

Abstract
Choline is an essential nutrient that plays an important role in lipid metabolism and DNA methylation. Studies in rodents suggest that choline may adversely affect glycemic control, yet studies in humans are lacking. Using the human hepatic and placental cells, HepG2 and BeWo, respectively, we examined the interaction between choline and glucose treatments. In HepG2 cells, choline supplementation (1 mM) increased global DNA methylation and DNA methyltransferase expression in both low-glucose (5 mM) and high-glucose (35 mM) conditions. Choline supplementation increased the expression of peroxisomal acyl-coenzyme A oxidase 1 (ACOX1), which mediates fatty acid β-oxidation, especially in the high-glucose condition. High-glucose exposure increased the transcription of the gluconeogenic gene phosphoenolpyruvate carboxykinase (PEPCK), while choline supplementation mitigated such increase. Compared to HepG2 cells, the placenta-derived BeWo cells were relatively unresponsive to either high-glucose or -choline treatment. In conclusion, choline and glucose interacted to affect macronutrient metabolic genes, yet there was no indication that choline may worsen glycemic control in these in vitro human cell culture models.
Keywords: choline, hyperglycemia, DNA methylation, gene expression
Introduction
The essential nutrient choline participates in various biological processes. It can be derived into phosphatidylcholine, a structural component of lipoproteins, as well as betaine, which provides methyl groups for transmethylation reactions.1 The demand for choline increases substantially during pregnancy. Increasing choline intake during pregnancy has been shown to improve cognitive functions of rodent offspring2,3 and improve maternal and placental angiogenesis and stress markers in humans.4,5
Despite the potential benefits of choline supplementation on improving pregnancy outcomes, several rodent studies suggest that choline may adversely affect glycemic control.6–9 Dietary choline restriction yields better glucose tolerance despite paradoxical exacerbation of fatty liver either in mice fed with high-fat diets or in mice with genetic mutations in choline or energy metabolic genes.6–9 In these rodent models, choline is suggested to increase glucagon-mediated gluconeogenesis7 and reduce fatty acid esterification, which allows for higher hepatic output of free fatty acids,8 a known contributor to insulin resistance. Additionally, the role of choline as a methyl donor may modify glycemic control. DNA cytosine-phosphate-guanine (CpG) hypermethylation is seen in leukocytes10 and pancreatic β cells11 in cases of insulin resistance or type 2 diabetes. Choline as a methyl group donor may further promote such elevation in CpG methylation. Nevertheless, in vitro or in vivo evidence in humans regarding the interaction between choline and glycemic control is still lacking.
Pregnancy is characterized by insulin resistance and increased risk of hyperglycemia.12 The placenta is implicated as a major mediator of gestational diabetes mellitus as the disease is resolved after the placenta is removed at delivery.13 In this study, we used a human placental trophoblast cell line BeWo to examine the influence of choline supplementation and high-glucose exposure on cell growth and macronutrient metabolism in this gestation-specific organ. Since the liver is the main location of choline metabolism and is sensitive to insulin and glucagon actions, we also used the human hepatocellular carcinoma cell line HepG2 to delineate hepatic metabolic changes in response to choline and glucose supplementation. As the enzyme betaine-homocysteine S-methyltransferase (BHMT), which transfers choline-derived methyl groups to the transmethylation cycle, is predominantly expressed in liver but not in placenta,14 study of the two aforementioned cell lines helps distinguish the influence of choline on macronutrient metabolism and epigenetic regulation in modulating glycemic control.
Methods
Cell culture and treatments
The human hepatocellular carcinoma cell line HepG2 was kindly provided by Dr. Jean Grassman (Brooklyn College), and the human choriocarcinoma cell line BeWo was retrieved from the American Type Culture Collection (ATCC). The BeWo cells retain important characteristics, such as hormone synthesis and morphology of first-trimester trophoblasts.15
HepG2 cells were maintained in Eagle’s minimum essential medium (MEM) with 2 mM l-glutamine and 10% fetal bovine serum (FBS). BeWo cells were maintained in Kaighn’s modification of Ham’s F-12 medium (ATCC® 30-2004™) with 10% FBS. All cells were incubated in 5% CO2–95% air at 37°C. For the experiments, both HepG2 and BeWo cells were cultured in six-well cell culture plates and grown in MEM containing 2.5% FBS. MEM contained 7 μM of choline as was specified by the manufacturer (Corning), and the 2.5% FBS contained 16 μM of choline as was reported previously.16 As such, total choline provided by the MEM and 2.5% FBS was 23 μM. Glucose concentration of the medium was 5 mM as was specified by the manufacturer. To examine the effects of glucose and choline supplementation, the low-glucose, low-choline (LG–LC) group was not provided with additional glucose or choline, the low-glucose, high-choline (LG–HC) group was supplemented with 1 mM of choline chloride, the high-glucose, low-choline (HG–LC) group was supplemented with 30 mM of glucose in addition to the 5 mM glucose in the MEM, and the high-glucose, high-choline (HG–HC) group was supplemented with both 30 mM of glucose and 1 mM of choline chloride. Cells were seeded at a starting amount of 2 × 105 cells (passages 5–15) per well and cultured for 72 hours for HepG2 cells and 96 hours for BeWo cells before harvest. The length of cell culture was determined based on previous studies16,17 and preliminary experiments, which suggest that the cells reached 70% confluence at those time points and the effect of choline and glucose on biomarkers of interest can be detected. Each experiment was repeated three times.
Cell count and viability
After 72 hours of cell culture for HepG2 and 96 hours of cell culture for BeWo, culture medium was removed, and the attached cells were trypsinized, centrifuged, and resuspended in phosphate-buffered saline. Cell counts were measured using a hemocytometer, and cell viability was assessed using the Trypan blue dye exclusion.
RNA extraction, reverse transcription, and quantitative real-time polymerase chain reaction (PCR)
Total RNA was extracted from HepG2 and BeWo cells using TRIzol® (Life Technologies) and chloroform. RNA concentration and integrity were checked using a NanoDrop2000 instrument (Thermo Fisher Scientific). Reverse transcription was conducted using the high-capacity cDNA reverse transcription kit (Life Technologies) as per the manufacturer’s instructions. Quantitative real-time PCR was conducted using SYBR-green detection in a MiniOpticon real-time PCR system (Bio-Rad). Expression of the following genes was analyzed: antigen identified by monoclonal antibody Ki-67 (MKI67), BHMT, caspase 3 (CASP3), choline dehydrogenase (CHDH), DNA methyltransferase 1 (DNMT1), DNA methyltransferase 3A (DNMT3A), fatty acid synthase (FAS), glucose transporter 2 (SLC2A2 or GLUT2), GLUT1 (SLC2A1), glucose-6-phosphate dehydrogenase (G6PD); glycerol-3-phosphate acyltransferase 2 (GPAT2), peroxisomal acyl-coenzyme A oxidase 1 (ACOX1), proliferating cell nuclear antigen (PCNA), and phosphoenolpyruvate carboxykinase (PEPCK). All primers were designed using GeneRunner Version 3.01 (http://www.softpedia.com) (Supplementary Table 1). The efficiency of each pair of primers was between 90% and 100%. Data were expressed as the fold changes in mRNA concentrations of the above genes relative to the housekeeping gene (β-glucuronidase, GUSB) using the ΔΔCt method.18
Global DNA methylation
DNA was extracted from HepG2 and BeWo cells using the GeneJET Genomic DNA Purification Kit (Thermo Fisher Scientific) as per the manufacturer’s instructions. DNA was treated with nuclease P1, alkaline phosphatase, and phosphodiesterase (Sigma-Aldrich).19 CpG methylation of the samples was quantified using a DNA methylation enzyme-linked immunosorbent assay (ELISA) kit (Cayman Chemical) as per the manufacturer’s instructions.
Statistical analysis
Analysis of variance tests followed by post hoc Bonferroni’s correction were conducted to assess the differences among the treatment groups. Data not meeting the normality assumption were log transformed. All analyses were performed using SPSS (release 22; IBM). Differences were considered as significant at P < 0.05. Values are presented as mean ± standard error.
Results
Differential effects of choline and glucose on HepG2 and BeWo cell growth
At 72 hours of culture, HepG2 cell counts did not differ between the HG or LG groups. However, choline supplementation significantly increased (P < 0.01) the cell counts regardless of glucose treatment (Fig. 1A). At 96 hours of culture, BeWo cell counts tended to increase (P = 0.07) moderately in the HG–LC (high-glucose control) group compared to the LG–LC (low-glucose control) group. However, choline supplementation in the HG–HC group seemed to alleviate the growth-promoting effect of glucose and normalize cell counts to the level of the LG–LC group (Fig. 1B).
Figure 1.

Cell numbers (A and B) and expression of cellular proliferative and apoptotic markers (C and D) of HepG2 and BeWo cells after 72 or 96 hours of cultivation in different choline and glucose concentrations. LG–LC (black bars): low-glucose, low-choline; LG–HC (hatched bars): low-glucose, high-choline; HG–LC (gray bars): high-glucose, low-choline; HG–HC (open bars): high-glucose, high-choline. Different letters indicate significant difference, P < 0.05; (b) P < 0.1.
Abbreviations: CASP3, caspase 3; KI67, antigen identified by monoclonal antibody Ki-67; PCNA, proliferating cell nuclear antigen.
In HepG2 cells, mRNA expression of the cellular proliferative marker KI67 was increased (P < 0.05) by high-glucose treatment. Choline supplementation further increased (P < 0.05) KI67 expression in both low- and high-glucose groups (Fig. 1C). In BeWo cells, KI67 expression was suppressed (P < 0.01) by high glucose despite the increased cell counts at 96 hours of culture, yet its expression was not altered by choline supplementation (Fig. 1D). The expression of another cellular proliferative marker, PCNA, and the cellular apoptotic marker, CASP3, was not altered by glucose or choline supplementation in BeWo cells.
Choline supplementation increased DNA meth-ylation of HepG2 cells
Expression of the DNMT1, which maintains existing DNA methylation marks, and DNMT3A, which mediates de novo DNA methylation, was examined. In HepG2, neither methyltransferases were altered by glucose treatment. However, choline supplementation significantly increased (P < 0.05) DNMT1 expression, and further, the HG–HC group had a higher (P < 0.01 for DNMT1 and P = 0.09 for DNMT3A) expression of those methyltransferases than the LG–HC group, suggesting that choline-mediated increase in methyltransferase expression was more pronounced in a high-glucose environment (Fig. 2A). Consistent with the higher DNMT expression, choline supplementation increased (P < 0.05) global CpG methylation (Fig. 2B). However, DNA methylation was not modified by glucose treatment in HepG2 cells. In BeWo cells, methyltransferase expression was not altered by glucose or choline treatment (Fig. 2C), and their global CpG methylation levels were below the reliable detection limit of the assay kit (data not shown).
Figure 2.

Methylation-related gene expression (A and C) and global CpG DNA methylation (B) in HepG2 and BeWo cells after 72 or 96 hours of cultivation in different choline and glucose concentrations. LG–LC (black bars): low-glucose, low-choline; LG–HC (hatched bars): low-glucose, high-choline; HG–LC (gray bars): high-glucose, low-choline; HG–HC (open bars): high-glucose, high-choline. Different letters indicate significant difference, P < 0.05.
Abbreviations: NS, not statistically significant; DNMT, DNA methyltransferase; BHMT, betaine-homocysteine S-methyltransferase; CHDH, choline dehydrogenase.
In order for choline to be used as a methyl donor, it is first oxidized to betaine, which is mediated by the rate-limiting enzyme CHDH. Betaine then donates its labile methyl group to homocysteine for its remethylation to methionine. After methionine is converted to the universal methyl donor S-adenosylmethionine, the choline- or betaine-derived methyl groups can be used in various methylation reactions. The methyl group transfer of betaine is mediated by the enzyme BHMT1, which is predominantly expressed in the liver. Glucose and choline treatments interact to affect CHDH and BHMT1 expression in HepG2 cells. Choline supplementation increases CHDH (LG–LC vs LG–HC, P = 0.01) and BHMT1 (LG–LC vs LG–HC, P < 0.01) expression in the low-glucose condition but not in the high-glucose condition (Fig. 2A).
BeWo cells did not express BHMT1 (data not shown), and CHDH expression was not altered by choline or glucose treatment (Fig. 2C). The lack of BHMT in BeWo may explain the ineffectiveness of choline serving as a methyl donor or influencing methyltransferase expression in these cells.
Choline supplementation altered lipid and glucose metabolic genes in HepG2 cells
In HepG2 cells, both choline and glucose supplementation increased (P < 0.05) the expression of ACOX1, a gene that encodes the ACOX1 protein, which mediates fatty acid β-oxidation in the peroxisome (Fig. 3A).20 The HG–HC group had the highest expression compared to the other three groups. However, neither FAS, which mediates de novo fatty acid synthesis, nor GPAT2, which mediates triglyceride synthesis, was affected by glucose or choline treatment. PEPCK, the rate-limiting enzyme in the process of gluconeogenesis, was increased (P < 0.05) in the HG–LC group compared to the LG–LC group. However, choline supplementation in the HG–HC group seemed to normalize the elevation of PEPCK due to high glucose (Fig. 3A). G6PD, the first enzyme in the pentose phosphate pathway, was upregulated in the LG–HC group compared to the LG–LC group, but this effect of choline was not seen in the high-glucose groups. The glucose transporter GLUT2, which mediates hepatic glucose uptake, was upregulated (P < 0.05) by high glucose regardless of choline supplementation.
Figure 3.
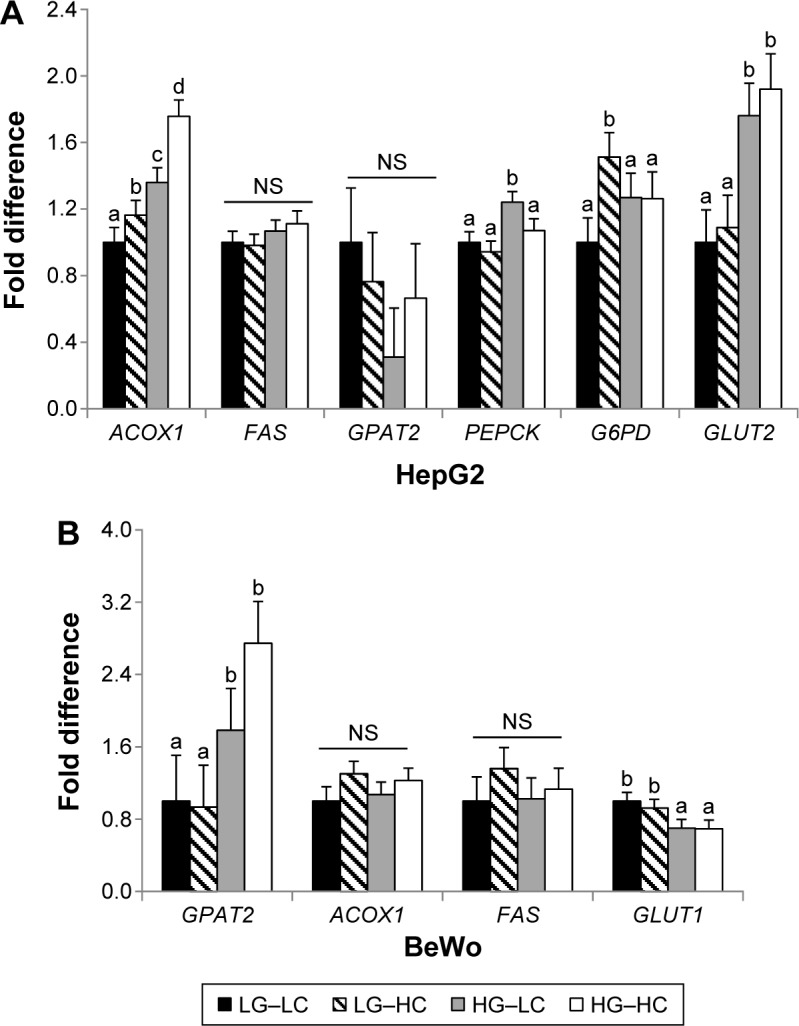
Glucose and fatty acid metabolic gene expression in HepG2 (A) and BeWo (B) cells after 72 or 96 hours of cultivation in different choline and glucose concentrations. LG–LC (black bars): low-glucose, low-choline; LG–HC (hatched bars): low-glucose, high-choline; HG–LC (gray bars): high-glucose, low-choline; HG–HC (open bars): high-glucose, high-choline. Different letters indicate significant difference, P < 0.05.
Abbreviations: NS, not statistically significant; ACOX1, peroxisomal acyl-coenzyme A oxidase 1; FAS, fatty acid synthase; GLUT, glucose transporter; G6PD, glucose-6-phosphate dehydrogenase; GPAT2, glycerol-3-phosphate acyltransferase 2; PEPCK, phosphoenolpyruvate carboxykinase.
No alterations in fatty acid or glucose metabolic gene expression as a result of choline supplementation were observed in BeWo cells (Fig. 3B). High glucose decreased (P < 0.01) the expression of the glucose transporter GLUT1 but increased (P < 0.05) the expression of GPAT2 regardless of choline supplementation. PEPCK was not expressed in the placenta (data not shown).
Discussion
We examined the interaction between choline and glucose supplementations in modifying global DNA methylation and expression of genes that mediate fatty acid and glucose metabolism in human placental and hepatic cell lines. In HepG2 cells, choline supplementation seemed to promote DNA methylation regardless of glucose concentrations, whereas the changes in fatty acid and glucose metabolic gene expression by choline supplementation may favor the mitigation of hyperglycemic stress. BeWo cells were relatively unresponsive to glucose or choline supplementation.
Effects of choline and glucose on cell growth differed between cell lines
In HepG2 cells, choline appeared to be a stronger promoting factor for cell growth and proliferation than glucose, as both cell counts and the expression of the cellular proliferative marker KI67 were increased by choline supplementation. In contrast, in vitro hyperglycemia did not alter HepG2 cell counts, which was consistent with the observation by Iyer et al.21 However, another study by Chandrasekaran et al22 suggests that when HepG2 cells were supplemented with 50 mM glucose, they demonstrated abnormal morphology, detachment from culture plates, and apoptosis, which were not observed in this study. The differed results may be due to the lower dose (35 mM) of glucose used in our experiment, which seems to be well tolerated by HepG2 cells. High glucose upregulated KI67, which was further amplified by choline cosupplementation. Previous studies suggest that hyperglycemia enhances liver stellate cell proliferation and exacerbates collagen production and connective tissue overgrowth, which is a potential mechanism of liver fibrosis.23,24 It is unclear whether the growth and proliferation promoting effect of choline also applies to other liver cell types and plays a role in liver damage. In vivo studies in animals and humans, however, generally suggest choline as a lipotrope that prevents the formation of fatty liver and normalizes liver enzymes.8,25
High-glucose treatment tended to increase BeWo cell counts modestly in the current study setting, although Weiss et al26 and Masumoto et al27 both reported that trophoblast growth was inhibited by in vitro hyperglycemia. Interestingly, expression of the cellular proliferative marker KI67 was significantly dampened in high glucose, whereas another proliferative marker PCNA was unchanged, despite the modest increase in cell counts. These results may indicate that after prolonged culture (96 hours) in high glucose, there was a larger proportion of cells in the high-glucose groups that were not proliferating compared to the low-glucose groups, since neither KI67 nor PCNA was expressed in resting (G0) cells. Choline supplementation seemed to partially normalize the overgrowth triggered by in vitro hyperglycemia, although it did not affect KI67 expression. Taken together, both glucose and choline have complicated roles in the growth and proliferation of BeWo cells.
Choline but not glucose treatment promoted DNA methylation in HepG2 cells
CpG methylation of DNA is a major epigenetic event that alters gene expression without changing the genetic sequence. There is mounting evidence suggesting that both global and site-specific CpG methylation levels are altered in patients with metabolic diseases, such as diabetes. Zhao et al10 reported that leukocyte global CpG methylation was positively associated with insulin resistance. Pancreatic and duodenal homeobox 1 (PDX1), a critical gene that promotes pancreatic cell maturation and islet function, was hypermethylated in pancreatic islet cells isolated from patients with type 2 diabetes.11 These findings highlight the potential role of epigenetics in regulating the pathogenesis and progression of diabetes. In a HepG2 cell culture model, Chiang et al.28 demonstrated that in vitro hyperglycemia increased DNMT3A expression and global CpG methylation, suggesting activation of the transmethylation cycle. Supplying additional methyl donors, such as choline, under the condition of hyperglycemia may exacerbate the already active transmethylation cycle. In this study, choline supplementation increased DNMT expression and global CpG methylation in HepG2 cells. However, in contrast to that reported by Chiang et al,28 global CpG methylation was not altered by glucose treatment. Cosupplementation of choline and glucose seems to have an additive effect on promoting DNMT expression but not on global CpG methylation levels. It should be noted that both CHDH and BHMT expression were decreased in the HG–HC group. This may serve as a feedback mechanism to prevent excess amounts of choline-derived methyl groups from entering the transmethylation cycle when both glucose and choline are in surplus, thereby maintaining CpG methylation levels despite the upregulation of DNMTs. Taken together, supplementing choline in HepG2 cell culture did not seem to exacerbate CpG hypermethylation under the condition of hyperglycemia.
In BeWo cells, DNMT expression was not altered by either choline or glucose. The lack of BHMT expression in these cells may explain the null effect of choline. However, in an in vivo system, choline-derived methyl groups can be transferred from the maternal compartment (eg, liver) to the placenta. A previous human study has shown that higher maternal choline intake increases both global and site-specific CpG methylation of the placenta.5 As such, in vivo studies are needed to further discern the interaction of glucose and choline on trophoblast CpG methylation in an integral whole-body system.
Choline supplementation was not associated with unfavorable alterations in fatty acid or glucose metabolism
Although fatty liver is associated with a higher risk of insulin resistance,29 recent studies suggest that fat accumulation in the liver itself does not always result in insulin resistance.8,30–32 While choline deficiency results in fatty liver, it improves insulin resistance and glucose tolerance in male animals.7,8 The dissociation between fatty liver and insulin resistance is proposed to be related to choline’s effect on promoting the triglyceride breakdown and efflux of free fatty acids from the liver to circulation.8 Circulating free fatty acids exacerbate peripheral insulin resistance. In this study, we found that choline supplementation enhanced the transcription of ACOX1 in HepG2 cells, especially in a high-glucose environment. ACOX1 mediates the first step of long-chain fatty acid β-oxidation in the peroxisome, and its increase may upregulate fatty acid catabolism.20 However, genes that mediate de novo fatty acid synthesis (eg, FAS) or triglyceride synthesis (eg, GPAT2) were not differentially expressed in the high- or low-choline groups. These findings do not support the role of choline in promoting triglyceride breakdown and fatty acid output but rather indicate its effect on reducing fatty acid content by enhancing β-oxidation.
Choline supplementation ameliorated the elevation in PEPCK expression under the condition of high glucose. Although glucose normally inhibits PEPCK expression and reduces gluconeogenesis, chronic hyperglycemia blunts its inhibitory effect,33,34 leading to PEPCK overexpression and an increased rate of gluconeogenesis, which is observed in patients with type 2 diabetes. Our HepG2 cell model suggests that choline supplementation may rectify PEPCK expression when hepatocytes are exposed to high glucose, which may in turn mitigate gluconeogenesis and hepatic glucose output. Choline supplementation did not change glucose or fatty acid metabolic gene expression in BeWo cells, likely due to the lack of PEPCK expression and gluconeogenesis in these cells.
Glucose treatment triggered differential responses in HepG2 and BeWo cells. While increased expression of GLUT2 in HepG2 cells may enhance glucose uptake, there was a decrease in GLUT1 expression in the high-glucose-treated BeWo cells, which may be a mechanism of the trophoblasts to prevent excessive glucose transfer under hyperglycemic stress. The elevation of GPAT2 in BeWo cells in the high-glucose groups may be an additional regulating mechanism to promote triglyceride or phospholipid synthesis and trap free fatty acids in the placenta, thereby preventing their transport to the fetus.
Taken together, these results suggest that choline supplementation do not adversely affect hepatic glucose and fatty acid metabolism in either HepG2 cells or BeWo cells. Choline may be used as a lipotropic supplementation to prevent or improve hepatic steatosis by improving fatty acid β-oxidation and may lower the risk of hyperglycemia by dampening gluconeogenesis. The neutral effect of choline on placental-derived BeWo cells discerns the concerns about its potential negative impacts on the metabolic balance of pregnant women and their fetuses. Nevertheless, clinical studies are needed to confirm the efficacy and safety of choline supplementation in modulating macronutrient homeostasis in humans.
Conclusion
Choline supplementation increased DNA methylation and modified the transcription of fatty acid and glucose metabolic genes in HepG2 cells but did not have a direct effect on BeWo cells. There was no indication that choline led to unfavorable changes in the above processes under the condition of in vitro hyperglycemia. In vivo studies are needed to further delineate the interaction between choline and glucose in special physiological states, such as pregnancy.
Supplementary Material
Supplementary table 1. Real-time PCR primers.
Footnotes
ACADEMIC EDITOR: Joseph Zhou, Editor in Chief
PEER REVIEW: Three peer reviewers contributed to the peer review report. Reviewers’ reports totaled 651 words, excluding any confidential comments to the academic editor.
FUNDING: This study was funded by PSC-CUNY Research Award TRADA-45-72. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert single-blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived and designed the experiments: XJ. Analyzed the data: XJ, EG, and CJ-R. Wrote the first draft of the article: XJ and EG. Contributed to the writing of the article: XJ, EG, and CJ-R. Agreed with manuscript results and conclusions: XJ, EG, and CJ-R. Jointly developed the structure and arguments for the article: XJ, EG, and CJ-R. Made critical revisions and approved the final version: XJ, EG, and CJ-R. All authors reviewed and approved the final article.
REFERENCES
- 1.Caudill MA. Pre- and postnatal health: evidence of increased choline needs. J Am Diet Assoc. 2010;110(8):1198–1206. doi: 10.1016/j.jada.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 2.McCann JC, Hudes M, Ames BN. An overview of evidence for a causal relationship between dietary availability of choline during development and cognitive function in offspring. Neurosci Biobehav Rev. 2006;30(5):696–712. doi: 10.1016/j.neubiorev.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Blusztajn JK, Mellott TJ. Neuroprotective actions of perinatal choline nutrition. Clin Chem Lab Med. 2013;51(3):591–599. doi: 10.1515/cclm-2012-0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang X, Bar HY, Yan J, et al. A higher maternal choline intake among third-trimester pregnant women lowers placental and circulating concentrations of the antiangiogenic factor fms-like tyrosine kinase-1 (sFLT1) FASEB J. 2013;27(3):1245–1253. doi: 10.1096/fj.12-221648. [DOI] [PubMed] [Google Scholar]
- 5.Jiang X, Yan J, West AA, et al. Maternal choline intake alters the epigenetic state of fetal cortisol-regulating genes in humans. FASEB J. 2012;26(8):3563–3574. doi: 10.1096/fj.12-207894. [DOI] [PubMed] [Google Scholar]
- 6.Wu G, Zhang L, Li T, et al. Choline supplementation promotes hepatic insulin resistance in phosphatidylethanolamine N-methyltransferase-deficient mice via increased glucagon action. J Biol Chem. 2013;288(2):837–847. doi: 10.1074/jbc.M112.415117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu G, Zhang L, Li T, Lopaschuk G, Vance DE, Jacobs RL. Choline deficiency attenuates body weight gain and improves glucose tolerance in ob/ob mice. J Obes. 2012(2012):319172. doi: 10.1155/2012/319172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raubenheimer PJ, Nyirenda MJ, Walker BR. A choline-deficient diet exacerbates fatty liver but attenuates insulin resistance and glucose intolerance in mice fed a high-fat diet. Diabetes. 2006;55(7):2015–2020. doi: 10.2337/db06-0097. [DOI] [PubMed] [Google Scholar]
- 9.Teng YW, Ellis JM, Coleman RA, Zeisel SH. Mouse betaine-homocysteine S-methyltransferase deficiency reduces body fat via increasing energy expenditure and impairing lipid synthesis and enhancing glucose oxidation in white adipose tissue. J Biol Chem. 2012;287(20):16187–16198. doi: 10.1074/jbc.M111.303255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao J, Goldberg J, Bremner JD, Vaccarino V. Global DNA methylation is associated with insulin resistance: a monozygotic twin study. Diabetes. 2012;61(2):542–546. doi: 10.2337/db11-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang BT, Dayeh TA, Volkov PA, et al. Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes. Mol Endocrinol. 2012;26(7):1203–1212. doi: 10.1210/me.2012-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barbour LA, McCurdy CE, Hernandez TL, Kirwan JP, Catalano PM, Friedman JE. Cellular mechanisms for insulin resistance in normal pregnancy and gestational diabetes. Diabetes Care. 2007;30(suppl 2):S112–S119. doi: 10.2337/dc07-s202. [DOI] [PubMed] [Google Scholar]
- 13.Lowe WL, Karban J. Genetics, genomics and metabolomics: new insights into maternal metabolism during pregnancy. Diabet Med. 2014;31(3):254–262. doi: 10.1111/dme.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pajares MA, Pérez-Sala D. Betaine homocysteine S-methyltransferase: just a regulator of homocysteine metabolism? Cell Mol Life Sci. 2006;63(23):2792–2803. doi: 10.1007/s00018-006-6249-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pattillo RA, Gey GO. The establishment of a cell line of human hormone-synthesizing trophoblastic cells in vitro. Cancer Res. 1968;28(7):1231–1236. [PubMed] [Google Scholar]
- 16.Jiang X, Jones S, Andrew BY, et al. Choline inadequacy impairs trophoblast function and vascularization in cultured human placental trophoblasts. J Cell Physiol. 2014;229(8):1016–1027. doi: 10.1002/jcp.24526. [DOI] [PubMed] [Google Scholar]
- 17.Vrablic AS, Albright CD, Craciunescu CN, Salganik RI, Zeisel SH. Altered mitochondrial function and overgeneration of reactive oxygen species precede the induction of apoptosis by 1-O-octadecyl-2-methyl-rac-glycero-3-phosphocholine in p53-defective hepatocytes. FASEB J. 2001;15(10):1739–1744. doi: 10.1096/fj.00-0300com. [DOI] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Shin W, Yan J, Abratte CM, Vermeylen F, Caudill MA. Choline intake exceeding current dietary recommendations preserves markers of cellular methylation in a genetic subgroup of folate-compromised men. J Nutr. 2010;140(5):975–980. doi: 10.3945/jn.110.121186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ouali F, Djouadi F, Merlet-Bénichou C, Riveau B, Bastin J. Regulation of fatty acid transport protein and mitochondrial and peroxisomal beta-oxidation gene expression by fatty acids in developing rats. Pediatr Res. 2000;48(5):691–696. doi: 10.1203/00006450-200011000-00023. [DOI] [PubMed] [Google Scholar]
- 21.Iyer VV, Yang H, Ierapetritou MG, Roth CM. Effects of glucose and insulin on HepG2-C3A cell metabolism. Biotechnol Bioeng. 2010;107(2):347–356. doi: 10.1002/bit.22799. [DOI] [PubMed] [Google Scholar]
- 22.Chandrasekaran K, Swaminathan K, Chatterjee S, Dey A. Apoptosis in HepG2 cells exposed to high glucose. Toxicol In Vitro. 2010;24(2):387–396. doi: 10.1016/j.tiv.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 23.Paradis V, Perlemuter G, Bonvoust F, et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: a potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology. 2001;34(4 pt 1):738–744. doi: 10.1053/jhep.2001.28055. [DOI] [PubMed] [Google Scholar]
- 24.Sugimoto R, Enjoji M, Kohjima M, et al. High glucose stimulates hepatic stellate cells to proliferate and to produce collagen through free radical production and activation of mitogen-activated protein kinase. Liver Int. 2005;25(5):1018–1026. doi: 10.1111/j.1478-3231.2005.01130.x. [DOI] [PubMed] [Google Scholar]
- 25.Corbin KD, Zeisel SH. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr Opin Gastroenterol. 2012;28(2):159–165. doi: 10.1097/MOG.0b013e32834e7b4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weiss U, Cervar M, Puerstner P, et al. Hyperglycaemia in vitro alters the proliferation and mitochondrial activity of the choriocarcinoma cell lines BeWo, JAR and JEG-3 as models for human first-trimester trophoblast. Diabetologia. 2001;44(2):209–219. doi: 10.1007/s001250051601. [DOI] [PubMed] [Google Scholar]
- 27.Masumoto A, Takamoto N, Masuyama H, Akahori Y, Inoue S, Hiramatsu Y. Effects of intermittent high glucose on BeWo choriocarcinoma cells in culture. J Obstet Gynaecol Res. 2011;37(10):1365–1375. doi: 10.1111/j.1447-0756.2011.01539.x. [DOI] [PubMed] [Google Scholar]
- 28.Chiang EP, Wang YC, Chen WW, Tang FY. Effects of insulin and glucose on cellular metabolic fluxes in homocysteine transsulfuration, remethylation, S-adenosylmethionine synthesis, and global deoxyribonucleic acid methylation. J Clin Endocrinol Metab. 2009;94(3):1017–1025. doi: 10.1210/jc.2008-2038. [DOI] [PubMed] [Google Scholar]
- 29.Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. 2014;510(7503):84–91. doi: 10.1038/nature13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rinella ME, Green RM. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol. 2004;40(1):47–51. doi: 10.1016/j.jhep.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 31.Stefan N, Staiger H, Häring HU. Dissociation between fatty liver and insulin resistance: the role of adipose triacylglycerol lipase. Diabetologia. 2011;54(1):7–9. doi: 10.1007/s00125-010-1938-y. [DOI] [PubMed] [Google Scholar]
- 32.Asai A, Chou PM, Bu HF, et al. Dissociation of hepatic insulin resistance from susceptibility of nonalcoholic fatty liver disease induced by a high-fat and high-carbohydrate diet in mice. Am J Physiol Gastrointest Liver Physiol. 2014;306(6):G496–G504. doi: 10.1152/ajpgi.00291.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Y, Liu S, Ferguson S, et al. Phosphoenolpyruvate carboxykinase overexpression selectively attenuates insulin signaling and hepatic insulin sensitivity in transgenic mice. J Biol Chem. 2002;277(26):23301–23307. doi: 10.1074/jbc.M200964200. [DOI] [PubMed] [Google Scholar]
- 34.Shao J, Qiao L, Janssen RC, Pagliassotti M, Friedman JE. Chronic hyperglycemia enhances PEPCK gene expression and hepatocellular glucose production via elevated liver activating protein/liver inhibitory protein ratio. Diabetes. 2005;54(4):976–984. doi: 10.2337/diabetes.54.4.976. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table 1. Real-time PCR primers.