis was performed at 110 milliAmps, 21 Volts for 45 minutes. Slides were then placed in DNA Precipitation Solution for 30 minutes at room temperature followed by 70% ethanol for 30 minutes. Samples were dried at 37°C and then diluted SYBR Gold was added to them. Slides were then briefly rinsed and dried completely. Slides were viewed by epifluorescence microscopy. At least 50 cells were counted and imaged. CometScore software (TriTek Corp, Sumerduck, VA) was used to quantify the average tail moment.
Immunofluorescence for Nuclear Foci
For phospho-H2AX and α tubulin: Cells plated on coverslips were fixed with 2% paraformaldehyde, permeabilized with 1% Triton X-100 and blocked with 3% bovine serum albumin (BSA) in PBS. Cells were stained with anti-phospho-H2AX (Cell Signaling, Danvers, MA) or α-tubulin (Sigma, St. Louis, MO), washed and incubated with a fluorophore-conjugated secondary antibody (Biotium, Hayward, CA).
For 53BP1, BRCA1, and DNA-PK: Cells were fixed with 3%, 4%, and 2% paraformaldehyde, respectively, then permeabilized with 0.2–1% Triton X-100 and blocked with 3% BSA in PBS. 10% BSA was used for BRCA1. Cells were stained with anti-53BP1 (Cell Signaling, Danvers, MA), anti-BRCA1 (Santa Cruz Biotechnologies, Santa Cruz, CA), or anti-DNA-PK (AbCam, Cambridge, MA) followed by fluorophore-conjugated secondary antibody.
For RAD51: Cells were fixed with 4% paraformaldehyde, permeabilized with 0.25% Triton X-100 and blocked in 1% goat serum in PBS. Anti-RAD51 (Santa Cruz) was used followed by a fluorophore-conjugated secondary antibody.
Slides were examined on a Zeiss fluorescence microscope. For each experiment the total number of foci per cell was determined in at least 100 cells.
Flow cytometry
A stable clone of MIAPaCa-2 cells transfected with the pDR-GFP plasmid has been previously characterized,41 and was used to measure HR activity. In order to measure NHEJ activity, stable clones of MIAPaCa-2 cells transfected with NHEJ-GFP-PEM1 plasmid (generously provided by V. Gorbunova), were created by transient transfection using Lipofectamine 2000 and selection with 1 mg/mL G418 according to a previously published protocol.53 NHEJ-GFP-PEM1 clones were selected based on identifying clones with the highest percentage of green fluorescent protein (GFP) positivity 24 hr after infection with the adenovirus AdNGUS24i using flow cytometry analysis. In addition, the NHEJ-GFP-PEM1 construct was validated in MIAPaCa2 cells by treatment with an inhibitor of NHEJ, NU-7441 (Selleckchem, Houston,TX) as described. For the experiments, MIAPaCa2-DR-GFP or –NHEJ-GFP-PEM1 cells were treated with adenovirus expressing the I-SceI restriction enzyme that induces double-strand DNA breaks in the recognition sequence within the reporter construct. Cells were also pretreated with DMSO or GSK212 using different variations in scheduling as described. Cells were then washed with ice-cold PBS and fixed in cold 1% formaldehyde solution for 30 minutes. Cells were then washed and I-SceI–induced homologous recombination (HR) and non-homologous end-joining (NHEJ) were measured by flow cytometry as the percentage of GFP positive cells. Cells were sorted through BD LSR II flow cytometer.
Data Analysis
Data are presented as the mean ± standard error of the mean (s.e.m.) for clonogenic survival and foci experiments. Statistical comparisons were made between the control and experimental conditions using the unpaired 2-tailed Student's t-test with significance assessed at p-values <0.05.
Funding Statement
This work was supported by the following grants: ASTRO Junior Faculty Career Research Training Award (TW), American Society for Radiation Oncology (Grant: JF2013-2), and the National Institutes of Health National Center for Advancing Translational Sciences (Grant: KL2TR001068 to TW).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Advancing Translational Sciences or the National Institutes of Health. The authors would like to thank M. Freeman for providing comments on this manuscript.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.American Cancer Society , 2014. [Google Scholar]
- 2. SEER Stat Fact Sheets: Pancreatic Cancer, 2014. [Google Scholar]
- 3.Duldulao MP, Lee W, Nelson RA, Li W, Chen Z, Kim J, Garcia-Aguilar J. Mutations in specific codons of the KRAS oncogene are associated with variable resistance to neoadjuvant chemoradiation therapy in patients with rectal adenocarcinoma. Ann Surg Oncol 2013; 20(7):p. 2166-71; PMID:23456389; http://dx.doi.org/ 10.1245/s10434-013-2910-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKenna WG, Muschel RJ, Gupta AK, Hahn SM, Bernhard EJ. The RAS signal transduction pathway and its role in radiation sensitivity. Oncogene 2003; 22(37):p. 5866-75; PMID:12947393; http://dx.doi.org/ 10.1038/sj.onc.1206699 [DOI] [PubMed] [Google Scholar]
- 5.Sklar MD, The ras oncogenes increase the intrinsic resistance of NIH 3T3 cells to ionizing radiation. Science 1988; 239(4840):p. 645-7; PMID:3277276; http://dx.doi.org/ 10.1126/science.3277276 [DOI] [PubMed] [Google Scholar]
- 6.Brunet A, Roux D, Lenormand P, Dowd S, Keyse S, Pouysségur J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J 1999; 18(3):p. 664-74; PMID:9927426; http://dx.doi.org/ 10.1093/emboj/18.3.664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 2006; 24(1):p. 21-44; PMID:16393692; http://dx.doi.org/ 10.1080/02699050500284218 [DOI] [PubMed] [Google Scholar]
- 8.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes & development 2003; 17(24):p. 3112-26; PMID:14681207; http://dx.doi.org/ 10.1101/gad.1158703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins MA, Bednar F, Zhang Y, Brisset JC, Galbán S, Galbán CJ, Rakshit S, Flannagan KS, Adsay NV, Pasca di Magliano M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. The Journal of clinical investigation 2012; 122(2):p. 639-53; PMID:22232209; http://dx.doi.org/ 10.1172/JCI59227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baer R, Cintas C, Dufresne M, Cassant-Sourdy S, Schönhuber N, Planque L, Lulka H, Couderc B, Bousquet C, Garmy-Susini B, et al.. Pancreatic cell plasticity and cancer initiation induced by oncogenic Kras is completely dependent on wild-type PI 3-kinase p110alpha. Genes Dev 2014; 28(23):p. 2621-35; PMID:25452273; http://dx.doi.org/ 10.1101/gad.249409.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Junttila MR, Devasthali V, Cheng JH, Castillo J, Metcalfe C, Clermont AC, Otter DD, Chan E, Bou-Reslan H, Cao T, et al.. Modeling Targeted Inhibition of MEK and PI3 Kinase in Human Pancreatic Cancer. Mol Cancer Ther 2015; 14(1):p. 40-7; PMID:25376606; http://dx.doi.org/ 10.1158/1535-7163.MCT-14-0030 [DOI] [PubMed] [Google Scholar]
- 12.Williams TM, Flecha AR, Keller P, Ram A, Karnak D, Galbán S, Galbán CJ, Ross BD, Lawrence TS, Rehemtulla A, et al.. Cotargeting MAPK and PI3K signaling with concurrent radiotherapy as a strategy for the treatment of pancreatic cancer. Molecular cancer therapeutics 2012; 11(5):p. 1193-202; PMID:22411900; http://dx.doi.org/ 10.1158/1535-7163.MCT-12-0098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM, Sellers WR, Lengauer C, Stegmeier F. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res 2009; 69(10):p. 4286-93; PMID:19401449; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-4765 [DOI] [PubMed] [Google Scholar]
- 14.Balmanno K, Chell SD, Gillings AS, Hayat S, Cook SJ. Intrinsic resistance to the MEK1/2 inhibitor AZD6244 (ARRY-142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines. Int J Cancer 2009; 125(10):p. 2332-41; PMID:19637312; http://dx.doi.org/ 10.1002/ijc.24604 [DOI] [PubMed] [Google Scholar]
- 15.Golding SE, Rosenberg E, Neill S, Dent P, Povirk LF, Valerie K. Extracellular signal-related kinase positively regulates ataxia telangiectasia mutated, homologous recombination repair, and the DNA damage response. Cancer Res 2007; 67(3):p. 1046-53; PMID:17283137; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-2371 [DOI] [PubMed] [Google Scholar]
- 16.Kharbanda S, Saleem A, Shafman T, Emoto Y, Weichselbaum R, Kufe D. Activation of the pp90rsk and mitogen-activated serine/threonine protein kinases by ionizing radiation. Proc Natl Acad Sci U S A 1994; 91(12):p. 5416-20; PMID:8202500; http://dx.doi.org/ 10.1073/pnas.91.12.5416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonner JA, Vroman BT, Christianson TJ, Karnitz LM. Ionizing radiation-induced MEK and Erk activation does not enhance survival of irradiated human squamous carcinoma cells. Int J Radiat Oncol Biol Phys 1998; 42(4):p. 921-5; PMID:9845123; http://dx.doi.org/ 10.1016/S0360-3016(98)00325-3 [DOI] [PubMed] [Google Scholar]
- 18.Chung EJ, Brown AP, Asano H, Mandler M, Burgan WE, Carter D, Camphausen K, Citrin D. In vitro and in vivo radiosensitization with AZD6244 (ARRY-142886), an inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2 kinase. Clin Cancer Res 2009; 15(9):p. 3050-7; PMID:19366835; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shannon AM, Telfer BA, Smith PD, Babur M, Logie A, Wilkinson RW, Debray C, Stratford IJ, Williams KJ, Wedge SR. The mitogen-activated protein/extracellular signal-regulated kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) enhances the radiation responsiveness of lung and colorectal tumor xenografts. Clin Cancer Res 2009; 15(21):p. 6619-29; PMID:19843666; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-2958 [DOI] [PubMed] [Google Scholar]
- 20.Lin SH, Zhang J, Giri U, Stephan C, Sobieski M, Zhong L, Mason KA, Molkentine J, Thames HD, Yoo SS, et al.. A high content clonogenic survival drug screen identifies mek inhibitors as potent radiation sensitizers for KRAS mutant non-small-cell lung cancer. J Thorac Oncol 2014; 9(7):p. 965-73; PMID:24922006; http://dx.doi.org/ 10.1097/JTO.0000000000000199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rinehart J, Adjei AA, Lorusso PM, Waterhouse D, Hecht JR, Natale RB, Hamid O, Varterasian M, Asbury P, Kaldjian EP, et al.. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004; 22(22):p. 4456-62; PMID:15483017; http://dx.doi.org/ 10.1200/JCO.2004.01.185 [DOI] [PubMed] [Google Scholar]
- 22.Lorusso PM, Adjei AA, Varterasian M, Gadgeel S, Reid J, Mitchell DY, Hanson L, DeLuca P, Bruzek L, Piens J, et al.. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol 2005; 23(23):p. 5281-93; PMID:16009947; http://dx.doi.org/ 10.1200/JCO.2005.14.415 [DOI] [PubMed] [Google Scholar]
- 23.Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, Hanson LJ, Gore L, Chow L, Leong S, et al.. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol 2008; 26(13):p. 2139-46; PMID:18390968; http://dx.doi.org/ 10.1200/JCO.2007.14.4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, Franke FA, Grinsted L, Zazulina V, Smith P, et al.. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol 2013; 14(1):p. 38-47; PMID:23200175; http://dx.doi.org/ 10.1016/S1470-2045(12)70489-8 [DOI] [PubMed] [Google Scholar]
- 25.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, et al.. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 2012; 367(18):p. 1694-703; PMID:23020132; http://dx.doi.org/ 10.1056/NEJMoa1210093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, et al.. Improved survival with MEK inhibition in BRAF-mutated melanoma. The New England journal of medicine 2012; 367(2):p. 107-14; PMID:22663011; http://dx.doi.org/ 10.1056/NEJMoa1203421 [DOI] [PubMed] [Google Scholar]
- 27.Kim KB, Kefford R, Pavlick AC, Infante JR, Ribas A, Sosman JA, Fecher LA, Millward M, McArthur GA, Hwu P, et al.. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol 2013; 31(4):p. 482-9; PMID:23248257; http://dx.doi.org/ 10.1200/JCO.2012.43.5966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, Rominger CM, Erskine S, Fisher KE, Yang J, et al.. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res 2011; 17(5):989-1000; PMID:21245089 [DOI] [PubMed] [Google Scholar]
- 29.Povirk LF, Biochemical mechanisms of chromosomal translocations resulting from DNA double-strand breaks. DNA Repair (Amst) 2006; 5(9-10):p. 1199-212; PMID:16822725; http://dx.doi.org/ 10.1016/j.dnarep.2006.05.016 [DOI] [PubMed] [Google Scholar]
- 30.Mladenov E, Magin S, Soni A, Iliakis G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front Oncol 2013; 3:p. 113; PMID:23675572; http://dx.doi.org/ 10.3389/fonc.2013.00113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res 1988; 175(1):p. 184-91; PMID:3345800; http://dx.doi.org/ 10.1016/0014-4827(88)90265-0 [DOI] [PubMed] [Google Scholar]
- 32.van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol 2009; 19(5):p. 207-17; PMID:19342239; http://dx.doi.org/ 10.1016/j.tcb.2009.03.001 [DOI] [PubMed] [Google Scholar]
- 33.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 1999; 146(5):p. 905-16; PMID:10477747; http://dx.doi.org/ 10.1083/jcb.146.5.905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jucha A, Wegierek-Ciuk A, Koza Z, Lisowska H, Wojcik A, Wojewodzka M, Lankoff A. FociCounter: a freely available PC programme for quantitative and qualitative analysis of gamma-H2AX foci. Mutat Res 2010; 696(1):p. 16-20; PMID:20018253; http://dx.doi.org/ 10.1016/j.mrgentox.2009.12.004 [DOI] [PubMed] [Google Scholar]
- 35.Deng CX, Wang RH. Roles of BRCA1 in DNA damage repair: a link between development and cancer. Hum Mol Genet 2003; 12 Spec No 1:p. R113-23; PMID:12668603; http://dx.doi.org/ 10.1093/hmg/ddg082 [DOI] [PubMed] [Google Scholar]
- 36.Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer 2012; 12(1):p. 68-78; http://dx.doi.org/ 10.1038/nrc3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Speit G, Trenz K. Chromosomal mutagen sensitivity associated with mutations in BRCA genes. Cytogenet Genome Res 2004; 104(1-4):p. 325-32; PMID:15162060; http://dx.doi.org/ 10.1159/000077511 [DOI] [PubMed] [Google Scholar]
- 38.Fabbro M, Rodriguez JA, Baer R, Henderson BR. BARD1 induces BRCA1 intranuclear foci formation by increasing RING-dependent BRCA1 nuclear import and inhibiting BRCA1 nuclear export. J Biol Chem 2002; 277(24):p. 21315-24; PMID:11925436; http://dx.doi.org/ 10.1074/jbc.M200769200 [DOI] [PubMed] [Google Scholar]
- 39.Haaf T, Golub EI, Reddy G, Radding CM, Ward DC. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc Natl Acad Sci U S A 1995; 92(6):p. 2298-302; PMID:7892263; http://dx.doi.org/ 10.1073/pnas.92.6.2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vispe S, Cazaux C, Lesca C, Defais M. Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res 1998; 26(12):p. 2859-64; PMID:9611228; http://dx.doi.org/ 10.1093/nar/26.12.2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC, Arumugarajah S, Hylander-Gans L, Morosini D, Simeone DM, et al.. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer research 2010; 70(12):p. 4972-81; PMID:20501833; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-3573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 1999; 13(20):p. 2633-8; PMID:10541549; http://dx.doi.org/ 10.1101/gad.13.20.2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meek K, Gupta S, Ramsden DA, Lees-Miller SP. The DNA-dependent protein kinase: the director at the end. Immunol Rev 2004; 200:p. 132-41; PMID:15242401; http://dx.doi.org/ 10.1111/j.0105-2896.2004.00162.x [DOI] [PubMed] [Google Scholar]
- 44.Mladenov E, Iliakis G. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mutat Res 2011; 711(1-2):p. 61-72; PMID:21329706; http://dx.doi.org/ 10.1016/j.mrfmmm.2011.02.005 [DOI] [PubMed] [Google Scholar]
- 45.Hansen LT, Lundin C, Helleday T, Poulsen HS, Sørensen CS, Petersen LN, Spang-Thomsen M. DNA repair rate and etoposide (VP16) resistance of tumor cell subpopulations derived from a single human small cell lung cancer. Lung Cancer 2003; 40(2):p. 157-64; PMID:12711116; http://dx.doi.org/ 10.1016/S0169-5002(03)00026-6 [DOI] [PubMed] [Google Scholar]
- 46.Beskow C, Skikuniene J, Holgersson A, Nilsson B, Lewensohn R, Kanter L, Viktorsson K. Radioresistant cervical cancer shows upregulation of the NHEJ proteins DNA-PKcs, Ku70 and Ku86. Br J Cancer 2009; 101(5):p. 816-21; PMID:19672258; http://dx.doi.org/ 10.1038/sj.bjc.6605201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shintani S, Mihara M, Li C, Nakahara Y, Hino S, Nakashiro K, Hamakawa H. Up-regulation of DNA-dependent protein kinase correlates with radiation resistance in oral squamous cell carcinoma. Cancer Sci 2003; 94(10):p. 894-900; PMID:14556663; http://dx.doi.org/ 10.1111/j.1349-7006.2003.tb01372.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee SW, Cho KJ, Park JH, Kim SY, Nam SY, Lee BJ, Kim SB, Choi SH, Kim JH, Ahn SD, et al.. Expressions of Ku70 and DNA-PKcs as prognostic indicators of local control in nasopharyngeal carcinoma. Int J Radiat Oncol Biol Phys 2005; 62(5):p. 1451-7; PMID:16029807; http://dx.doi.org/ 10.1016/j.ijrobp.2004.12.049 [DOI] [PubMed] [Google Scholar]
- 49.Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol 2000; 151(7):p. 1381-90; PMID:11134068; http://dx.doi.org/ 10.1083/jcb.151.7.1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint. Science 2002; 298(5597):p. 1435-8; PMID:12364621; http://dx.doi.org/ 10.1126/science.1076182 [DOI] [PubMed] [Google Scholar]
- 51.Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero RD, Motoyama N, et al.. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol 2002; 4(12):p. 993-7; PMID:12447390; http://dx.doi.org/ 10.1038/ncb884 [DOI] [PubMed] [Google Scholar]
- 52.Mochan TA, Venere M, DiTullio RA Jr, Halazonetis TD. 53BP1 and NFBD1/MDC1-Nbs1 function in parallel interacting pathways activating ataxia-telangiectasia mutated (ATM) in response to DNA damage. Cancer Res 2003; 63(24):p. 8586-91; PMID:14695167 [PubMed] [Google Scholar]
- 53.Seluanov A, Mao Z, Gorbunova V. Analysis of DNA double-strand break (DSB) repair in mammalian cells. J Vis Exp 2010; 8(43):pii: 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Okada S, Ouchi T. Cell cycle differences in DNA damage-induced BRCA1 phosphorylation affect its subcellular localization. J Biol Chem 2003; 278(3):p. 2015-20; PMID:12427729; http://dx.doi.org/ 10.1074/jbc.M208685200 [DOI] [PubMed] [Google Scholar]
- 55.Chen BP, Uematsu N, Kobayashi J, Lerenthal Y, Krempler A, Yajima H, Löbrich M, Shiloh Y, Chen DJ. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J Biol Chem 2007; 282(9):p. 6582-7; PMID:17189255; http://dx.doi.org/ 10.1074/jbc.M611605200 [DOI] [PubMed] [Google Scholar]
- 56.Thelander L, Eriksson S, Akerman M. Ribonucleotide reductase from calf thymus. Separation of the enzyme into two nonidentical subunits, proteins M1 and M2. J Biol Chem 1980; 255(15):p. 7426-32; PMID:6993487 [PubMed] [Google Scholar]
- 57.Rubin EH, Cory JG. Differential turnover of the subunits of ribonucleotide reductase in synchronized leukemia L1210 cells. Cancer Res 1986; 46(12 Pt 1):p. 6165-8; PMID:3536076 [PubMed] [Google Scholar]
- 58.Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 1997; 277(5331):p. 1501-5; PMID:9278512; http://dx.doi.org/ 10.1126/science.277.5331.1501 [DOI] [PubMed] [Google Scholar]
- 59.Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 1997; 277(5331):p. 1497-501; PMID:9278511; http://dx.doi.org/ 10.1126/science.277.5331.1497 [DOI] [PubMed] [Google Scholar]
- 60.Hofmann I, Weiss A, Elain G, Schwaederle M, Sterker D, Romanet V, Schmelzle T, Lai A, Brachmann SM, Bentires-Alj M, et al.. K-RAS Mutant Pancreatic Tumors Show Higher Sensitivity to MEK than to PI3K Inhibition In Vivo. PloS one 2012; 7(8):p. e44146; PMID:22952903; http://dx.doi.org/ 10.1371/journal.pone.0044146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao C, Xiao H, Wu X, Li C, Liang G, Yang S, Lin J. Rational combination of MEK inhibitor and the STAT3 pathway modulator for the therapy in K-Ras mutated pancreatic and colon cancer cells. Oncotarget 2015; 6(16):p. 14472-87; PMID:25961376; http://dx.doi.org/ 10.18632/oncotarget.3991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget 2014; 5(15):p. 6512-25; PMID:25156567; http://dx.doi.org/ 10.18632/oncotarget.2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carter S, Auer KL, Reardon DB, Birrer M, Fisher PB, Valerie K, Schmidt-Ullrich R, Mikkelsen R, Dent P. Inhibition of the mitogen activated protein (MAP) kinase cascade potentiates cell killing by low dose ionizing radiation in A431 human squamous carcinoma cells. Oncogene 1998; 16(21):p. 2787-96; PMID:9652746; http://dx.doi.org/ 10.1038/sj.onc.1201802 [DOI] [PubMed] [Google Scholar]
- 64.Maacke H, Jost K, Opitz S, Miska S, Yuan Y, Hasselbach L, Lüttges J, Kalthoff H, Stürzbecher HW. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene 2000; 19(23):p. 2791-5; PMID:10851081; http://dx.doi.org/ 10.1038/sj.onc.1203578 [DOI] [PubMed] [Google Scholar]
- 65.Sebolt-Leopold JS. Advances in the development of cancer therapeutics directed against the RAS-mitogen-activated protein kinase pathway. Clin Cancer Res 2008; 14(12):p. 3651-6; PMID:18559577; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-0333 [DOI] [PubMed] [Google Scholar]
- 66.Rachagani S, Senapati S, Chakraborty S, Ponnusamy MP, Kumar S, Smith LM, Jain M, Batra SK. Activated KrasG(1)(2)D is associated with invasion and metastasis of pancreatic cancer cells through inhibition of E-cadherin. Br J Cancer 2011; 104(6):p. 1038-48; PMID:21364589; http://dx.doi.org/ 10.1038/bjc.2011.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Park JT, Johnson N, Liu S, Levesque M, Wang YJ, Ho H, Huso D, Maitra A, Parsons MJ, Prescott JD, et al.. Differential in vivo tumorigenicity of diverse KRAS mutations in vertebrate pancreas: a comprehensive survey. Oncogene 2015; 34(21):p. 2801-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Venkitaraman AR. Functions of BRCA1 and BRCA2 in the biological response to DNA damage. J Cell Sci 2001; 114(Pt 20):p. 3591-8; PMID:11707511 [DOI] [PubMed] [Google Scholar]
- 69.Yan Y, Spieker RS, Kim M, Stoeger SM, Cowan KH. BRCA1-mediated G2/M cell cycle arrest requires ERK1/2 kinase activation. Oncogene 2005; 24(20):p. 3285-96; PMID:15735702; http://dx.doi.org/ 10.1038/sj.onc.1208492 [DOI] [PubMed] [Google Scholar]
- 70.Abbott DW, Holt JT. Mitogen-activated protein kinase kinase 2 activation is essential for progression through the G2/M checkpoint arrest in cells exposed to ionizing radiation. J Biol Chem 1999; 274(5):p. 2732-42; PMID:9915804; http://dx.doi.org/ 10.1074/jbc.274.5.2732 [DOI] [PubMed] [Google Scholar]
- 71.Zhang YW, Jones TL, Martin SE, Caplen NJ, Pommier Y. Implication of checkpoint kinase-dependent up-regulation of ribonucleotide reductase R2 in DNA damage response. The Journal of biological chemistry 2009; 284(27):p. 18085-95; PMID:19416980; http://dx.doi.org/ 10.1074/jbc.M109.003020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.D'Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, Saraf A, Florens L, Washburn MP, Pagano M. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 2012; 149(5):p. 1023-34; PMID:22632967; http://dx.doi.org/ 10.1016/j.cell.2012.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yoshida Y, Tsunoda T, Doi K, Tanaka Y, Fujimoto T, Machida T, Ota T, Koyanagi M, Takashima Y, Sasazuki T, et al.. KRAS-mediated up-regulation of RRM2 expression is essential for the proliferation of colorectal cancer cell lines. Anticancer research 2011; 31(7):p. 2535-9; PMID:21873171 [PubMed] [Google Scholar]
- 74.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. RNA interference targeting the M2 subunit of ribonucleotide reductase enhances pancreatic adenocarcinoma chemosensitivity to gemcitabine. Oncogene 2004; 23(8):p. 1539-48; PMID:14661056; http://dx.doi.org/ 10.1038/sj.onc.1207272 [DOI] [PubMed] [Google Scholar]
- 75.Williams TM, Flecha AR, Keller P, Ram A, Karnak D, Galbán S, Galbán CJ, Ross BD, Lawrence TS, Rehemtulla A, et al.. Cotargeting MAPK and PI3K signaling with concurrent radiotherapy as a strategy for the treatment of pancreatic cancer. Mol Cancer Ther 2012; 11(5):p. 1193-202; PMID:22411900; http://dx.doi.org/ 10.1158/1535-7163.MCT-12-0098 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.