

In the presence of physical injury or genotoxic stress, the developing tissue activates a series of repair and regenerative responses that ultimately lead to distinct cellular outcomes such as the commitment to apoptosis accompanied by simultaneous proliferation. Such adapted cellular response is a result of complex interactions among different signaling pathways that play pleiotropic roles during development and stress response. How cells integrate the instructions from the pleiotropic signaling factors such as JNK, p53 or pRb to generate a specific response is one of the enduring questions in cell biology. For example, in the developing Drosophila wing, JNK, p53 or pRb can alternatively induce apoptosis, proliferation or apoptosis-induced proliferation (AiP) depending on the cellular context and stimuli. AiP is the process by which the apoptotic machinery induces the expression of mitogens, which in turn triggers the proliferation of the neighboring cells. This process is relevant to regeneration and has been proposed to be important in tumor repopulation after irradiation.1 Molecular mechanisms of AiP were first discovered and greatly studied in Drosophila imaginal tissues that have a high regenerative capacity, such as the wing and the eye discs. AiP in Drosophila channels through the apoptotic machinery, where Dronc/Caspase-9 activates a feedback loop involving JNK and p53.2,3 However, the underlying mechanisms of AiP lack apparent molecular details. It has been proposed that the molecular mechanisms that distinguish between apoptosis and AiP may rely on specific p53 isoforms (Dp53 or DΔNp53 expression,4 Fig. 1). While full-length Dp53 induces solely apoptosis via the activation of rpr, DΔNp53 favors Hid induction and results in the expression of Wingless/Wnt mitogen and AiP. Other studies have depicted the role of JNK signaling as an important partner of p53 in apoptosis and AiP.5
Figure 1.
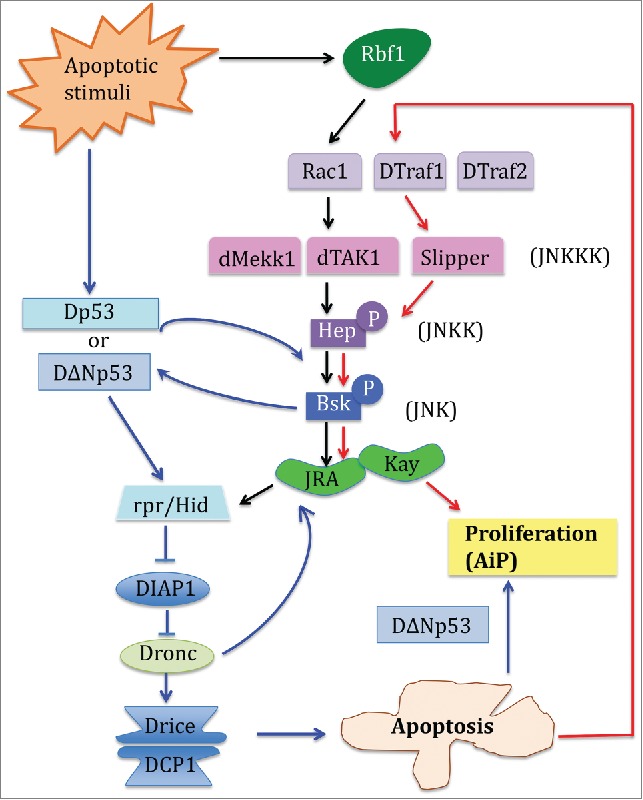
In the Drosophila developing imaginal tissues, 3 major classes of signaling molecules mediate Apoptosis induced Proliferation (AiP). Previous reports have depicted that Drosophila p53 (Dp53) and the JNK signaling pathways form a positive amplification loop with the apoptotic machinery to induce apoptosis and AiP. The blue solid lines and arrows delineate this pathway. In the study by Clavier et al published in this tissue, the authors identified that downstream of Rb, specific JNK pathway adaptors can be used in distinct combinations to generate distinct cellular outcome. The black solid arrows indicate the JNK pathway that leads to apoptosis, and the red solid arrows show the JNK pathway that mediates AiP.
In the current study, Clavier et al.6 (this issue of Cell cycle) used Drosophila developing wing disc to dissect how the genetic interaction between the specific components of the JNK signaling pathways and fly pRb specifically elicits a regenerative cellular response. pRb is well-known regulator of cell cycle but its function in inducing apoptosis or AiP after genotoxic stress is less characterized. The authors first overexpressed rbf1 (the fly pRb) in the Drosophila wing imaginal disc, which mimics pRb stabilization during the induction of apoptosis after DNA damage. While Rbf1 induced apoptosis, Rbf1D253A, a mutated form of Rbf1 that alters the conserved caspase cleavage site, leads to both apoptosis and AiP, with an overgrowth phenotype due to mitogen signaling that are induced by apoptotic cells. They further hypothesized that specific JNK signaling components acting in combination may be responsible for the different cellular responses obtained by Rbf1 or Rbf1D253A expression. Indeed, JNK is a complex network with multiple JNKKs which themselves are activated by a number of JNKKKs.7 They found that 2 different JNK signaling pathways are required for the induction of apoptosis or AiP. While Rbf1-mediated apoptosis required Rac1-dTak1-dMekk1, Rbf1D253A-mediated AiP relied on dTraf1 and Slipper (Fig. 1). Interestingly, another study has recently identified overlapping but also different components of JNK signaling in AiP.2 Like in Clavier et al., Fan et al. found that JNK/bsk and JNKK/hep mediated AiP through Hid, but the upstream adaptors involved dTraf2 instead of dTraf1, and dTak1 instead of Slp. The difference between the results may be explained as the following: first, the 2 studies utilized different molecular means to generate the apoptotic stimuli: Rbf1 versus Hid; secondly, these studies were conducted in different tissues, namely the wing vs. eye disc, in which AiP was induced. Together these results indicate that a specific JNK signaling pathway mediates apoptosis and AiP but that the kinase and corresponding adaptors are tissue specific and can be differentially recruited depending on the apoptotic stimulus. More studies will be needed to determine how specific JNK signaling pathways induced by different apoptotic stimuli integrate with its partner p53 in the regulation of AiP.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We apologize to colleagues to which work was not cited due to space limitation.
Funding
BM was supported by a grant from the ARC foundation
References
- 1.Mollereau B, et al.. Cell Death Differ 2013; 20:181; PMID:22722336; http://dx.doi.org/ 10.1038/cdd.2012.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fan Y, et al. PLoS Genet 2014; 10:e1004131; PMID:24497843; http://dx.doi.org/ 10.1371/journal.pgen.1004131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mollereau B, Ma D. Apoptosis 2014; 19:1421-9; PMID:25217223; http://dx.doi.org/ 10.1007/s10495-014-1035-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dichtel-Danjoy ML, et al.. Cell Death Differ 2013; 20:108-16; PMID:22898807; http://dx.doi.org/ 10.1038/cdd.2012.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shlevkov E, Morata G. Cell Death Differ 2012; 19:451-60; PMID:21886179; http://dx.doi.org/ 10.1038/cdd.2011.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clavier A, et al. Cell Cycle 2016. (in press); http://dx.doi.org/ 10.1080/15384101.2015.1100776 [DOI] [Google Scholar]
- 7.Stronach B. Dev Dyn 2005; 232:575-84; PMID:15704176; http://dx.doi.org/ 10.1002/dvdy.20283 [DOI] [PubMed] [Google Scholar]