

Abstract
Polo-like kinase 1 (Plk1) plays a critical role in proper M-phase progression and cell proliferation. Plk1 is overexpressed in a broad spectrum of human cancers and is considered an attractive anticancer drug target. Although a large number of inhibitors targeting the catalytic domain of Plk1 have been developed, these inhibitors commonly exhibit a substantial level of cross-reactivity with other structurally related kinases, thus narrowing their applicable dose for patient treatment. Plk1 contains a C-terminal polo-box domain (PBD) that is essentially required for interacting with its binding targets. However, largely due to the lack of both specific and membrane-permeable inhibitors, whether PBD serves as an alternative target for the development of anticancer therapeutics has not been rigorously examined. Here, we used an intracellularly expressed 29-mer-long PBIP1-derived peptide (i.e., PBIPtide), which can be converted into a “suicidal” PBD inhibitor via Plk1-dependent self-priming and binding. Using this highly specific and potent system, we showed that Plk1 PBD inhibition alone is sufficient for inducing mitotic arrest and apoptotic cell death in cancer cells but not in normal cells, and that cancer cell–selective killing can occur regardless of the presence or absence of oncogenic RAS mutation. Intriguingly, PBD inhibition also effectively prevented anchorage-independent growth of malignant cancer cells. Thus, targeting PBD represents an appealing strategy for anti-Plk1 inhibitor development. Additionally, PBD inhibition–induced cancer cell–selective killing may not simply stem from activated RAS alone but, rather, from multiple altered biochemical and physiological mechanisms, which may have collectively contributed to Plk1 addiction in cancer cells.
Keywords: anchorage-independent growth, Plk1, Polo-box domain, T78 peptide
Abbreviations
- CBB
Coomassie brilliant blue
- GFP
green fluorescent protein
- KT-MT
kinetochore-microtubule
- PBD
polo-box domain
- PBIPtide
PBIP1-derived peptide
- Plk1
Polo-like kinase 1
Introduction
Polo-like kinase 1 (Plk1) is a Ser/Thr protein kinase that plays pivotal roles in the regulation of the cell cycle and cell proliferation.1,2 Not surprisingly, Plk1 is highly overexpressed in a broad spectrum of human cancers, and its overexpression is closely associated with the aggressiveness and poor prognosis of these cancers.[reviewed in refs.3,4] Intriguingly, cancer cells are addicted to a high level of Plk1.5,6 thus making them more vulnerable to Plk1 interrogation than normal cells. Consistent with this notion, interference with Plk1 function readily induces apoptotic cell death in most tumor cells but not in normal cells, and it reduces tumor growth in mouse xenograft models.3 Thus, Plk1 appears to be an attractive target for therapeutic intervention against human cancers.
Over the years, efforts have been made to develop ATP-competitive inhibitors that are designed to block the catalytic activity of Plk1.3,7 Among these inhibitors are a dihydropteridinone derivative, BI2536,8 and its pharmacologically optimized analog, BI6726,9 and a thiophene inhibitor, GSK461364A,10,11 all of which have been used successfully to antagonize Plk1 function in vitro and in animal models. While all 3 inhibitors have been tested in clinical trials, BI6726 appears to be the most clinically advanced anti-Plk1 inhibitor and is currently under phase III development, with promising results in clinical studies.12-15 However, these inhibitors exhibit somewhat limited specificity against Plk1, mainly because of a large number (> 500) of protein kinases in mammalian cells and the high degree of structural conservation among the ATP-binding pockets within their catalytic domains. For instance, BI6727, by far the most promising anti-Plk1 inhibitor for clinical applications, exhibits only ˜6- and ˜60-fold selectivity over the 2 closely related kinases, Plk2 and Plk3, respectively.9
It is now well appreciated that the C-terminal, non-catalytic polo-box domain (PBD) is critically required for various Plk1-dependent biochemical and cellular processes.16,17 At the molecular level, PBD forms a phosphoepitope-recognition module that binds to a p-Ser/p-Thr-containing motif with high affinity.18,19 Remarkably, the Plk1 PBD– dependent interaction appears to be highly specific, since the targets that interact with Plk1 PBD do not significantly interact with Plk2 and Plk3 PBDs.17,20,21 Furthermore, several studies suggested that Plk1 PBD inhibition by either small-molecule compounds or peptide-derived inhibitors leads to mitotic arrest and apoptotic cell death in cultured mammalian cells.20,22-25 These findings suggest that, distinctively from the prevailing strategy of targeting the catalytic domain of Plk1, blocking the PBD-dependent protein–protein interaction may represent an alternative and highly specific means of inhibiting Plk1 function. However, small-molecule inhibitors reported to date exhibit only a sub-optimal level of PBD-binding affinity,26 whereas all peptide-derived inhibitors suffer greatly from poor membrane permeability, albeit their superb binding affinity and specificity against Plk1 PBD.23,24 As a result of these limitations, an accurate assessment on the applicability of Plk1 PBD inhibition in various biological systems has been greatly thwarted.
In this study, we took advantage of the unique ability of Plk1 to phosphorylate and generate its own docking site on the T78 residue of a kinetochore protein, PBIP1 (also known as MLF1IP, KLIP1, CENP-50 or CENP-U),27-31 and to bind to the resulting p-T78 motif.27,32,33 This mechanism, termed self-priming and binding, allowed us to develop a conserved, 29-mer-long PBIP1 T78 motif–containing peptide (referred to hereafter as PBIPtide), which, when phosphorylated by Plk1's catalytic activity, induces a suicidal inhibition of its own PBD. This PBIPtide-based suicidal inhibition is highly specific because the Plk1 PBD inhibition can occur only after Plk1-dependent specific phosphorylation onto its target, PBIPtide, and ensuing PBD-dependent interaction with the resulting phosphoepitope (i.e., p-T78 PBIPtide). With this highly specific and potent suicidal system, here, we demonstrated that Plk1 PBD inhibition is sufficient for effectively imposing mitotic arrest and apoptotic cell death on cancer cells but not their isogenic normal cells, and for inhibiting anchorage-independent growth of malignant cancer cells. Thus, we propose that targeting PBD represents an attractive alternative anti-Plk1 therapeutic approach for cancer therapy.
Results
PBIPtide-based suicidal inhibition of Plk1 PBD induces mitotic block and apoptotic cell death
It has been shown that Plk1 phosphorylates the T78 motif of a kinetochore component, PBIP1, and binds to the resulting p-T78 motif with a high affinity and specificity.27,32,33 To examine whether we can take advantage of this unique self-priming and binding mechanism to induce suicidal inhibition of Plk1, we utilized PBIPtide, which, when phosphorylated by endogeneous Plk1, can lead to the production of PBD-binding p-T78 PBIPtide (Fig. 1A).
Figure 1.
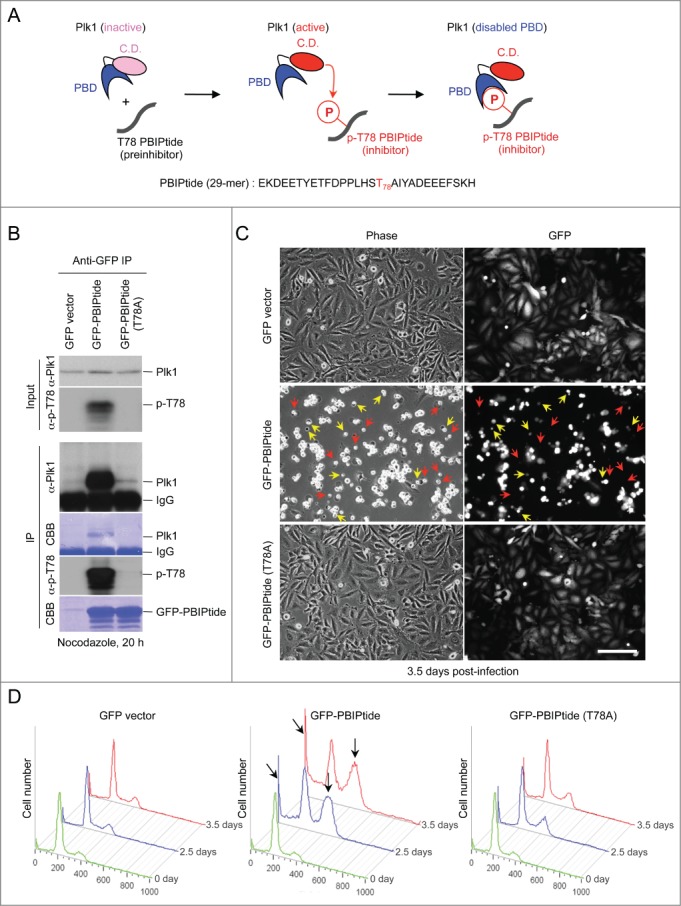
PBIPtide, but not the PBIPtide (T78A) mutant, efficiently induces mitotic block and apoptotic cell death in HeLa cells. (A) The schematic diagram illustrates that, upon being activated, the catalytic domain (C.D.) of Plk1 phosphorylates the T78 residue of PBIPtide and permits its PBD to bind to the resulting p-T78 motif through a self-priming and binding mechanism. As a consequence of p-T78 binding, the PBD of Plk1 becomes disabled. (B) HeLa cells infected with lentiviruses expressing the indicated constructs were treated with nocodazole for 20 h and subjected to immunoprecipitation and immunoblotting analyses. The resulting membranes were stained with Coomassie brilliant blue (CBB). (C) Representative images of HeLa cells expressing the indicated lentiviral constructs are shown. Phase-contrast and widefield green fluorescent protein (GFP) fluorescent images were taken 3.5 days after virus infection. Dying and already dead cells with unusually small (yellow) and hardly detectable (red) GFP signals, respectively, are indicated. Bars, 100 μm. (D) Flow cytometry analyses were carried out using HeLa cells infected with lentiviruses expressing the indicated constructs, and the cells were harvested at 2.5 or 3.5 days after infection. Apoptotic cells with less than G1 (2N) DNA content (slanted arrows) and cells arrested in G2/M with 4N DNA content (vertical arrows) are indicated.
To explore the suitability of PBIPtide-based Plk1 inhibition, we first examined whether PBIPtide (Fig. 1A and S1A) is effectively phosphorylated by intracellular Plk1 activity and whether it also binds to Plk1. To this end, HeLa cells were infected with lentiviruses expressing control green fluorescent protein (GFP), GFP-PBIPtide, or GFP-PBIPtide (T78A), and subjected to immunoprecipitation analysis. As expected, cells expressing GFP-PBIPtide exhibited the p-T78 epitope, whereas cells expressing the GFP-PBIPtide (T78A) mutant did not (Fig. 1B, input). Consistent with this result, immunoprecipitation of GFP-PBIPtide, but not the GFP-PBIPtide (T78A) mutant, coprecipitated endogenous Plk1 to a level that is easily detectable by Coomassie brilliant blue (CBB) staining (Fig. 1B, panel 4). Judging from the levels of Coomassie-stained protein bands, the binding of endogenous Plk1 to the exogenous GFP-PBIPtide appeared to be highly efficient (Fig. 1B, compare the 2 CBB-labeled gels). In line with the p-T78 motif requirement for the PBIPtide-Plk1 interaction,27 the p-T78 epitope was detected in the PBIPtide ligand, but not in the PBIPtide (T78A) mutant (Fig. 1B, panel 5). These findings suggest that endogenous Plk1 activity is sufficient for converting a PBD-binding-incompetent PBIPtide (i.e., preinhibitor) into a PBD-binding-competent p-T78 PBIPtide (i.e., active inhibitor), which can then interact with the PBD of Plk1 in a suicidal fashion. A shorter, 18-mer T78-containing fragment (YETFDPPLHST78AIYADEE; Fig. S1A) failed to bind to Plk1 significantly, suggesting that a longer PBIPtide is needed for proper Plk1-dependent T78 phosphorylation and subsequent PBD binding.
To estimate the amount of PBIPtide needed to effectively bind to and inhibit endogenous Plk1, we carried out an enzyme-linked immunosorbent assay (ELISA)-based analysis,33 using 250 ng of purified recombinant GST-PBIPtide and various amounts of Plk1. As expected, the level of the p-T78 epitope generated on immobilized GST-PBIPtide was proportional to the amount of Plk1 provided, and the increase in the level of bound Plk1 (half maximal binding at 13.8 ng of Plk1) closely mirrored that in the level of produced p-T78 epitope (half maximal p-T78 production at 4.4 ng of Plk1) (Fig. S1B). Providing that all PBIPtides immobilized on ELISA wells were functional, approximately 18-fold (13.8 ng/250 ng) overexpression of PBIPtide appeared to be sufficient to bind to and incapacitate a half of intracellular Plk1 molecules.
We then investigated whether the expression of the 29-mer PBIPtide can inhibit the function of Plk1 and induce mitotic abnormalities, using HeLa cells generated in Figure 1B. The expression of GFP-fused forms of PBIPtide allowed us to conveniently assess their expression levels, thus minimizing the concern of imposing varying degrees of Plk1 inhibition among different experimental groups. We observed that the expression of GFP-PBIPtide efficiently induced a large fraction of cells with a rounded-up morphology, whereas the expression of either the control GFP or the Plk1 PBD–binding-defective GFP-PBIPtide (T78A) mutant did not (Fig. 1C). Among the rounded-up cells, a large fraction exhibited either unusually small or hardly detectable GFP signals (indicated by yellow or red arrows, respectively, in Fig. 1C), which are suggestive of dying or already dead cells. Consistent with this view, flow cytometry analysis showed that the expression of GFP-PBIPtide, but not GFP-PBIPtide (T78A), induced a large fraction of cells in either G2/M or sub-G1 DNA content (Fig. 1D). Given that Plk1 is critical for proper M-phase progression,1,34 these findings suggest that inhibiting PBD sufficiently interferes with Plk1-dependent mitotic processes.
PBIPtide efficiently delocalizes Plk1 from mitotic centrosomes and kinetochores, and induces a potent pre-anaphase arrest with misaligned chromosomal morphologies
To further investigate whether the phenotype associated with the expression of GFP-PBIPtide is the consequence of inhibiting the function of Plk1 PBD in vivo, we closely examined whether PBD-dependent subcellular localization of Plk1 is compromised by PBIPtide expression. Studies have shown that Plk1 is hardly detectable at the early stage of the cell cycle but becomes manifest at late-interphase centrosomes and prekinetochores, and reaches its highest abundance at prometaphase centrosomes and kinetochores.35-37 Similarly, Plk1 activity progressively increases as cells go through the cell cycle, sharply peaking in early mitosis.36,38 Therefore, we chose to examine the effect of PBIPtide expression on Plk1 localization using late interphase (which can be defined by clear, nuclear-localized Plk1 signals) and prometaphase cells.
We observed that the expression of GFP-PBIPtide, but not the GFP-PBIPtide (T78A) mutant, in HeLa cells induced an almost complete delocalization of Plk1 from centrosomes and prekinetochores/kinetochores (Fig. 2A), and potent mitotic arrest (Fig. 2B). Notably, a large fraction of the arrested cells showed aberrant bipolar spindles with misaligned chromosomal morphologies, although a low degree of cells with monopolar or multipolar spindles, and cells with lagging chromosomes, were also observed (Fig. 2C). Thus, distinct from a monopolar spindle morphology observed with Plk1 inhibition or depletion,37,39 PBIPtide-dependent inhibition of PBD function leads to pre-anaphase arrest with aberrant bipolar spindles, suggesting that PBIPtide interferes with a subset of Plk1-dependent functions. The quantification of Plk1 signals in Figure 2A showed that PBIPtide expression modestly (˜2-fold) diminished Plk1 signals at late-interphase (i.e., G2) centrosomes and prekinetochores, whereas it drastically (˜5–6 fold) diminished Plk1 signals at prometaphase centrosomes and kinetochores (Fig. 2D-E). Under the same conditions, the expression of the control GFP vector or the PBD-binding-defective GFP-PBIPtide (T78A) mutant failed to significantly alter the level of Plk1 localization at these sites (Fig. 2D-E). Since Plk1 becomes activated by AurA as cells enter mitosis,40,41 more efficient PBIPtide-induced Plk1 delocalization at mitotic centrosomes and kinetochores than at their respective interphase counterparts is likely attributed to increased Plk1 kinase activity in mitotic cells.
Figure 2.
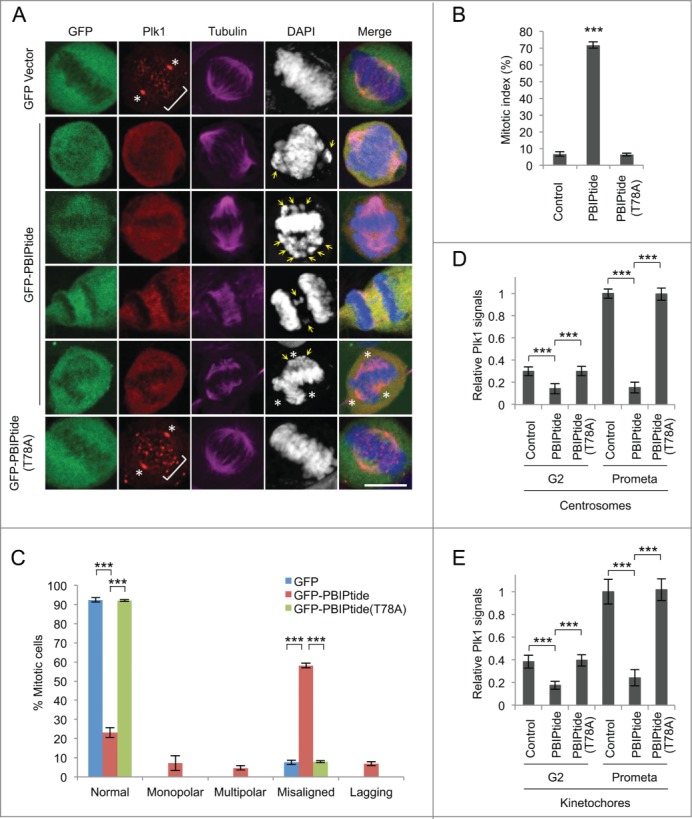
PBIPtide expression delocalizes Plk1 and induces potent prometaphase arrest in HeLa cells. (A) Confocal microscopic analysis was carried out for HeLa cells expressing GFP control, GFP-PBIPtide, or GFP-PBIPtide (T78A), and stained with anti-Plk1 (red) and anti-tubulin (magenta) antibodies. The second and third panels show cells with misaligned chromosomes (arrows), while the fourth and fifth panels show cells with lagging chromosomes (arrows) and multipolar spindles, respectively. Asterisks, centrosomes; brackets, punctate kinetochores. Bars, 10 μm. (B–E) Quantifications of mitotic populations (B), mitotic cells with aberrant chromosome morphologies (C), and Plk1 fluorescence signals at centrosomes (D) and kinetochores (E) were carried out with the samples obtained in (A). For each of 3 independent experiments, n > 500 for (B), n > 200 for (C), and n > 70 for (D and E). G2 cells in (D and E) were identified by accumulated Plk1 signals in the nucleus, whereas prometaphase cells were recognized by typical prometaphase DNA morphologies. Statistics in (B–E): ***, p < 0.001 (unpaired 2-tailed t-test). Bars, standard deviation.
To better understand the nature of PBIPtide-dependent mitotic arrest, we closely examined the level of phosphorylations on multiple mitotic regulators, including several proteins (BubR1, Bub1, Cdc25C, and Wee1A), all of which are known to be regulated by PBD-dependent Plk1 function.42-46 To this end, HeLa cells expressing either control GFP alone, GFP-PBIPtide, or GFP-PBIPtide (T78A) were arrested at the G1/S boundary by double thymidine treatment and then released into medium containing nocodazole to trap the cells in the following mitosis (Fig. 3). The results showed that the expression of GFP-PBIPtide, but not GFP alone or the GFP-PBIPtide (T78A) mutant, significantly diminished the level of BubR1 phosphorylation, while mildly altering Bub1 phosphorylation (Fig. 3). Under the same conditions, the expression of GFP-PBIPtide did not detectably influence the level of phosphorylations on various other mitotically phosphorylated proteins (Cdc25C, Wee1A, and Mps1) (Fig. 3). In all 3 sets of experiments, the level of phospho-histone H3 increased significantly 12 h after double thymidine release (Fig. 3, bottom panels), suggesting that the expression of PBIPtide did not cause any unexpected cell cycle delay during the early stages of the cell cycle where Plk1 is not abundantly expressed.36,47 Since Plk1-dependent BubR1 phosphorylation appears to be critical for establishing stable kinetochore-microtubule (KT-MT) attachment,42,43 the PBIPtide-induced mitotic arrest observed in Fig. 1C-D could be in part attributed to spindle checkpoint activation caused by improper Plk1-dependent BubR1 phosphorylation. The absence of any detectable effect of the Plk1 PBD–binding-defective GFP-PBIPtide (T78A) mutant (Fig. 3) suggests that the effect of PBIPtide on BubR1 is specific. Since GFP has been shown to dimerize at high concentrations,48,49 which could induce an unanticipated consequence, we also examined the effect of expressing a Flag epitope–fused.50 PBIPtide or PBIPtide (T78A) form. Consistent with the results in Figure. 3, that Flag-PBIPtide, but not the Flag-PBIPtide (T78A) mutant, induced drastic dephosphorylation of BubR1, while dephosphorylating Bub1 at a somewhat reduced level (Fig. S2). These results suggest that among various Plk1 substrates that we tested (BubR1, Bub1, Cdc25C, and Wee1),42-46 BubR1 is the most sensitive to PBD-mediated phosphorylation by Plk1. Importantly, Mps1 is thought to be a major kinase that phosphorylates BubR1 in vivo.51 However, expression of PBIPtide did not appear to significantly alter the level of Mps1 autophosphorylation (Fig. 3 and S2).
Figure 3.
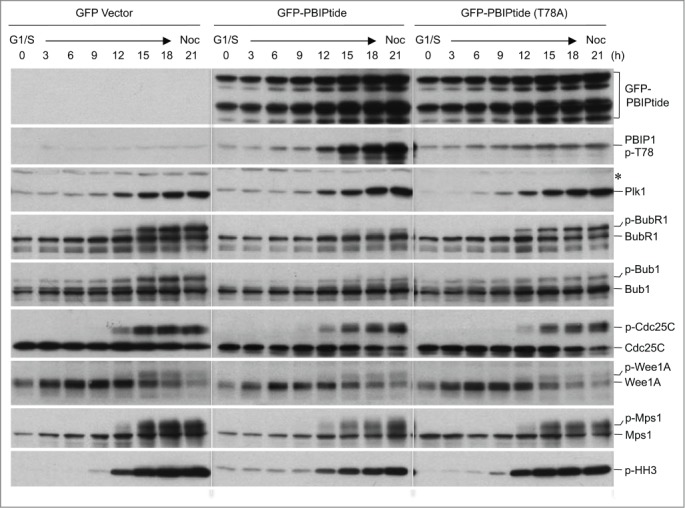
PBIPtide inhibits proper BubR1 phosphorylation in HeLa cells. Immunoblotting analyses were performed using HeLa cells released synchronously from a double thymidine block (G1/S) into nocodazole (Noc)-containing medium. Samples were harvested at the indicated time points and analyzed. Asterisk, cross-reacting protein; p-HH3, phospho-histone H3.
Normal cells are less susceptible to PBIPtide-mediated mitotic arrest and apoptotic cell death than cancer cells
Having observed that cervical cancer–derived HeLa cells were highly susceptible to PBIPtide-based Plk1 PBD inhibition (Fig. 1), we then further investigated the effect of PBIPtide expression in other independently derived cancer or normal cell lines. The results showed that, similar to the findings in HeLa cells (Fig. 1C-D), the expression of PBIPtide induced a drastic mitotic block and apoptotic cell death in osteosarcoma-derived U2OS cells (Fig. 4A). In contrast to this observation, PBIPtide expression in near-diploid human mammary epithelial MCF-10A cells or normal diploid human fetal lung MRC-5 fibroblasts failed to induce any noticeable cell cycle defect under the conditions where GFP fluorescence of PBIPtide in these cells was at a level similar to that in U2OS cells (Fig. 4B-C). Under similar conditions, both control GFP and the GFP-PBIPtide (T78A) mutant lacking the self-priming and binding site failed to exhibit any significant level of cell cycle arrest and apoptotic cell death in all the cell lines examined (Fig. 4). Comparable to the effect of PBIPtide expression shown in Figure. 4, treatment of cells with a small molecule-based Plk1 catalytic activity inhibitor, BI6727,52 showed that cancer-derived U2OS cells were substantially more susceptible to Plk1 inhibition than MCF-10A and MRC-5 cells (Fig. S3A) (note a drastic sub-G1 accumulation in U2OS cells; a slanted arrow). However, BI6727 treatment also induced a substantial level of mitotic arrest in MCF-10A and MRC-5 cells (Fig. S3A; vertical arrows). It is possible that inhibiting the catalytic activity of Plk1 is less selective between normal and cancer cells than inhibiting a wide array of Plk1 PBD-dependent interactions, as discussed recently.53
Figure 4.

PBIPtide induces mitotic arrest and apoptotic cell death in U2OS cells but not in near-diploid MCF-10A and normal MRC-5 cells. (A–C) The indicated cells were infected with lentiviruses expressing control GFP, GFP-PBIPtide, or GFP-PBIPtide (T78A). Representative images of phase-contrast and widefield GFP fluorescence were taken 3.5 days after lentivirus infection. Flow cytometry analyses were performed with cells harvested at the indicated time points after infection. Bars, 100 μm. Arrows in (A) indicate apoptotic cell populations with less than G1 (2N) DNA content (slanted) and cells arrested in G2/M with 4N DNA content (vertical).
To investigate whether a faster cell proliferation rate could account for the PBIPtide-induced phenotype observed in U2OS cells, we comparatively analyzed doubling times for U2OS, MCF-10A, and MRC-5 cells. Interestingly, U2OS cells (a doubling time of 18.2 h) proliferated much slower than MCF-10A cells (a doubling time of 13.7 h), while they did much faster than MRC-5 cells (a doubling time of 27.4 h) (Fig. S3B). These findings suggest that the susceptibility of U2OS cells to PBIPtide-induced inhibition may likely stem from uncharacterized Plk1-dependent biochemical processes rewired in these cancer cells, rather than simply from an altered rate of cell cycle progression. In line with this view, an ELISA-based Plk1 kinase activity assay.33 revealed that cancer-derived HeLa and U2OS cells contained several-fold higher levels of overall Plk1 activities per given amount of total cellular lysates than MCF-10A and MRC-5 cells (Fig. S3C). Thus, a drastic Plk1 inhibition phenotype observed in cancer-derived HeLa and U2OS cells is likely due to several-fold increased intracellular Plk1 activities in these cells.
PBIPtide selectively inhibits the cell division and growth of activated HRAS-transformed premalignant or malignant cells
We next investigated the effect of PBIPtide expression during different stages in the carcinogenic process, using a set of human breast–derived cell lines from a single cell lineage: i.e., normal epithelial (MCF-10A; M-I), premalignant epithelial (M-II), low-grade carcinoma (M-III), and high-grade metastatic carcinoma (M-IV) lines.54 To do this, cells were first infected with lentiviruses expressing control GFP, GFP-PBIPtide, or GFP-PBIPtide (T78A), then selected with puromycin to eliminate uninfected cells, and then analyzed after arresting the cells with nocodazole for 20 h. Results showed that both GFP-PBIPtide and GFP-PBIPtide (T78A) were expressed at similar levels in all M-I, M-II, M-III, and M-IV cells, whereas control GFP was expressed at levels several fold higher than GFP-PBIPtides (Fig. 5A). Under these conditions, the expression of PBIPtide, but not the PBIPtide (T78A) mutant, completely abrogated the slow-migrating, phosphorylated BubR1 form in all 4 cell lines (Fig. 5A). This observation suggests that, in all of these cells, Plk1-mediated BubR1 phosphorylation is equally sensitive to PBD inhibition.
Figure 5.
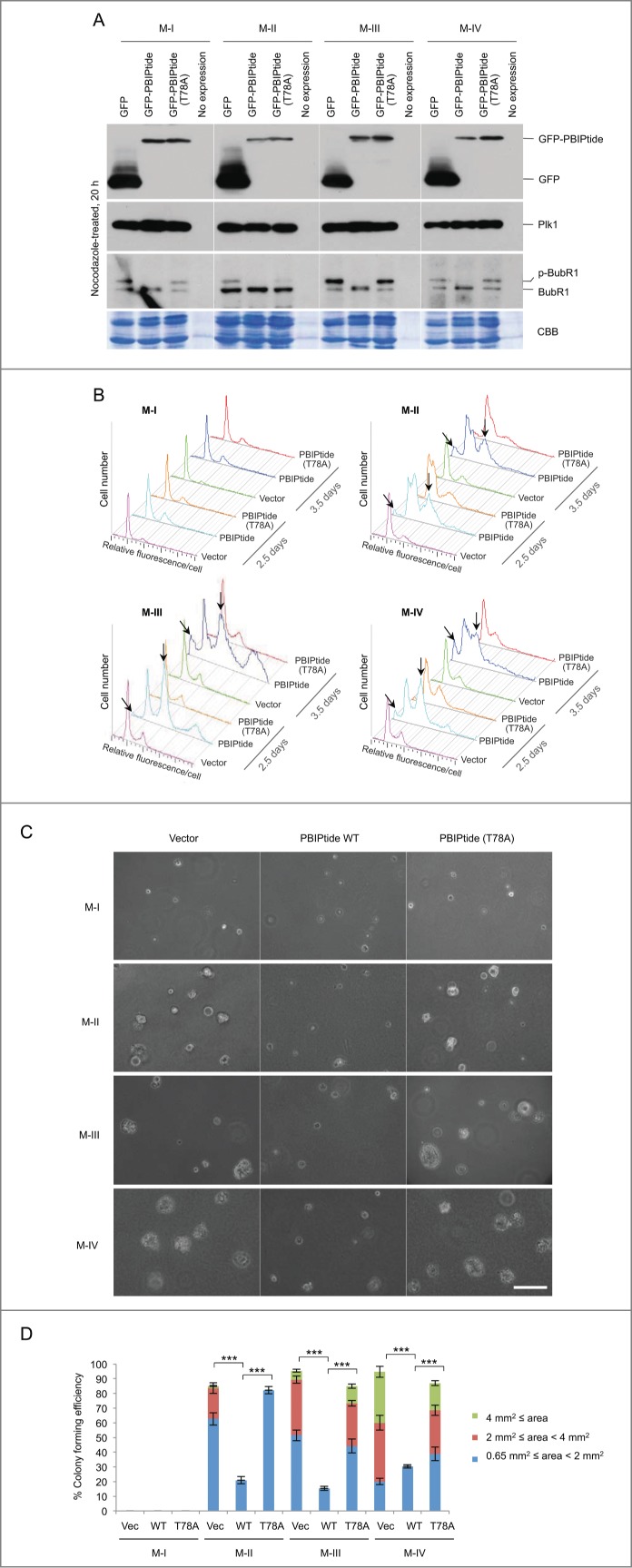
PBIPtide induces mitotic block and apoptotic cell death, and effectively inhibits anchorage-independent cell growth in HRAS-transformed premalignant M-II and malignant M-III and M-IV cells. (A) Immunoblotting was carried out in M-I, M-II, M-III, and M-IV cells expressing the indicated construct using a lentiviral system. CBB, Coomassie brilliant blue stain. (B–D) Cells in (A) were subjected to flow cytometry analysis (B) and soft agar growth assay (C and D). Arrows in (B) indicate apoptotic cell populations with sub-G1 DNA content (slanted) and cells arrested in G2/M with 4N DNA content (vertical). Bars in (C), 100 μm. (D) Colony size is arbitrarily classified into 3 different groups based on the image area calculated by Zeiss ZEN software. Statistics: ***, p < 0.001 (unpaired 2-tailed t-test). Bars, standard deviation.
Next, to investigate whether PBIPtide expression alters cell cycle progression, we performed flow cytometry analysis using the cells obtained in Figure. 5A. Since intracellular PBIPtide can be visualized using a GFP fusion form, a GFP-PBIPtide-based method provides us with an advantage to assess the differential effect of Plk1 PBD inhibition among different cell types with similar levels of GFP-PBIPtide expression. The results showed that GFP-PBIPtide expression induced a large fraction of cells with G2/M and sub-G1 DNA contents in both activated HRAS-transformed premalignant (M-II) and malignant (M-III and M-IV) cells. Consistent with the results in Figure. 4B, GFP-PBIPtide failed to induce any detectable level of cell cycle defect in normal epithelial M-I cells (Fig. 5B). Under these conditions, neither the control GFP vector nor the PBD-binding-defective GFP-PBIPtide (T78A) mutant appeared to alter cell cycle progression (Fig. 5B), thus corroborating the results shown in Figs. 1 and 4. Notably, the level of the sub-G1 population in M-II, M-III, and M-IV cells was higher at 3.5 days post-infection than at 2.5 days post-infection, whereas the level of the G2/M-arrested population remained largely unchanged (Fig. 5B). These observations suggest that PBIPtide-dependent PBD inhibition efficiently induces mitotic arrest and apoptotic cell death in these cells, as observed in HeLa and U2OS cells (Figs. 1 and 4A), and that apoptotic cell death likely occurs as a consequence of prolonged mitotic arrest.
Since the ability of anchorage-independent growth has been shown to closely correlate with tumorigenicity in animal models,55,56 we employed a soft agar growth assay to determine potential antitumorigenic activity of PBIPtide. To this end, we examined whether the expression of PBIPtide alters the M-I, M-II, M-III, and M-IV cells' ability to proliferate in the absence of adhesion to extracellular matrix proteins. As expected, normal epithelial M-I cells failed to form noticeable-sized colonies in soft agar (Fig. 5C). Under these conditions, HRAS-transformed premalignant (M-II) and malignant (M-III and M-IV) cells formed sizable colonies several days after plating the cells (Fig. 5C). The high-grade metastatic carcinoma line (M-IV) formed colonies most efficiently, while premalignant epithelial M-II cells formed colonies significantly less efficiently (Fig. 5C-D). These results are consistent with the previous finding that the in vitro anchorage-independent growth ability of tumor cells correlates with their in vivo metastatic potential.56 Under these conditions, the expression of PBIPtide, but not the PBIPtide (T78A) mutant, almost completely annihilated the M-II, M-III, and M-IV cells' ability to form colonies (Fig. 5C-D). These findings suggest that inhibition of Plk1 PBD function is sufficient for interfering with the ability of oncogenic HRAS-transformed malignant cancer cells to grow in an anchorage-independent fashion and to metastasize to other tissues in vivo.
Discussion
PBIPtide-dependent suicidal inhibition of Plk1 PBD: a novel method for targeting Plk1
Plk1 is considered one of the most attractive targets for anticancer therapies. Although some of the inhibitors targeting the ATP-binding motif of Plk1 yielded various degrees of success in preclinical and clinical studies,8-11 they commonly exhibit an unintended toxicity largely because of their limited specificity in the presence of many other structurally related kinases. Plk1 offers, within one molecule, 2 functionally distinct but interconnected drug targets—the N-terminal catalytic domain and the C-terminal noncatalytic PBD essential for mediating interactions with various Plk1 binding targets. Therefore, given the unique nature of protein–protein interactions, targeting the PBD could be a viable approach for the development of highly specific anti-Plk1 inhibitors. Although substantial progress has been made in developing PBD inhibitors, this class of inhibitors has suffered greatly from poor binding affinity, stability, and/or membrane permeability. Therefore, whether targeting the PBD of Plk1 may constitute an alternative strategy to interrogate Plk1 function in cancer cells has not been closely evaluated.
Here, our results demonstrated that an intracellularly expressed PBIP1-derived PBIPtide can efficiently induce suicidal inhibition of PBD via a Plk1-mediated self-priming and binding mechanism (Fig. 1A).27,32,33 Using this unique system, we addressed 2 major questions: 1) whether PBD inhibition is sufficient for interfering with Plk1 function and, if so, 2) whether it can induce a cancer cell–selective killing effect, as suggested previously with the depletion or inhibition of Plk1.5,6,57,58 Using a PBIPtide-based highly specific suicidal system, we unequivocally demonstrated that the interrogation of Plk1 PBD function is an appealing strategy for effectively antagonizing Plk1-mediated mitotic progression in cancer cells. Furthermore, under the conditions where an equal level of PBIPtide is expressed, we showed that the inhibition of Plk1 PBD function is sufficient for imposing cancer cell–selective mitotic arrest and killing.
Unlike Plk1 PBD inhibition by currently available small-molecule compounds or peptide-derived inhibitors, PBIPtide-mediated suicidal inhibition confers multiple advantages. First, PBIPtide-based inhibition is highly specific because it requires not only Plk1-dependent phosphorylation at the T78 residue of PBIPtide (i.e., preinhibitor), but also PBD-dependent interaction with the resulting p-T78 PBIPtide (i.e., active inhibitor). Second, since the suicidal p-T78 PBIPtide inhibitor is generated by catalytically active Plk1, the intracellular level of p-T78 PBIPtide would be proportional to that of Plk1 activity, therefore imposing more potent PBD inhibition on actively dividing cells with higher Plk1 activity, such as most cancer cells. Third, since the inhibitory p-T78 PBIPtide can be generated only if an active form of Plk1 is present, and its level would gradually diminish as Plk1 is being inhibited, this suicidal mechanism is autoregulated. By nature, this autoregulatory system could provide a biochemical safeguard, under which cells are prevented from being completely deprived of Plk1 activity, thus allowing a submaximal inhibition of Plk1 PBD function. In contrast to many cancer cells addicted to non-oncogenic Plk1, their isogenic normal cells exhibited much less dependency on Plk1.5,6,57 In fact, a low level (∼20%) of Plk1 was shown to be sufficient for maintaining cellular proliferation in various normal organs of adult mice.59 These findings suggest that PBIPtide-based submaximal inhibition of Plk1 may help minimize any detrimental effect on normal cells and tissues, while also imposing mitotic arrest and apoptotic cell death on cancer cells. On the contrary, since an ATP analog inhibitor would inhibit Plk1 in a dose-dependent manner, catastrophic killing of both normal and cancer cells may not be avoidable when intracellular Plk1 is completely inhibited.
Mechanism underlying PBIPtide-induced cancer cell–selective killing
Earlier studies have suggested that cancer cells are addicted not only to oncogenes, but also to many non-oncogenes for their viability.60,61 A genome-wide study revealed that Plk1 is the only non-oncogenic kinase critically required for the viability of activated RAS-containing cells, but not for otherwise isogenic WT cells.61 This finding suggests that biochemical pathways in oncogenic RAS-containing cancer cells are likely rewired in such a way that they are addicted to a high level of Plk1 for proper proliferation. Consistent with this view, we demonstrated that oncogenic HRAS-transformed premalignant (M-II) and malignant carcinoma (M-III and M-IV) lines are sensitive to Plk1 PBD inhibition for proper cell cycle progression and proliferation, whereas their isogenic normal epithelial (M-I) cells are not (Fig. 5B). Furthermore, all 3 cell lines (M-II, M-III, and M-IV) failed to grow in an anchorage-independent manner under the conditions where Plk1 PBD was inhibited by PBIPtide expression (Fig. 5C). These results provide the proof of principle that the inhibition of PBD function is sufficient for interfering with the proliferation of oncogenic RAS-containing premalignant and malignant cells. They also raise the possibility that PBIPtide serves as a tool for effectively inhibiting the function of Plk1 PBD in various biological systems. In this regard, it will be interesting to explore a new avenue of developing PBIPtide-based inhibitors that can be applied to clinically relevant experimental systems.
At present, the mechanism of how PBIPtide-dependent Plk1 PBD inhibition selectively induces differential killing of both premalignant epithelial (M-II) and malignant (M-III and M-IV) carcinoma lines (Fig. 5) remains unknown. Notably, we found that PBIPtide expression selectively interfered with proper BubR1 phosphorylation (Fig. 3 and S2). Since Plk1-dependent BubR1 phosphorylation is important for establishing stable KT-MT attachment,42,43 improper BubRI phosphorylation may have contributed to PBIPtide-induced mitotic arrest and apoptotic cell death, as observed in Figure. 5. However, PBIPtide abrogated proper BubR1 phosphorylation in all M-I, M-II, M-III, and M-IV cells. This finding suggests that, although impaired BubR1 phosphorylation could impose more mitotic stress on the oncogenic HRAS-containing M-II, M-III, and M-IV cells than on M-I cells, it is not likely the primary reason for the preferential killing of these cells. Interestingly, PBIPtide can efficiently induce mitotic arrest and apoptotic cell death in other independently derived cancer lines, such as HeLa and U2OS cells (Figs. 1 and 4), which are reported as not having oncogenic RAS mutations. Therefore, an oncogenic RAS mutation is not likely the single driver for cancer cells' sensitivity to PBIPtide-dependent Plk1 PBD inhibition. Rather, altered biochemical and physiological mechanisms may collectively contribute to Plk1 addiction and PBIPtide-dependent cancer cell–selective killing. In line with this view, the detrimental effect of Plk1 interrogation can also be synergized by the loss of p53 in cancer cells.6,57,58 Cancer cells' addiction to Plk1 is likely the consequence of an amplified Plk1-dependent process(es). Therefore, identifying and characterizing a cancer cell–enriched PBD-binding protein will be a key step in better understanding the underlying mechanism of how PBIPtide-dependent Plk1 PBD inhibition can induce cancer cell–selective killing.
Materials and Methods
Plasmid construction
Lentiviral constructs expressing Flag-GFP alone (pKM2203), Flag-GFP-PBIPtide (pKM2051), or Flag-GFP-PBIPtide (T78A) (pKM2081) were constructed by inserting an AgeI-BglII (both end-filled) fragment containing Flag-GFP alone, Flag-GFP-PBIPtide WT, or its respective (T78A) mutant into a pHR′-CMV-SV-puro vector.27 digested with EcoRI and end-filled. The lentiviral constructs expressing Flag alone (pKM4067), Flag-PBIPtide (pKM2050), or Flag-PBIPtide (T78A) (pKM2079) were generated similarly as above, except that an AgeI-KpnI (both end-filled) fragment containing Flag alone, Flag-PBIPtide WT, or its respective (T78A) mutant was inserted into a pHR′-CMV-SV-puro vector digested with EcoRI and end-filled. PBIPtide contains 4 tandem repeats of the 29-mer-long T78 peptide shown in Figure 1A.
Cell culture and transfection
HEK293T, HeLa, U2OS, MCF-10A, and MRC-5 cells were cultured as recommended by American Type Culture Collection (Manassas, VA). MCF-10A (M-I)-derived premalignant epithelial (M-II), low-grade carcinoma (M-III), and high-grade metastatic carcinoma (M-IV) lines were cultured as previously described.54
To carry out the G1/S-release experiment in Figure 4, HeLa cells were first infected with either control GFP, GFP-PBIPtide, or GFP-PBIPtide (T78A) for one day. The cells were then treated with 2 mM thymidine (Sigma-Aldrich, St. Louis, MO) for 18 h, released for 9 h, and treated with another 2 mM thymidine for 16 h. The resulting cells were released into medium containing 660 nM of nocodazole, and samples were harvested at the indicated time points. To enrich the mitotic cell population, single nocodazole treatment was carried out for 20 h.
For the experiment shown in Figure. S2, HeLa cells were treated essentially the same way except that they were infected with lentiviruses expressing the control Flag vector, Flag-PBIPtide, or Flag-PBIPtide (T78A).
Lentivirus generation and infection
For the production of lentiviruses, transfection was performed by a calcium phosphate coprecipitation method.62 Lentiviruses were generated by cotransfecting HEK293T cells with pHR′-CMVΔR8.2Δvpr-, pHR′-CMV-VSV-G- (protein G of vesicular stomatitis virus), and pHR′-CMV-SV-puro-based constructs containing a target gene.
Cells infected with lentiviruses expressing the control vector, PBIPtide, or PBIPtide (T78A) were selected with 2 μg/ml of puromycin (Sigma-Aldrich) for 1 day, continuously cultured for the indicated length of time, and then analyzed.
Immunoprecipitation and immunoblotting
Cells were lysed in TBSN buffer [50 mM Tris-Cl (pH 8.0), 120 mM NaCl, 0.5% NP-40, 5 mM EGTA, 1.5 mM EDTA, 1% p-nitrophenyl phosphate + protease inhibitors]. After centrifugation (14,000 × g, 15 min), the resulting supernatant was subjected to immunoprecipitation, which was performed in a similar manner as was previously described.27
For immunoblotting analysis, samples were electrophoresed on a 10% SDS-PAGE (15% to detect phospho-histone H3), transferred to a PVDF membrane, and then detected with the indicated antibodies using the enhanced chemiluminescence detection system (Pierce, Rockford, IL).
Confocal and widefield fluorescence microscopy
For confocal microscopic analysis, cells were immunostained essentially as previously described,63 using the indicated primary and appropriate secondary antibodies. Confocal images were obtained using the Zeiss LSM 780 system mounted on a Zeiss Observer Z1 microscope. To capture phase-contrast and fluorescence images of Flag-GFP-PBIPtide-expressing cells, widefield fluorescence microscopy was performed using a Zeiss Axiovert S100.
Confocal images were acquired at 512 × 512 pixels and 12-bit resolution using a Zeiss LSM 710 system mounted on a Zeiss Axiovert 100M microscope. For the quantification of fluorescence signal intensities, images of unsaturated fluorescence signals were used. The fluorescence intensity for a particular signal was determined after subtracting the background signal intensity using Zeiss AIM confocal software.
Flow cytometry analysis
Cultured cells were harvested, washed 1X with PBS + 1% fetal bovine serum (FBS), 1X with PBS + 0.1% glucose, and then finally resuspended in 200 μl of PBS + 0.1% glucose. The resulting samples were added with 5 ml of 70% ethanol (−20°C) dropwise, and kept at −20°C for more than 30 min. Samples were then harvested, washed 2X with PBS + 0.1% glucose + 1% FBS, then resuspended in 0.5 ml of 40 mM sodium citrate buffer (pH 7.4) containing 70 μM propidium iodide. After the addition of RNase to the final concentration of 50 μg/ml, samples were incubated at 37°C for 30 min and analyzed by a FACScan cell sorter (Becton Dickinson, San Jose, CA). Obtained data were analyzed by the FlowJo program (Tree Star, San Carlos, CA).
Soft agar growth assay
M-I, M-II, M-III, and M-IV cells infected with lentiviruses expressing the indicated constructs were selected with puromycin. The resulting cells were subjected to a 2-layer soft agar assay in 6-well plates. After solidifying a bottom layer of 2 ml of 1% Sepharose in its respective 1X culture medium,54 the resulting cells (1.5 × 104) were mixed with 2 ml of 1X culture medium with 10% FBS and 0.33% agarose, cooled to 42°C, and plated on top of the bottom layer. Every 2 days, 1 ml of growth medium was added into the wells. One week later, bright-field images were taken using a Zeiss Axiovert S100. The number of colonies grown in soft agar was quantified, and the colony size (i.e., image area) was calculated using Zeiss ZEN software. Colonies were arbitrarily categorized into 3 groups based on their sizes. The colony-forming efficiency was determined by dividing the number of colonies in each class with the total number of cells.
Acknowledgments
We are grateful to Dr. Lalage Wakefield for providing MCF-10A-derived M-I, M-II, M-III, and M-IV cells; Dr. Johng S. Rhim for various helpful advice; and Dr. Hye-Ran Kim for technical assistance.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was supported in part by National Cancer Institute intramural grants awarded to K.S.L.
References
- 1.Archambault V, Glover DM. Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol 2009; 10:265-75; PMID:19305416; http://dx.doi.org/ 10.1038/nrm2653 [DOI] [PubMed] [Google Scholar]
- 2.Petronczki M, Lénárt P, Peters JM. Polo on the Rise-from Mitotic Entry to Cytokinesis with Plk1. Dev Cell 2008; 14:646-59; PMID:18477449; http://dx.doi.org/ 10.1016/j.devcel.2008.04.014 [DOI] [PubMed] [Google Scholar]
- 3.Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov 2010; 9:643-60; PMID:20671765; http://dx.doi.org/ 10.1038/nrd3184 [DOI] [PubMed] [Google Scholar]
- 4.Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer 2006; 6:321-30; PMID:16557283; http://dx.doi.org/ 10.1038/nrc1841 [DOI] [PubMed] [Google Scholar]
- 5.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009; 137:835-48; PMID:19490893; http://dx.doi.org/ 10.1016/j.cell.2009.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sur S, Pagliarini R, Bunz F, Rago C, Diaz LAJ, Kinzler KW, Vogelstein B, Papadopoulos N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A 2009; 106:3964-9; PMID:19225112; http://dx.doi.org/ 10.1073/pnas.0813333106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garuti L, Roberti M, Bottegoni G. Polo-like kinases inhibitors. Curr Med Chem 2012; 19:3937-48; PMID:22709006; http://dx.doi.org/ 10.2174/092986712802002455 [DOI] [PubMed] [Google Scholar]
- 8.Steegmaier M, Hoffmann M, Baum A, Lénárt P, Petronczki M, Krssák M, Gürtler U, Garin-Chesa P, Lieb S, Quant J, et al.. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol 2007; 17:316-22; PMID:17291758; http://dx.doi.org/ 10.1016/j.cub.2006.12.037 [DOI] [PubMed] [Google Scholar]
- 9.Rudolph D, Steegmaier M, Hoffmann M, Grauert M, Baum A, Quant J, Haslinger C, Garin-Chesa P, Adolf GR. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res 2009; 15:3094-102; PMID:19383823; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-2445 [DOI] [PubMed] [Google Scholar]
- 10.Gilmartin AG, Bleam MR, Richter MC, Erskine SG, Kruger RG, Madden L, Hassler DF, Smith GK, Gontarek RR, Courtney MP, et al.. Distinct concentration-dependent effects of the polo-like kinase 1-specific inhibitor GSK461364A, including differential effect on apoptosis. Cancer Res 2009; 69:6969-77; PMID:19690138; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-0945 [DOI] [PubMed] [Google Scholar]
- 11.Emmitte KA, Andrews CW, Badiang JG, Davis-Ward RG, Dickson HD, Drewry DH, Emerson HK, Epperly AH, Hassler DF, Knick VB, et al.. Discovery of thiophene inhibitors of polo-like kinase. Bioorg Med Chem Lett 2009; 19:1018-21; PMID:19097784; http://dx.doi.org/ 10.1016/j.bmcl.2008.11.041 [DOI] [PubMed] [Google Scholar]
- 12.Janning M, Fiedler W. Volasertib for the treatment of acute myeloid leukemia: a review of preclinical and clinical development. Future Oncol 2014; 10:1157-65; PMID:24947257; http://dx.doi.org/ 10.2217/fon.14.53 [DOI] [PubMed] [Google Scholar]
- 13.Gjertsen BT, Schöffski P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia 2015; 29:11-9; PMID:25027517; http://dx.doi.org/ 10.1038/leu.2014.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rudolph D, Impagnatiello MA, Blaukopf C, Sommer C, Gerlich DW, Roth M, Tontsch-Grunt U, Wernitznig A, Savarese F, Hofmann MH, et al.. Efficacy and mechanism of action of volasertib, a potent and selective inhibitor of Polo-like kinases, in preclinical models of acute myeloid leukemia. J Pharmacol Exp Ther 2015; 352:579-89; PMID:25576074; http://dx.doi.org/ 10.1124/jpet.114.221150 [DOI] [PubMed] [Google Scholar]
- 15.Lin CC, Su WC, Yen CJ, Hsu CH, Su WP, Yeh KH, Lu YS, Cheng AL, Huang DC, Fritsch H, et al.. A phase I study of two dosing schedules of volasertib (BI 6727), an intravenous polo-like kinase inhibitor, in patients with advanced solid malignancies. Br J Cancer 2014; 110:2434-40; PMID:24755882; http://dx.doi.org/ 10.1038/bjc.2014.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lowery DM, Mohammad DH, Elia AE, Yaffe MB. The Polo-box domain: a molecular integrator of mitotic kinase cascades and Polo-like kinase function. Cell Cycle 2004; 3:128-31; PMID:14712072; http://dx.doi.org/ 10.4161/cc.3.2.660 [DOI] [PubMed] [Google Scholar]
- 17.Park JE, Soung NK, Johmura Y, Kang YH, Liao C, Lee KH, Park CH, Nicklaus MC, Lee KS. Polo-box domain: a versatile mediator of polo-like kinase function. Cell Mol Life Sci 2010; 67:1957-70; PMID:20148280; http://dx.doi.org/ 10.1007/s00018-010-0279-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng KY, Lowe ED, Sinclair J, Nigg EA, Johnson LN. The crystal structure of the human polo-like kinase-1 polo box domain and its phospho-peptide complex. EMBO J 2003; 22:5757-68; PMID:14592974; http://dx.doi.org/ 10.1093/emboj/cdg558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elia AE, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB. The molecular basis for phospho-dependent substrate targeting and regulation of Plks by the polo-box domain. Cell 2003; 115:83-95; PMID:14532005; http://dx.doi.org/ 10.1016/S0092-8674(03)00725-6 [DOI] [PubMed] [Google Scholar]
- 20.Yun SM, Moulaei T, Lim D, Bang JK, Park JE, Shenoy SR, Liu F, Kang YH, Liao C, Soung NK, et al.. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nat Struct Mol Biol 2009; 16:876-82; PMID:19597481; http://dx.doi.org/ 10.1038/nsmb.1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reindl W, Gräber M, Strebhardt K, Berg T. Development of high-throughput assays based on fluorescence polarization for inhibitors of the polo-box domains of polo-like kinases 2 and 3. Anal Biochem 2009; 395:189-94; PMID:19716361; http://dx.doi.org/ 10.1016/j.ab.2009.08.031 [DOI] [PubMed] [Google Scholar]
- 22.Reindl W, Yuan J, Krämer A, Strebhardt K, Berg T. Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem Biol 2008; 15:459-66; PMID:18482698; http://dx.doi.org/ 10.1016/j.chembiol.2008.03.013 [DOI] [PubMed] [Google Scholar]
- 23.Liu F, Park JE, Qian WJ, Lim D, Gräber M, Berg T, Yaffe MB, Lee KS, Burke TR Jr. Serendipitous alkylation of a Plk1 ligand uncovers a new binding channel. Nat Chem Biol 2011; 7:595-601; PMID:21765407; http://dx.doi.org/ 10.1038/nchembio.614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qian WJ, Park JE, Lim D, Lai CC, Kelley JA, Park SY, Lee KW, Yaffe MB, Lee KS, Burke TR Jr. Mono-anionic phosphopeptides produced by unexpected histidine alkylation exhibit high plk1 polo-box domain-binding affinities and enhanced antiproliferative effects in hela cells. Biopolymers 2014; 102:444-55; PMID:25283071; http://dx.doi.org/ 10.1002/bip.22569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe N, Sekine T, Takagi M, Iwasaki J, Imamoto N, Kawasaki H, Osada H. Deficiency in chromosome congression by the inhibition of Plk1 polo box domain-dependent recognition. J Biol Chem 2009; 284:2344-53; PMID:19033445; http://dx.doi.org/ 10.1074/jbc.M805308200 [DOI] [PubMed] [Google Scholar]
- 26.Liao C, Park JE, Bang JK, Nicklaus MC, Lee KS. Exploring Potential Binding Modes of Small Drug-like Molecules to the Polo-Box Domain of Human Polo-like Kinase 1. ACS Med Chem Lett 2010; 1:110-4; PMID:20625469; http://dx.doi.org/ 10.1021/ml100020e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang YH, Park JE, Yu LR, Soung NK, Yun SM, Bang JK, et al.. Self-regulation of Plk1 recruitment to the kinetochores is critical for chromosome congression and spindle checkpoint signaling. Mol Cell 2006; 24:409-22; PMID:17081991; http://dx.doi.org/ 10.1016/j.molcel.2006.10.016 [DOI] [PubMed] [Google Scholar]
- 28.Hanissian SH, Akbar U, Teng B, Janjetovic Z, Hoffmann A, Hitzler JK, Iscove N, Hamre K, Du X, Tong Y, et al.. cDNA cloning and characterization of a novel gene encoding the MLF1-interacting protein MLF1IP. Oncogene 2004; 23:3700-7; PMID:15116101; http://dx.doi.org/ 10.1038/sj.onc.1207448 [DOI] [PubMed] [Google Scholar]
- 29.Pan HY, Zhang YJ, Wang XP, Deng JH, Zhou FC, Gao SJ. Identification of a novel cellular transcriptional repressor interacting with the latent nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J Virol 2003; 77:9758-68; PMID:12941884; http://dx.doi.org/ 10.1128/JVI.77.18.9758-9768.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foltz DR, Jansen LE, Black BE, Bailey AO, Yates JRI, Cleveland DW. The human CENP-A centromeric nucleosome-associated complex. Nat Cell Biol 2006; 8:458-69; PMID:16622419; http://dx.doi.org/ 10.1038/ncb1397 [DOI] [PubMed] [Google Scholar]
- 31.Minoshima Y, Hori T, Okada M, Kimura H, Haraguchi T, Hiraoka Y, et al.. The constitutive centromere component CENP-50 is required for recovery from spindle damage. Mol Cell Biol 2005; 25:10315-28; PMID:16287847; http://dx.doi.org/ 10.1128/MCB.25.23.10315-10328.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee KS, Park JE, Kang YH, Zimmerman W, Soung NK, Seong YS, Kwak SJ, Erikson RL. Mechanisms of mammalian polo-like kinase 1 (Plk1) localization: Self- versus non-self-priming. Cell Cycle 2008; 7:141-5; PMID:18216497; http://dx.doi.org/ 10.4161/cc.7.2.5272 [DOI] [PubMed] [Google Scholar]
- 33.Park J-E, Li L, Park J, Knecht R, Strebhardt K, Yuspa SH, Lee KS. Direct quantification of polo-like kinase 1 activity in cells and tissues using a highly sensitive and specific ELISA assay. Proc Natl Acad Sci USA 2009; 106:1725-30; PMID:19181852; http://dx.doi.org/ 10.1073/pnas.0812135106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barr FA, Sillje HH, Nigg EA. Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol 2004; 5:429-40; PMID:15173822; http://dx.doi.org/ 10.1038/nrm1401 [DOI] [PubMed] [Google Scholar]
- 35.Golsteyn RM, Schultz SJ, Bartek J, Ziemiecki A, Ried T, Nigg EA. Cell cycle analysis and chromosomal localization of human Plk1, a putative homologue of the mitotic kinases Drosophila polo and Saccharomyces cerevisiae Cdc5. J Cell Sci 1994; 107:1509-17; PMID:7962193 [DOI] [PubMed] [Google Scholar]
- 36.Lee KS, Yuan Y-L, Kuriyama R, Erikson RL. Plk is an M-phase-specific protein kinase and interacts with a kinesin-like protein, kinesin-like protein, CHO1/MKLP-1. Mol Cell Biol 1995; 15:7143-51; http://dx.doi.org/ 10.1128/MCB.15.12.7143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahonen LJ, Kallio MJ, Daum JR, Bolton M, Manke IA, Yaffe MB, Stukenberg PT, Gorbsky GJ. Polo-like kinase 1 creates the tension-sensing 3F3/2 phosphoepitope and modulates the association of spindle-checkpoint proteins at kinetochores. Curr Biol 2005; 15:1078-89; PMID:15964272; http://dx.doi.org/ 10.1016/j.cub.2005.05.026 [DOI] [PubMed] [Google Scholar]
- 38.Golsteyn RM, Mundt KE, Fry AM, Nigg EA. Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. J Cell Biol 1995; 129:1617-28; PMID:7790358; http://dx.doi.org/ 10.1083/jcb.129.6.1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol 2007; 17:304-15; PMID:17291761; http://dx.doi.org/ 10.1016/j.cub.2006.12.046 [DOI] [PubMed] [Google Scholar]
- 40.Macůrek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008; 455:119-23; PMID:18615013; http://dx.doi.org/ 10.1038/nature07185 [DOI] [PubMed] [Google Scholar]
- 41.Seki A, Coppinger JA, Jang CY, Yates JR, Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008; 320:1655-8; PMID:18566290; http://dx.doi.org/ 10.1126/science.1157425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elowe S, Hümmer S, Uldschmid A, Li X, Nigg EA. Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore microtubule interactions. Genes Dev 2007; 21:2205-19; PMID:17785528; http://dx.doi.org/ 10.1101/gad.436007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsumura S, Toyoshima F, Nishida E. Polo-like kinase 1 facilitates chromosome alignment during prometaphase through BubR1. J Biol Chem 2007; 282:15217-27; PMID:17376779; http://dx.doi.org/ 10.1074/jbc.M611053200 [DOI] [PubMed] [Google Scholar]
- 44.Toyoshima-Morimoto F, Taniguchi E, Nishida E. Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep 2002; 3:341-8; PMID:11897663; http://dx.doi.org/ 10.1093/embo-reports/kvf069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qi W, Tang Z, Yu H. Phosphorylation- and Polo-Box-dependent Binding of Plk1 to Bub1 Is Required for the Kinetochore Localization of Plk1. Mol Biol Cell 2006; 17:3705-16; PMID:16760428; http://dx.doi.org/ 10.1091/mbc.E06-03-0240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, Osada H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci U S A 2004; 101:4419-24; PMID:15070733; http://dx.doi.org/ 10.1073/pnas.0307700101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Golsteyn RM, Mundt KE, Fry AM, Nigg EA. Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. J Cell Biol 1995; 129:1617-28; PMID:7790358; http://dx.doi.org/ 10.1083/jcb.129.6.1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang F, Moss LG, Phillips GNJ. The molecular structure of green fluorescent protein. Nat Biotechnol 1996; 14:1246-51; PMID:9631087; http://dx.doi.org/ 10.1038/nbt1096-1246 [DOI] [PubMed] [Google Scholar]
- 49.Terry BR, Matthews EK, Haseloff J. Molecular characterisation of recombinant green fluorescent protein by fluorescence correlation microscopy. Biochem Biophys Res Commun 1995; 217:21-7; PMID:8526912; http://dx.doi.org/ 10.1006/bbrc.1995.2740 [DOI] [PubMed] [Google Scholar]
- 50.Hopp TP, Prickett KS, Price VL, Libby RT, March CJ, P CD, et al.. A short polypeptide marker sequence useful for recombinant protein identification and purification. Bio/Technology 1988; 6 1204-10; http://dx.doi.org/ 10.1038/nbt1088-1204 [DOI] [Google Scholar]
- 51.Huang H, Hittle J, Zappacosta F, Annan RS, Hershko A, Yen TJ. Phosphorylation sites in BubR1 that regulate kinetochore attachment, tension, and mitotic exit. J Cell Biol 2008; 183:667-80; PMID:19015317; http://dx.doi.org/ 10.1083/jcb.200805163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rudolph D, Steegmaier M, Hoffmann M, Grauert M, Baum A, Quant J, Haslinger C, Garin-Chesa P, Adolf GR. BI 6727, A Polo-like Kinase Inhibitor with Improved Pharmacokinetic Profile and Broad Antitumor Activity. Clin Cancer Res 2009; 15:3094-102; PMID:19383823; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-2445 [DOI] [PubMed] [Google Scholar]
- 53.Lee KS, Burke TR Jr, Park JE, Bang JK, Lee E. Recent advances and new strategies in targeting Plk1 for anticancer therapy. Trends in pharm sci 2015; pii: S0165-6147(15)00183-2. doi: 10.1016/j.tips.2015.08.013. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tang B, Vu M, Booker T, Santner SJ, Miller FR, Anver MR, Wakefield LM. TGF-β switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest 2003; 112:1116-24; PMID:14523048; http://dx.doi.org/ 10.1172/JCI200318899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Freedman VH, Shin SI. Cellular tumorigenicity in nude mice: correlation with cell growth in semi-solid medium. Cell 1974; 3:355-9; PMID:4442124; http://dx.doi.org/ 10.1016/0092-8674(74)90050-6 [DOI] [PubMed] [Google Scholar]
- 56.Cifone MA, Fidler IJ. Correlation of patterns of anchorage-independent growth with in vivo behavior of cells from a murine fibrosarcoma. Proc Natl Acad Sci U S A 1980; 77:1039-43; PMID:6928659; http://dx.doi.org/ 10.1073/pnas.77.2.1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu X, Lei M, Erikson RL. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol 2006; 26 2093-108; PMID:16507989; http://dx.doi.org/ 10.1128/MCB.26.6.2093-2108.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Degenhardt Y, Greshock J, Laquerre S, Gilmartin AG, Jing J, Richter M, Zhang X, Bleam M, Halsey W, Hughes A, et al.. Sensitivity of cancer cells to Plk1 inhibitor GSK461364A is associated with loss of p53 function and chromosome instability. Mol Cancer Ther 2010; 9:2079-89; PMID:20571075; http://dx.doi.org/ 10.1158/1535-7163.MCT-10-0095 [DOI] [PubMed] [Google Scholar]
- 59.Raab M, Kappel S, Krämer A, Sanhaji M, Matthess Y, Kurunci-Csacsko E, Calzada-Wack J, Rathkolb B, Rozman J, Adler T, et al.. Toxicity modelling of Plk1-targeted therapies in genetically engineered mice and cultured primary mammalian cells. Nat Commun 2011; 2:395-405; PMID:21772266; http://dx.doi.org/ 10.1038/ncomms1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weinstein IB. Addiction to oncogenes–the Achilles heal of cancer. Science 2002; 297:63-4; PMID:12098689; http://dx.doi.org/ 10.1126/science.1073096 [DOI] [PubMed] [Google Scholar]
- 61.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 2009; 136:823-37; PMID:19269363; http://dx.doi.org/ 10.1016/j.cell.2009.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol 1987; 7:2745-52; PMID:3670292; http://dx.doi.org/ 10.1128/MCB.7.8.2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seong YS, Kamijo K, Lee JS, Fernandez E, Kuriyama R, Miki T, et al.. A spindle checkpoint arrest and a cytokinesis failure by the dominant-negative polo-box domain of Plk1 in U-2 OS cells. J Biol Chem 2002; 277:32282-93; PMID:12034729; http://dx.doi.org/ 10.1074/jbc.M202602200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.