

Abstract
p21WAF1 is a well-characterized mediator of cell cycle arrest and may also modulate chemotherapy-induced cell death. The role of p21WAF1 in drug-induced cell cycle arrest and apoptosis of acute lymphoblastic leukemia (ALL) cells was investigated using p53-functional patient-derived xenografts (PDXs), in which p21WAF1 was epigenetically silenced in T-cell ALL (T-ALL), but not in B-cell precursor (BCP)-ALL PDXs. Upon exposure to diverse cytotoxic drugs, T-ALL PDX cells exhibited markedly increased caspase-3/7 activity and phosphatidylserine (PS) externalization on the plasma membrane compared with BCP-ALL cells. Despite dramatic differences in apoptotic characteristics between T-ALL and BCP-ALL PDXs, both ALL subtypes exhibited similar cell death kinetics and were equally sensitive to p53-inducing drugs in vitro, although T-ALL PDXs were significantly more sensitive to the histone deacetylase inhibitor vorinostat. Transient siRNA suppression of p21WAF1 in the BCP-ALL 697 cell line resulted in a moderate depletion of the cell fraction in G1 phase and marked increase in PS externalization following exposure to etoposide. Furthermore, stable lentiviral p21WAF1 silencing in the BCP-ALL Nalm-6 cell line accelerated PS externalization and cell death following exposure to etoposide and vorinostat, supporting previous findings. Finally, the Sp1 inhibitor, terameprocol, inhibited p21WAF1 expression in Nalm-6 cells exposed to vorinostat and also partially augmented vorinostat-induced cell death. Taken together, these findings demonstrate that p21WAF1 regulates the early stages of drug-induced apoptosis in ALL cells and significantly modulates their sensitivity to vorinostat.
Keywords: childhood acute lymphoblastic leukemia, etoposide, p21WAF1, patient derived xenograft models, terameprocol, vorinostat
Introduction
As a cyclin-dependent kinase (CDK) inhibitor, p21WAF1 can cause cell cycle arrest at both the G1-S and G2-M cell cycle phases.1,2 p21WAF1 has been characterized as a tumor suppressor3 primarily due to its role in mediating cell cycle arrest by both p53-dependent and p53-independent processes.4 Under certain conditions p21WAF1 can also function as an anti-apoptotic factor. Up-regulation of p21WAF1 can efficiently protect various cell types from apoptosis,5,6 whereas suppression of p21WAF1 results in increased sensitivity to apoptosis induced by a range of cytotoxic drugs and γ-irradiation.7-9 Specifically, in malignant hematopoietic cells inhibition of p21WAF1 by various means, including antisense oligonucleotide transfection, increased sensitivity to cytotoxic drugs such as paclitaxel or cytarabine.10,11 Conversely, overexpression of p21WAF1 in a range of leukemia and lymphoma cell lines conferred some resistance to cytarabine or etoposide.10,12,13 However, in a recent study deletion of cdkn1a in a murine Eµ-Myc lymphoma model did not sensitize lymphoma cells to histone deacetylase inhibitor (HDAC)-induced apoptosis,14 highlighting that questions remain over the proposed strength of the anti-apoptotic role of p21WAF1 in hematopoietic cells.
The interplay between cell cycle inhibition and apoptosis initiation is regulated in certain settings by the p53 protein, which ultimately determines the relative sensitivity of tumor cells to chemotherapy induced cell death.15 One mechanism that has been recently demonstrated to control the switch between cell cycle arrest and death under DNA damaging conditions is the binding of DNA-dependent protein kinase, catalytic subunit (DNA-PKcs) to p53 and their recruitment to p53-responsive elements on the p21WAF1 promoter leading to rapid loss of p21WAF1 transcription and increased apoptosis.16 TP53 mutations represent another mechanism that leads to drug resistance and perturbed initiation of apoptosis in cancer cells,17 although in diseases such as childhood acute lymphoblastic leukemia (ALL),18,19 apoptosis can also be inhibited by alternative mechanisms, including ATM inactivation,20 overexpression of Hdm-2,21 expression of anti-apoptotic proteins,22 or dysfunction in the p53-p21WAF1 axis.23,24 Upregulation of p21WAF1 and disruption of the cytotoxic response can occur irrespective of TP53 gene mutations.20,24,25 In addition, the induction of p21WAF1 that occurs after exposure to various cytotoxic stimuli can inhibit the apoptosis process in malignant hematopoietic cells26-28 and solid tumor cells.5,29 In the clinical setting, elevated p21WAF1 expression has been associated with chemotherapy resistance and poor prognosis in acute myeloid leukemia,30,31 while an association with p21WAF1 induction and poor clinical outcome in ALL has been proposed.32
Mechanisms proposed to explain the anti-apoptotic role of p21WAF1 include transcriptional regulation of anti-apoptotic genes,33 inhibition of CDKs that are involved in activation of caspases integral to apoptosis downstream of mitochondrial disruption,9 or direct inhibition of pro-apoptotic proteins, such as procaspase-3, caspase-8 or apoptosis signal-regulating kinase 1.3,33 Inhibition of CDKs has also been demonstrated to negatively affect caspase activation9 and chromatin condensation.34
Cell death pathways induced by chemotherapy drugs include apoptotic and non-apoptotic processes. Apoptosis is influenced by caspase activity, establishing the necessary characteristics of the early stages of apoptosis such as phosphatidylserine (PS) externalization and condensed nuclei.35 Though caspase-independent forms of cell death exist, the induction of apoptosis is thought to be the predominant pathway to cancer cell destruction. However, caspase activity is not always necessary for apoptosis, and other death pathways are initiated depending on cell type and cytotoxic stimuli.36,37 For example the apoptosis executioner, caspase-3 may stimulate the repopulation of cancer cells by increasing inflammatory signals and activating pro-survival pathways in other malignant cells.38,39
With p21WAF1 having an anti-apoptotic role in response to certain cytotoxic agents, inhibition of p21WAF1 has been considered as a strategy for cancer treatment to sensitize cells toward apoptosis after chemotherapy exposure.40 The Sp1 inhibitor, terameprocol, has been previously demonstrated to inhibit p21WAF1 expression41 and can be utilized to demonstrate any impact of p21WAF1 inhibition on cell death.
This study examines the influence of p21WAF1 on the cell death pathways of ALL cells after exposure to chemotherapeutic drugs. Various models of ALL were utilized, including patient-derived xenografts (PDXs) with epigenetically silenced p21WAF1 in p53-functional T-ALL samples, transient siRNA and stable lentiviral knockdown of p21WAF1 expression in BCP-ALL cell lines and pharmacological modulation of p21WAF1 induction by terameprocol.41 Our results show that p21WAF1 exerts a significant influence on the kinetics of apoptosis mediated by chemotherapeutic drugs, but does not markedly influence in vitro sensitivity to p53-inducing agents or non-apoptotic characteristics of cell death.
Results
T-ALL cells exhibit increased apoptotic characteristics compared with BCP-ALL cells following exposure to cytotoxic drugs
While p21WAF1 induction has been demonstrated to inhibit apoptosis in various cancer histotypes with a range of sensitivity to cytotoxic stimuli,5-8,10-14,28,42 the anti-apoptotic role of p21WAF1 in p53-functional, chemotherapy-sensitive hematopoietic cells has still not been fully elucidated. We assessed the in vitro apoptotic responses of T-ALL and BCP-ALL PDXs and cell lines following exposure to etoposide or vorinostat. The p53 status and p21WAF1 responses of the PDXs used in this study have previously been determined and are summarized in Table 1.23 To evaluate the apoptotic response of lymphoid leukemia cells to cytotoxic agents that induce p21WAF1, we assessed caspase-3/7 activity and PS externalization in PDX cells and ALL cell lines exposed to 5 µM of etoposide or vorinostat, with or without prior incubation with the pan-caspase inhibitor z-VAD-FMK for up to 48 h of drug exposure. Caspase-3/7 activity of 2 T-ALL PDXs increased to 2-5 OD450 units/mg protein from <0.6 OD450 units/mg protein within 6 h of exposure to etoposide (Fig. 1A-B) or vorinostat (Fig. S1A-B), while caspase activity in the 2 BCP-ALL PDXs and 1 BCP-ALL cell line (Nalm-6; data not shown) remained equal to or below 2 OD450 units/mg protein after drug exposure (Fig. 1C-D and Fig. S1C-D). Caspase-3/7 activity remained high in the T-ALL PDXs for 48 h, while exhibiting much reduced activity in the BCP-ALL PDXs (Fig. 1A-D and Fig. S1A-D). z-VAD-FMK, effectively inhibited caspase-3/7 activity in all PDX lines tested (Fig. 1A-D and Fig. S1A-D) .
Figure 1.
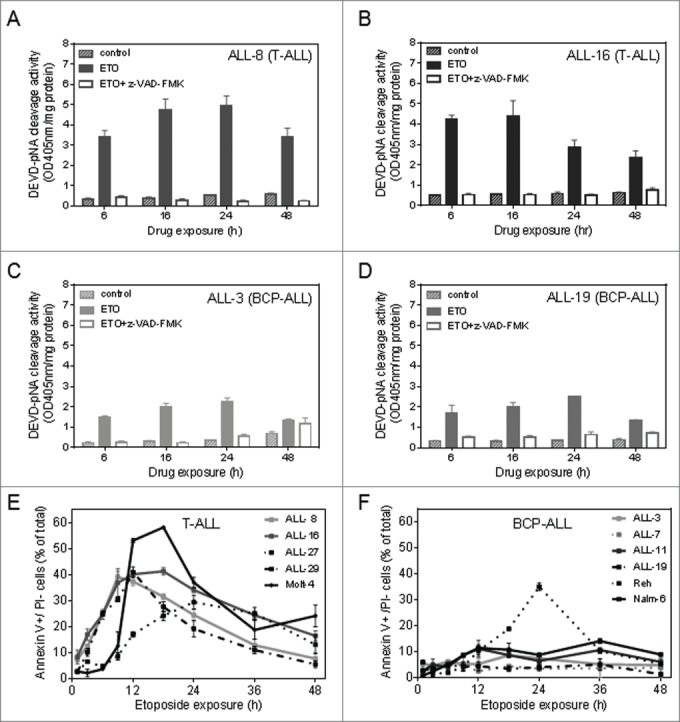
The effect of etoposide on caspase activity and PS externalization in T-ALL and BCP-ALL PDXs and cell lines. T-ALL PDX cells [A, ALL-8; B, ALL-16] and BCP-ALL PDX cells [C, ALL-3; D, ALL-19] were exposed to solvent control or etoposide (ETO) for 6 h, 16 hr, 24 h and 48 hr, with or without pre-treatment with z-VAD-FMK (75 µM, 16 h). Caspase-3/7 activity is expressed as DEVD-pNA cleavage activity. Each bar represents the mean caspase activity ± SEM of 3 independent assays. PS externalization on the outer membrane of cells was determined by staining cells with annexin V-FITC and PI. Collated data of annexin V+/PI- cells over 48 h of etoposide exposure are shown in E (T-ALL samples) and F (BCP-ALL samples). Each point represents the mean % of annexin V+/PI− cells ± SEM as calculated from 3 biological replicates.
Consistent with the difference in caspase-3/7 activity between T-ALL and BCP-ALL PDXs, PS externalization, as assessed by annexin V binding on PI− cells, was also increased in T-ALL PDXs and cell lines (Fig. 1E-F, Fig. S1E-F, Fig. S2A-D). T-ALL PDXs and the Molt-4 cell line exhibited rapid (within 6-12 h) PS externalization in up to 60% of cells, whereas BCP-ALL PDXs and Nalm-6 cells showed <25% of PS externalization over the 48 h test period, and all with delayed kinetics compared with T-ALL cells. An exception was the Reh cell line, where ∼35% of cells exhibited PS externalization following etoposide exposure (Fig. 1F), albeit with delayed kinetics. Despite dramatic differences in caspase-3/7 activation and PS externalization, both B- and T-cell lineages appeared equally sensitive to drug-induced cell death as evidenced by the proportion of PI+ cells. Cell death kinetics in T-ALL PDXs was only marginally accelerated compared to BCP-ALL PDXs over the first 12 h of exposure to 5 µM of each drug, but was almost identical thereafter (Fig. S3).
We next compared the apoptotic responses between the 2 ALL lineages exposed to cytotoxic drugs that do not specifically induce p21WAF1 expression. ALL-8 (T-ALL PDX) cells exhibited a prominent increase in caspase-3/7 activity and PS externalization compared with ALL-3 (BCP-ALL PDX) cells following exposure to the glucocorticoid dexamethasone and the kinase inhibitor staurosporine (Fig. S4A-D). However there was no substantial difference in the rate of loss of cell viability (Fig. S4E-F). Thus, T-ALL cells exhibit increased apoptotic characteristics compared with BCP-ALL cells when exposed to a broad range of cytotoxic drugs, despite demonstrating comparable kinetics of cell death.
To determine whether the differences in apoptotic characteristics exhibited by T-ALL and BCP-ALL cells were associated with subtle variations in drug responses, we tested their sensitivity to a range of concentrations of etoposide, vorinostat and the Hdm-2 inhibitor nutlin-3 using the MTT cytotoxicity assay. While equally sensitive to etoposide and nutlin-3, the IC50 values of T-ALL PDXs treated with vorinostat were significantly lower than BCP-ALL PDXs (p = 0.03) (Table 1, Fig. S5), demonstrating increased sensitivity of T-ALL PDXs to the cell death inducing effects of vorinostat.
Table 1.
Biological data and in vitro drug response of ALL PDXs and laboratory established cell lines.
| IC50 values (Mean ± SEM, μM) |
||||||
|---|---|---|---|---|---|---|
| Xenograft/ cell line | ALL subtype | p53 status | p21WAF1 status | Etoposide | Nutlin-3 | Vorinostat |
| ALL-2 | BCP-ALL | wt | Responsive | 9.20 ± 1.16 | 6.49 ± 1.09 | 7.76 ± 0.71 |
| ALL-3 | BCP-ALL | wt | Responsive | 0.74 ± 0.05 | 1.15 ± 0.07 | 0.57 ± 0.03 |
| ALL-7 | BCP-ALL | wt | Responsive | 0.64 ± 0.05 | 1.90 ± 0.51 | 0.61 ± 0.03 |
| ALL-8 | T-ALL | wt | Silenced | 0.36 ± 0.01 | 1.73± 0.53 | 0.38 ± 0.02 |
| ALL-11 | BCP-ALL | wt | Responsive | 1.02 ± 0.18 | 3.23 ± 0.55 | 1.68 ± 0.23 |
| ALL-16 | T-ALL | wt | Silenced | 0.31 ± 0.02 | 1.49 ± 0.04 | 0.33 ± 0.03 |
| ALL-19 | BCP-ALL | wt | Responsive | 2.14 ± 0.28 | 3.63 ± 0.27 | 2.48 ± 0.34 |
| ALL-27 | T-ALL | wt | Silenced | 1.22 ± 0.19 | 5.16 ± 0.89 | 0.48 ± 0.08 |
| ALL-29 | T-ALL | wt | Partial | 0.46 ± 0.09 | 2.06 ± 0.21 | 0.35 ± 0.03 |
| ALL-30 | T-ALL | wt | Silenced | 0.54 ± 0.04 | 1.41 ± 0.22 | 0.39 ± 0.06 |
| ALL-31 | T-ALL | wt | Silenced | 1.14 ± 0.11 | 2.69 ± 0.07 | 1.94 ± 0.13 |
| Reh | BCP-ALL | wt | Responsive | 0.49 ± 0.11 | 6.65 ± 1.03 | 2.94 ± 0.46 |
| Nalm-6 | BCP-ALL | wt | Responsive | 1.44 ± 0.25 | 4.62 ± 0.31 | 1.88 ± 0.13 |
| Molt-4 | T-ALL | wt | Silenced | 0.41 ± 0.03 | 3.90 ± 0.15 | 2.52 ± 0.61 |
p21WAF1 status was defined by p21WAF1 protein induction after etoposide or vorinostat exposure, as described in Davies et al., 2011. Mean IC50 values (µM) were determined in this study from 3 independent MTT assays.
p21WAF1 knockdown enhances drug-induced apoptosis in BCP-ALL cells
To determine whether the previously reported difference in p21WAF1 expression and induction between T-ALL and BCP-ALL PDXs accounted for the divergence in apoptotic responses to cytotoxic drugs, we used knockdown approaches in BCP-ALL cell lines. First, we utilized siRNA to efficiently knockdown p21WAF1 expression in the 697 cell line for a 48 h time period (Fig. 2A). p21WAF1 knockdown resulted in a marked increase in early apoptotic (annexin V+/PI−) cells following etoposide treatment (Fig. 2B) although, consistent with the PDX data described above, the proportion of non-viable (PI+) cells remained unaffected (Fig. 2C). p21WAF1 knockdown also had little impact on etoposide-induced accumulation of cells in G2/M or depletion from S phase, although it did result in a modest depletion of cells from G1 and increase in the sub-G1 fraction (Fig. 2D) .
Figure 2.
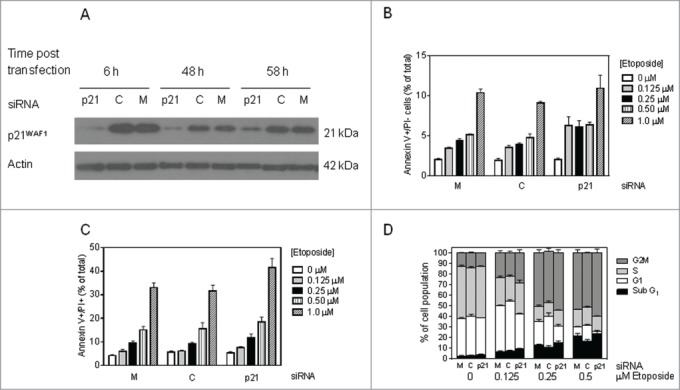
siRNA knockdown of p21WAF1 in 697 cells accelerates drug-induced apoptosis. (A) p21WAF1 expression by western blotting in 697 cells at various time points post siRNA electroporation of p21WAF1 siRNA (labeled p21), control siRNA against the MLL-AF4 fusion protein (MLL-AF4 not present in 697 cells) (C) and mock transfection (M). (B-D) 697 cells were electroporated with the siRNA constructs shown in A, and 16 h later exposed to etoposide (0.125, 0.25 , 0.5, 1 µM) for 48 hr. The proportion of early apoptotic (Annexin V+/PI-), (B); non-viable (Annexin V+/PI+) cells (C); and cell cycle distribution, (D); were determined by flow cytometry.
We also tested the efficiency of microRNAs cloned into a lentiviral vector for stable p21WAF1 knockdown in Nalm-6 cells (Table S1). Lentiviral transduction with miRNA #2 mediated the most effective knockdown of p21WAF1 protein in Nalm-6 cells (Fig. 3A) and this construct was used in subsequent experiments. Mean intracellular p21WAF1 protein levels assessed by flow cytometry were increased by 5.5–6.0 fold following etoposide exposure (5 μM, 6 h) in Nalm-6 cells transduced with GFP or scrambled constructs (Fig. 3B-C). However, cells transduced with p21WAF1 miRNA #2 showed a reduction in basal p21WAF1 protein expression (0.9 mean fold change), and complete inhibition of p21WAF1 induction after etoposide exposure (Fig. 3D-E) .
Figure 3.
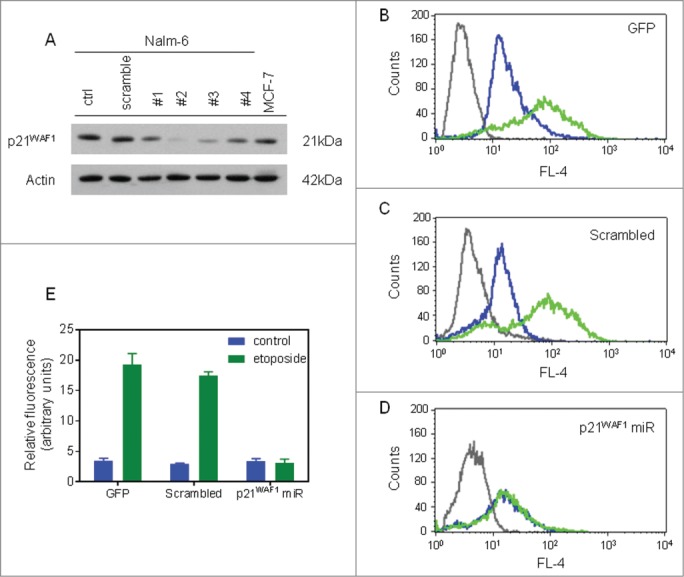
Lentiviral knockdown of p21WAF1 in Nalm-6 cells. (A) Western blot of lysates prepared from mass-cultures of lentiviral vector-transduced Nalm-6 cells. Numbers represent the different miRNA constructs encoded in the lentiviral constructs. The MCF-7 lysate was included as a positive control for p21WAF1 expression. (B-D) Transduced Nalm-6 cells were treated with DMSO or etoposide for 6 h and then prepared for detection of p21WAF1 expression by dual color flow cytometry. Gray lines, isotype control; blue lines, solvent-treated cells; green lines, etoposide-treated cells. (B) GFP or pLent6/V5-EmGFP transduced cells; (C) Scrambled or pLenti6/EmGFP-neg-pre-miR (encoding control miRNA) transduced cells; (D) p21WAF1 miRNA or pLenti6/EmGFP-p21-pre-miRNA #2 (p21WAF1 miRNA) transduced cells. (E) Graphical representation of the quantified relative fluorescence index of transduced cells with and without etoposide exposure from 3 independent experiments (error bars =mean ± SEM).
To evaluate the contribution of p21WAF1 to drug-induced cell death in ALL cells, transduced Nalm-6 cells were exposed to vorinostat and etoposide, and PS externalization and cell viability were assessed. After 9 h treatment, PS externalization was maximal for etoposide-treated T-ALL PDX cells (Fig. S4C). At this time point, p21WAF1 knockdown resulted in a significant increase in PS externalization in etoposide and vorinostat treated cells, as evidenced by an increase in the population of early apoptotic (annexin V+/PI−) and non-viable (annexin V+/PI+) cells (Fig. 4A-B, respectively). However, the differences in PS externalization and cell death elicited by p21WAF1 knockdown were not as pronounced following 24 h exposure to etoposide or vorinostat (Fig. 4C-D). Moreover, p21WAF1 knockdown resulted in only a marginal sensitization of Nalm-6 cells to 48 h of vorinostat and etoposide as assessed by MTT assay. For vorinostat exposure, knockdown of p21WAF1 resulted in a statistically significant difference to GFP and scrambled cells in IC50 values as determined by one-way ANOVA with multiple comparisons (ANOVA Multiplicity adjusted P-value vs. GFP = 0.023, vs. scrambled = 0.002) (Fig. 4E-F, Table 2). After etoposide exposure, Nalm-6 cells with p21WAF1 knockdown did not demonstrate a significant difference compared to GFP or scramble-transduced cells (ANOVA; Multiplicity adjusted P-value vs. GFP = 0.154, vs. scrambled = 0.117), highlighting that inhibition of p21WAF1 expression does not influence sensitivity of cells to p53-inducing agents and re-affirming the results revealed in vorinostat sensitivity for the p21WAF1-defective TALL PDXs cells.
Figure 4.
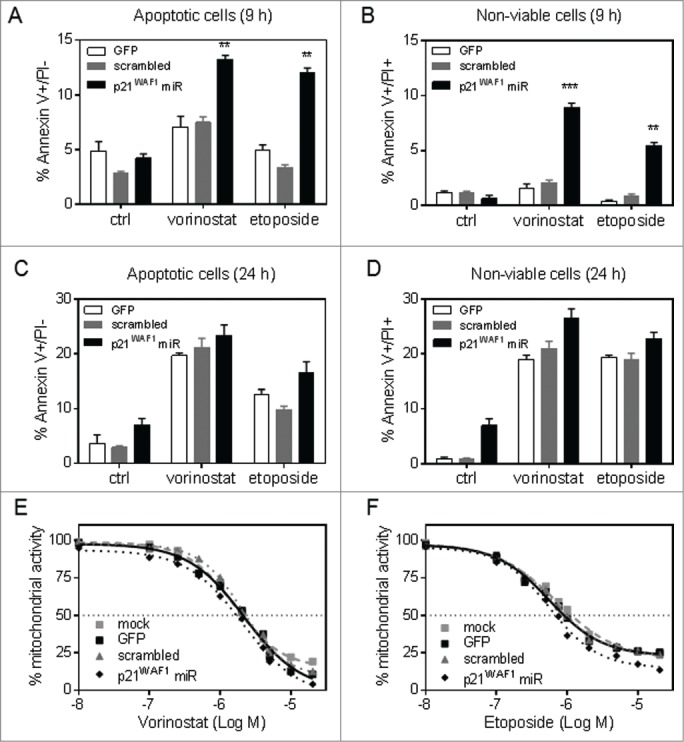
Knockdown of p21WAF1 expression sensitizes Nalm-6 cells to vorinostat- and etoposide-induced apoptosis. Transduced Nalm-6 cells were analyzed for PS externalization and membrane integrity by annexin V binding and PI staining after exposure to vorinostat (5 μM), etoposide (5 μM) or solvent control. (A) Early apoptotic (Annexin-V+/PI−PtdIns-) cells at 9 hr. (B) Non-viable cells (Annexin V+/PI+) at 9 hr. (C) Early apoptotic (Annexin-V+/PI−) cells at 24 hr. (D) Non-viable cells (Annexin V+/PI+) at 24 hr. Bars represent the mean ± SEM of 3 experiments. Significantly greater in p21WAF1 miRNA transduced cells than GFP and scrambled transduced cells (* = p<0.05) by one-way ANOVA with Tukey's multiple comparison test. (E, F) Transduced Nalm-6 cells were also treated with vorinostat (E) or etoposide (F) for 48 h and mitochondrial activity assessed relative to a vehicle treated control using the MTT assay. Results are the mean ± SEM of 3 independent experiments.
Table 2.
Impact of p21WAF1 repression on the sensitivity of Nalm-6 cells to vorinostat and etoposide.
| IC50 values (Mean ± SEM, μM) |
||
|---|---|---|
| Condition | Vorinostat | Etoposide |
| Mock | 2.05 ± 0.11 | 1.51 ± 0.11 |
| GFP-transduced | 1.99 ± 0.04 | 0.91 ± 0.04 |
| Scramble-transduced | 2.10 ± 0.06 | 0.97 ± 0.06 |
| p21WAF1-pre-miR transduced | 1.71 ± 0.07a,b | 0.70 ± 0.07 |
Statistical analysis of the IC50 values generated from each experiment of transduced cells (GFP, Scramble, p21WAF1 miRNA) was conducted by one-way ANOVA with Tukey's multiple comparison test: a = p21WAF1 miRNA transduced significant difference compared to GFP transduced cells (p<0.05); b = p21WAF1 miRNA transduced significant difference to scramble transduced cells (p<0.005).
Finally, to assess the effects of pharmacological inhibition of p21WAF1, the Sp1 inhibitor terameprocol which has previously been demonstrated to inhibit p21WAF1 induction, was utilized to inhibit p21WAF1 expression after vorinostat and etoposide exposure. Terameprocol inhibited CDKN1A mRNA induction after vorinostat exposure in Nalm-6 cells, but had little effect on CDKN1A mRNA induction by etoposide (Fig. 5A). Consistent with these differential effects on p21WAF1 induction by vorinostat and etoposide, terameprocol sensitized Nalm-6 cells to vorinostat to a greater extent than to etoposide (Fig. 5B) .
Figure 5.
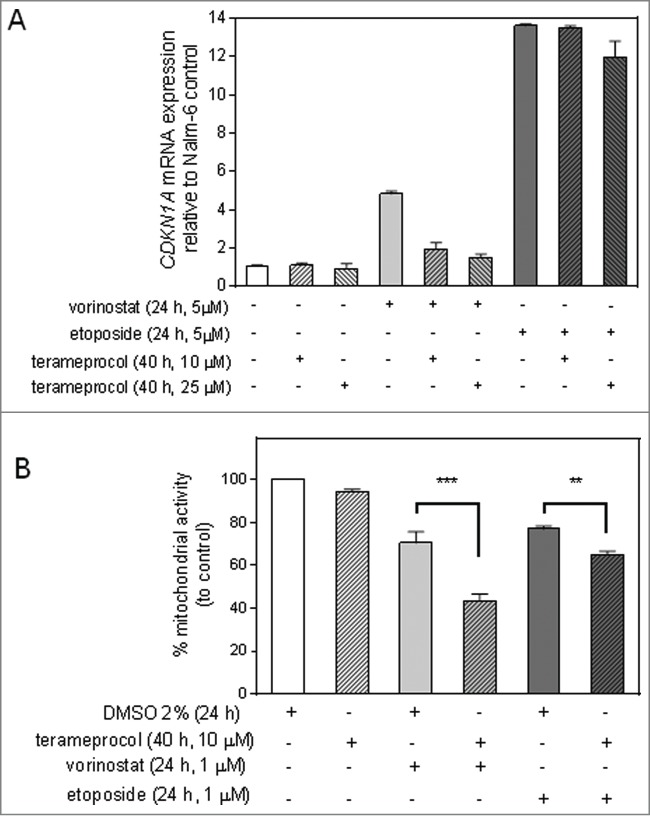
Terameprocol inhibits p21WAF1 induction by vorinostat and augments vorinostat-induced cell death in Nalm-6 cells. (A) Nalm-6 cells were pre-treated with terameprocol or vehicle control for 16 hr, and then exposed to either etoposide or vorinostat (5 μM, 24 hr). The p21WAF1 transcriptional activity was measured by quantitative real time RT-PCR. Bars represent the mean value of CDKN1A mRNA ± range of 2 independent experiments. (B) Nalm-6 cells were pre-treated with terameprocol or vehicle control for 16 hr, and then exposed to either etoposide or vorinostat (1 µM, 24 hr). Mitochondrial activity was assessed by the MTT cytotoxicity assay and normalized to cells exposed to DMSO solvent. Bars represent the mean % mitochondrial activity of ± SEM of 3 independent experiments. Statistical analysis of the mitochondrial activity after exposure to cytotoxic agents was conducted by one-way ANOVA with Tukey's multiple comparison test and significant differences between vorinostat or etoposide alone and in combination with terameprocol are presented (** = p<0.05, *** = p<0.005).
Discussion
The anti-apoptotic functions of p21WAF1 have been proposed in various models. Yet the influence of p21WAF1 on apoptosis has not been fully characterized in p53-functional hematopoietic cells that are inherently sensitive to cytotoxic stimuli. The investigations contained within evaluated the impact of p21WAF1 expression on the apoptotic processes of leukemia cells with a wild-type p53 response to DNA damaging agents and demonstrated a lineage specific response in T-ALL versus BCP-ALL cell death characteristics. As the cell systems tested in these experiments contained functional p53, this data also highlights more broadly the anti-apoptotic role of p21WAF1 in p53-functional cells. Previously the two cell lineages demonstrated specific modulation of p21WAF1 expression after p53 induction or HDAC inhibition.23 In the experiments described here, greater caspase-3/7 activity was exhibited by p21WAF1-defective T-ALL PDXs after exposure to etoposide and vorinostat in comparison to p21WAF1-functional BCP-ALL cells. Maximal caspase activity also occurred earlier in T-ALL cells than in BCP-ALL PDXs, demonstrating increased apoptotic kinetics in T-ALL samples.
PS externalization was another apoptotic characteristic measured in these experiments that also followed a lineage specific trend. PS externalization occurred in T-ALL cells with an intact membrane, yet was only detected in a small proportion of viable BCP-ALL cell samples. The only BCP-lineage cells that exhibited PS externalization comparable to T-ALL after vorinostat and etoposide exposure was the Reh cell line. The Reh cell line was the most sensitive BCP-ALL sample to the effects of etoposide, demonstrated by the lowest IC50 value. Although Nalm-6 cells also exhibited PS externalization after drug exposure, this did not occur to the same degree as seen in T-ALL samples.
For T-ALL PDXs, the rate and extent of PS externalization after drug exposure reflected cell sensitivity for this cell lineage. An initial sharp increase in PS externalization on cells with an intact membrane was a characteristic of drug sensitive T-ALL cells, as demonstrated by lower IC50 values from MTT assays.
PS externalization has been suggested to be a caspase-dependent feature of apoptosis.35 Early increase in caspase activity and extensive PS externalization observed in T-ALL PDX complies with this hypothesis. Three BCP-ALL samples showed proportionately less caspase activity and a later time to reach maximal effect, and further demonstrated minimal PS externalization over their untreated controls. An association between high caspase activity and PS externalization was demonstrated in T-ALL PDXs, while high PS externalization was seen in all T-ALL samples (both PDXs and cell lines) tested.
T-ALL cells exhibited increased apoptotic characteristics compared to BCP-ALL when exposed to a range of cytotoxic drugs, despite revealing comparable cell death kinetics. While BCP-ALL cells showed an increase in caspase activity and PS externalization over untreated controls, these results demonstrate that not all forms of cell death necessarily require a robust caspase-3 effect. Caspase activity can be inhibited by a range of cellular processes, such as high expression of Melanoma Inhibitor of Apoptosis Protein, overexpression of survivin, or even by binding of p21WAF1 to procaspase-3.43-45
T-ALL PDX cells were also sensitive to the death inducing effects of vorinostat, as determined by significantly lower IC50 values compared to BCP-ALL PDXs. In comparison, the p21WAF1-defective T-ALL and p21WAF1-functional BCP-ALL PDXs demonstrated generally similar sensitivities to the p53-inducing agents, etoposide and nutlin-3. As both T-ALL and BCP-ALL PDXs demonstrated wild-type p53 activity,23 it would be understandable that both lineages would be equally sensitive to DNA damage and Mdm-2 inhibition. While other perturbations in the p53 pathway besides TP53 gene mutations do exist, this observation suggests that their impact on the in vitro sensitivity of PDX ALL cells would be minimal.
To test whether the defective p21WAF1 induction observed in the majority of T-ALL cell samples could have an effect on the apoptotic pathway and their increased sensitivity to the death inducing effects of drugs, p21WAF1 expression was suppressed in Nalm-6 cells by transduction with an miRNA construct and by transfection in 697 cells with siRNA. The siRNA-mediated suppression of p21WAF1 protein in 697 cells led to an increase in the sub-G1 fraction after etoposide exposure, which has been observed previously with other agents.10,11,28 Further, this suppression of p21WAF1 led to an increase in sensitivity to the death inducing effects of etoposide in 697 cells.
p21WAF1-silenced Nalm-6 cells demonstrated an increase in PS externalization following exposure to vorinostat and etoposide compared to GFP or scramble-transduced cells. Silencing of p21WAF1 also increased sensitivity to vorinostat and etoposide, yet between the p21WAF1 knockdown cells and either the GFP or scramble-transduced cells, a significant decrease in mean IC50 was only seen after vorinostat exposure. Furthermore, greater differences in drug sensitivity were observed between the p21WAF1-defective T-ALL PDXs and the p21WAF1-expressing BCP-ALL cells. Although a weak effect on cell death was noted for p21WAF1 in this hematopoietic cell system, other in vitro models utilizing different genetic settings or cell types have demonstrated an important role for p21WAF1 in inhibiting cell death. A number of studies evaluating the impact of p21WAF1 on cell death have been conducted in epithelial derived tumor cells such as MCF-7 or HCT116 cell lines.6-9,46,47 In general, most of the tumor cell lines showed inherent resistance to the effects of certain cytotoxic stimuli.7,46 Furthermore, enhanced apoptotic responses of p21WAF1-deficient HCT116 cells to DNA damage occurred after 24 h exposure.9,46,47 Cell death was observed within 6 h after drug exposure in both T-ALL and BCP-ALL PDXs and cell lines, demonstrating the increased cytotoxic sensitivity of leukemia cells compared to epithelial derived cell lines.
Only a limited number of studies have reported on the antiapoptotic effect of p21WAF1 of hematopoietic cells with many conducted in cells with either a p53-null or mutant genotype.10,11,13,28 One study used a p53 expressing leukemia cell line (HL-60), and here the disruption of p21WAF1 function did not appreciably sensitize the cells to cytarabine.48 The study herein is the first to report on the anti-apoptotic role of p21WAF1 in a BCP-lineage leukemia cell line that was previously shown to have an intact p53 response to DNA damaging agents.23
Reports of p21WAF1 attenuating apoptosis in myelomonocytic and promyelocytic leukemia cells have demonstrated that inactivation of p21WAF1 renders an immediate effect on apoptotic characteristics and drug sensitivity. For example, in the U-937 promyelocytic cell line, increased apoptosis was observed within 4 h of drug exposure.11 Wang et al.11 surmised that the mechanism by which p21WAF1 dysregulation rendered leukemia cells more sensitive to drugs at earlier time points would differ fundamentally from the mechanisms in colon cancer cells.7,9,47,49 In our study, p21WAF1 silenced Nalm-6 cells demonstrated increased apoptosis, presented in the marker of PS externalization, after 9 h of exposure to etoposide and vorinostat. As silencing of p21WAF1 in these cells increased early exposure of PS externalization, this supports the hypothesis that p21WAF1 can interact with the initial phase of the apoptotic pathway. As some studies have reported on the interaction of p21WAF1 with caspases and have a direct, negative, impact on the apoptotic pathway,9,50 it is feasible to propose that in the early stages of apoptosis in leukemia cells, caspase activity can also be perturbed by p21WAF1. If p21WAF1 is low or not expressed, caspase activity is increased. When p21WAF1 is present, cell death still occurs, but with reduced level of apoptotic characteristics. The kinetics of this form of cell death may be slower than classical apoptotic cell death.
In previous studies terameprocol has demonstrated biological activity to inhibit Sp1-mediated gene transcription.41 HDAC inhibitors such as vorinostat have been shown to induce p21WAF1 through Sp1 activation.51 In these experiments, the selectivity of terameprocol in modulating p21WAF1 expression through Sp1 was confirmed as this drug inhibited p21WAF1 induction mediated by vorinostat but had no effect on etoposide mediated p21WAF1 expression. In combination with vorinostat, terameprocol augmented cell death in leukemia cells that express p21WAF1. These results lead us to propose that attenuation of p21WAF1 sensitizes leukemia cells with functional p21WAF1 to HDAC inhibitors. In addition to regulating p21WAF1 expression, terameprocol can regulate the expression of a range of proteins involved in cell cycle arrest, apoptosis and angiogenesis such as cyclin-dependent kinase 1, survivin and Vascular Endothelial Growth Factor.52 Therefore, further research with terameprocol should also encompass the influence of these genes to specify the mechanism by which terameprocol can enhance cell death induced by vorinostat. Irrespective of those studies, the results from the present investigations provide proof of the principle that p21WAF1 modulation by pharmacological methods may be used to enhance cell death kinetics.
In this study, knockdown of p21WAF1 protein sensitized Nalm-6 cells to the apoptotic-inducing effects of vorinostat and etoposide in a time-dependent manner. At early time points, p21WAF1 silenced cells exhibited a significant increase in vorinostat and etoposide induced PS externalization, yet at the 24 h time point, there were minimal differences. Chemotherapy-induced cell death still occurred in both T-ALL and BCP-ALL cell populations, demonstrating that high caspase activity was not essential for in vitro cell death. The differences in sensitivity observed between the p21WAF1-expressing and p21WAF1-silenced Nalm-6 cells were not as dramatic as that seen between p21WAF1-expressing BCP-ALL cell lines and PDXs and p21WAF1-defective T-ALL PDXs cells, highlighting that other characteristics besides differences in p21WAF1 induction could contribute to the drug sensitivity demonstrated between ALL cell lineages. The unique genetic characteristics that leukemia cells from different lineages possess could be one of the contributing factors that influence the sensitivity and apoptotic characteristics of the leukemia cell lineages. Previous work with T-ALL and BCP-ALL PDXs have established distinct gene expression profiles for the 2 leukemia lineages,53 while a subset-of the T-ALL and BCP-ALL PDXs tested have demonstrated differential expression of certain p53-target genes in response to etoposide.23 A group of genes that have been demonstrated to influence the apoptotic response to cytotoxic drugs is the Bcl-2 gene family. The basal expression levels of pro-apoptotic or anti-apoptotic Bcl-2 genes were presented for T-ALL and BCP-ALL PDXs in a recent publication.53 These results did not show a distinct homogeneous pattern of expression within lineages for individual members of this gene family and therefore basal gene expression was unable to explain simply the cell lineage differences in apoptotic response and drug sensitivity.
While the differences between T-ALL and BCP-ALL PDX sensitivity to vorinostat in vitro were modest, when previously tested in vivo by our group vorinostat was universally ineffective against the same pediatric ALL PDXs used in this study.54 However, the second-generation inhibitor of Class I and II HDACs, quisinostat (JNJ-26481585), exerted profoundly increased in vivo anti-leukemic efficacy against 2 T-ALL PDXs (ALL-8 and -16) compared with 6 BCP-ALL PDXs (including ALL-2, -3, -7, -11 and -19).55 Therefore, epigenetic silencing of p21WAF1 may provide a biomarker for the enhanced in vivo sensitivity of T-ALL to second-generation HDAC inhibitors.
Patients, Materials and Methods
All experimental studies were conducted with approval from the Human Research Ethics Committee and the Animal Care and Ethics Committee of the University of New South Wales (Sydney, Australia).
In vitro cell culture
Leukemia cell lines [697 (or PreB-697), Reh, Nalm-6, Molt-4] were obtained from the American Type Culture Collection (Manassas, VA, USA) or the European Collection of Cell Cultures (Salisbury, UK), and maintained in Roswell Park Memorial Institute (RPMI-1640) media supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL), streptomycin (100 µg/mL), and L-glutamine (2 mM). 293FT cells were purchased from Invitrogen (Catalog Number (#) R700-07) and maintained in Dulbecco's Modified Eagle media (DMEM) with the same supplements. PDX cells were prepared for in vitro culture as described previously.56 Lineage subtype and other details of PDXs and cell lines are shown in Table 1.
Patient clinical samples
Leukemia cells were obtained from the Centre for Children's Cancer and Blood Disorders at Sydney Children's Hospital from children presenting with ALL that were enrolled in Australia and New Zealand Children's Cancer Study Group (ANZCCSG) Study VII (1998-2001) or Study VIII (2002 to present). Protocols for processing samples, harvesting, cryopreservation and establishing PDX cells are detailed in previous work.57
In vitro cytotoxicity assays
In vitro drug sensitivity was assessed using the colorimetric methyl-thiazolyl-tetrazolium (MTT) assay, and is described elsewhere.56 The cells were exposed to etoposide (Sigma-Aldrich, #E1383), vorinostat (donation from Merck Research Laboratories), terameprocol (donation from Erimos Pharmaceuticals) or an equivalent volume of dimethyl sulfoxide (DMSO) vehicle (Sigma-Aldrich).
Caspase activity assays
Caspase activity was measured using the Caspase-3 Colorimetric Assay (Sigma-Aldrich, #CASP3C) following the manufacturer's instructions. Vorinostat (5 μM), etoposide (5 μM) or staurosporine (1 μM) or an equivalent volume of DMSO were added to cells for 0, 6, 16 and 24 h. Mean absorbance at A405nm on a Biotrack II spectrophotometer plate reader (Amersham Biosciences, Piscataway, NJ, USA) from triplicate samples was compared to a series of pnitroaniline (pNA) standard concentrations, and caspase activity expressed as an amount of Ac-DEVD-pNA released.
PS externalization assays
PS translocation from the inner surface of the plasma membrane to the outer leaflet was detected using Annexin V binding assay. PDX cells and ALL cell lines were treated over 48 h with cytotoxic stimuli (5 μM etoposide, 5 μM vorinostat, 1 μM dexamethasone, 1 μM staurosporine) or an equivalent volume of DMSO. At predetermined time points, the cells were washed with phosphate buffered saline (PBS), then incubated with 5 μL annexin V solution (FITC or APC, BD PharMingen, San Diego, CA, USA) and 250 ng propidium iodide (PI) in 200 μL of Annexin-V binding buffer (BD PharMingen, #556454). Cells were analyzed and data collected on a BD FacsCanto flow cytometer (BD PharMingen San Diego, CA, USA) using CellQuest software. 697 cells were treated over 24 h with etoposide (0.125-1 μM). Cells (3 x 105) were then washed twice in PBS followed by suspension in 250 μL of binding buffer containing 125 ng Annexin V (Abcam, #ab14082) and 100 ng PI. Cells were incubated for 20 min and a further 250 μL of binding buffer was then added prior to analysis on a BD FACScan flow cytometer (BD PharMingen) using CellQuest software.
Cell cycle analysis
697 cells were treated over 24 h with etoposide (0.125-1 μM). Cells (3 x 105) were re-suspended in 100 µL of 0.25 M sucrose, 40 mM sodium citrate (pH 7.6), 400 µL of DNA staining buffer (20 µg/mL PI, 0.5% NP-40 and 0.5 mM EDTA in PBS) and 28.8 µL of 44 mg/mL DNase free RNase A. Samples were incubated at 4°C in the dark for 30 min prior to analysis with BD FACScan cytometer (BD PharMingen) using CellQuest software.
Quantitative RT-PCR
RNA extraction and real-time quantitative reverse transcription polymerase chain reaction (RT2-PCR) was carried out as previously described.56 Primers and probes for CDKN1A (p21WAF1 gene) were purchased from Applied Biosystems (Hs00355782_m1). The Elongation factor-1a (EF-1α) gene was an internal standard in each reaction (primers EF-1αF, 5′-CTGAACCATCCAGGCCAAAT-3′; EF-1αR, 5′-GCCGTGTGGCAATCCAAT-3′; Probe, 5′-VIC-AGCGCCGGCTATGCCCCTG-TAMRA-3′). RNA levels were normalized to EF-1α values, expressed as fold differences relative to CDKN1A mRNA expression in Nalm-6 cells.
siRNA transfection
The 697 cell line was transfected with siRNA construct p21 SHS1.Hs01.00025255 (Sigma-Aldrich) or control siRNA construct siMA6 against the MLL-AF4 fusion, not present in 697 cells (sense construct 5′-AAGAAAAGCAGACCUACUCCA-3′) (Invitrogen). Cells (790 µL of 1 x 107/mL) were placed into an electroporation cuvette and 10 µL of either 10 µM p21WAF1 siRNA, 10 µM control (siMA6) siRNA (final siRNA concentration 250 nM) or water (mock control) were added and electroporated at 350V, 1200µF, 10ms with an EPI 2500 Electroporation Pulse Generator and then left for 15 min before diluting in 9 mL of culture media. Cells were incubated at 37°C, 5% CO2 for 16 h, from siRNA transfection, prior to addition of drug.
Lentiviral transductions
CDKN1A gene (p21WAF1) knockdown was performed using pLenti6/EmGFP lentiviral constructs (Invitrogen, #V496-10) in Nalm-6 cells. DNA oligonucleotides were designed using the RNAi Designer software available from Invitrogen to encode a target pre-microRNA (pre-miRNA) for CDKN1A as described in the BLOCK-iT™ Inducible Pol II miR RNAi Expression Vector instruction manual. Briefly, 4 double-stranded oligonucleotides encoding the engineered pre-miRNA were prepared having, among other structural features, a 5′G + short 21 nucleotide antisense sequence (mature miRNA) targeting the CDKN1A gene followed by 19 nucleotides to form the terminal loop and a short sense target sequence with 2 nucleotides removed to create an internal loop. The double-stranded DNA oligonucleotides were prepared from the set of single stranded oligonucleotides (Table S1) by PCR, annealed and ligated into an intermediary pcDNA plasmid construct. Lentiviral vectors encoding the green fluorescence protein (GFP) and a p21WAF1 pre-miRNA cassette driven by a CMV promoter were constructed utilizing Gateway→ recombination technology from the pcDNA plasmid constructs to produce pLenti6/EmGFP p21WAF1 microRNA vector. DNA was isolated from bacterial cultures using Purelink HQ Mini Plasmid Purification kit (Invitrogen, #K2100-15). Sequencing of plasmid DNA was conducted at Sydney University Prince Alfred Molecular Analysis Center (University of Sydney, NSW). Plasmids and PCR products were digested with restriction enzymes; BamHI and HindIII (Promega, #R6021 and #R6041) were used to linearize pcDNA constructs for verification, while EagI (New England Biolabs Inc., #R0505S) was used to linearize pcDNA6.2-GW/EmGFP-p21miR constructs before cloning.
Lentiviral infections were carried according to standard procedures for silencing experiments. 293FT cells were transfected using Lipofectamine (Invitrogen, #11668-019) as per the manufacturer's protocol with Virapower Packaging mix (Invitrogen, #K4975-00) and 3 μg of lentiviral plasmid in 3 mL Opti-MEM containing 10% FCS. The next day, media containing the DNA-Lipofectamine complexes was removed and replaced with 10 mL of complete DME media without antibiotics. The virus-containing media (VCM) was harvested from the 293FT cells after 24 h, and replaced with fresh complete DME media. VCM was centrifuged at 490 x g for 15 min at 4°C to pellet debris and filtered through a 0.45 μm filter. Thereafter, every 12 h, VCM was harvested from 293FT cells for infection of the Nalm-6 leukemia cell line.
Nalm-6 cells (2 x 105) were transduced with VCM prepared from pLenti6/EmGFP p21WAF1 miRNA vector (p21WAF1 miRNA), pLenti6/EmGFP vector (GFP-transduced) or pLenti6/EmGFP-scrambled vector (scramble) on 3 consecutive days at a Multiplicity of Infection (MOI) of 30 in a minimal volume of media with polybrene (8 μg/mL). After incubation in VCM for 72 h, Nalm-6 cells were expanded in complete RPMI media.
Analysis of protein expression
The preparation and separation of whole cell lysates from PDX cells and leukemia cell lines (Nalm6 and 697) have been described previously.23,56 697 cell line lysates were prepared using lysis buffer supplied by Cell Signaling Technology (New England Biolabs, #9803), containing a protease inhibitor cocktail (Roche, UK). Membranes were probed with mouse antibodies for p21WAF1 (clone SX118BD: BD Transduction Laboratories, #556430 or clone 70: BD Biosciences, #610234) and rabbit or mouse antibodies for actin (Sigma-Aldrich #A3853 or Clone JLA20, Calbiochem, #MABT219), followed by horseradish peroxidase (HRP) conjugated donkey anti-rabbit, sheep anti–mouse secondary antibodies (GE Healthcare, #RPN4301 and #RPN4201), or goat anti-mouse or anti-rabbit secondary antibodies (Dako, #P044701-2 and #P044801-2). Bound secondary antibodies were detected by chemiluminescence and visualized by autoradiography detection and phosphoimaging as described previously.23
Cellular p21WAF1 protein expression was measured in Nalm-6 cells after lentiviral transduction by flow cytometry. Following treatment with etoposide (5 μM, 6 h), transduced cells were fixed in 1% formaldehyde in PBS for 15 min, washed in PBS and permeabilized with 200 µL 0.1% Triton X-100. Fixed cells were incubated with anti-human p21WAF1 antibody (Clone SX118), and then a secondary anti-mouse IgG Cy5-conjugated antibody (Invitrogen, #A10524).
Statistical Comparison and Data Analysis
All data were compiled, the mean and standard error of mean (SEM) of data sets calculated using GraphPad Prism software (version 6.02). Mann-Whitney U tests (for non-normally distributed data) were utilized to compare differences between groups. Significance level was set at p < 0.05. Analysis of multiple samples was conducted by one-way ANOVA with Tukey's multiple comparison test to examine results between groups.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Vorinostat was generously provided by Merck, Sharpe and Dohme, Corp. and the National Cancer Institute, National Institutes of Health. Terameprocol was generously provided by Erimos Pharmaceuticals.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was supported by the Children's Cancer Institute Australia for Medical Research, the JGW Pattern Foundation (Newcastle upon Tyne) (to L.H, Grant ID: 6895), a fellowship (to R.B.L.) and grants from The Australian National Health and Medical Research Council (Grant ID: 1059804, 568703) and an Australian Postgraduate Award (to C.D.). Children's Cancer Institute is affiliated with the University of New South Wales and the Sydney Children's Hospitals Network.
References
- 1.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497-501; PMID:9822382; http://dx.doi.org/ 10.1126/science.282.5393.1497 [DOI] [PubMed] [Google Scholar]
- 2.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993; 75:805-16; PMID:8242751; http://dx.doi.org/ 10.1016/0092-8674(93)90499-G [DOI] [PubMed] [Google Scholar]
- 3.Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res 2005; 65:3980-5; PMID:15899785; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-3995 [DOI] [PubMed] [Google Scholar]
- 4.Gartel AL, Tyner AL. Transcriptional regulation of the p21(WAF1/CIP1) gene. Exp Cell Res 1999; 246:280-9; PMID:9925742; http://dx.doi.org/ 10.1006/excr.1998.4319 [DOI] [PubMed] [Google Scholar]
- 5.Burgess AJ, Pavey S, Warrener R, Hunter LJ, Piva TJ, Musgrove EA, Saunders N, Parsons PG, Gabrielli BG. Upregulation of p21(WAF1/CIP1) by histone deacetylase inhibitors reduces their cytotoxicity. Mol Pharmacol 2001; 60:828-37; PMID:11562446 [PubMed] [Google Scholar]
- 6.Wendt J, Radetzki S, von Haefen C, Hemmati PG, Guner D, Schulze-Osthoff K, Dörken B, Daniel PT. Induction of p21CIP/WAF-1 and G2 arrest by ionizing irradiation impedes caspase-3-mediated apoptosis in human carcinoma cells. Oncogene 2006; 25:972-80; PMID:16331277; http://dx.doi.org/ 10.1038/sj.onc.1209031 [DOI] [PubMed] [Google Scholar]
- 7.Javelaud D, Besancon F. Inactivation of p21WAF1 sensitizes cells to apoptosis via an increase of both p14ARF and p53 levels and an alteration of the Bax/Bcl-2 ratio. J Biol Chem 2002; 277:37949-54; PMID:12151395; http://dx.doi.org/ 10.1074/jbc.M204497200 [DOI] [PubMed] [Google Scholar]
- 8.Tian H, Wittmack EK, Jorgensen TJ. p21WAF1/CIP1 antisense therapy radiosensitizes human colon cancer by converting growth arrest to apoptosis. Cancer Res 2000; 60:679-84; PMID:10676653 [PubMed] [Google Scholar]
- 9.Sohn D, Essmann F, Schulze-Osthoff K, Jänicke RU. p21 blocks irradiation-induced apoptosis downstream of mitochondria by inhibition of cyclin-dependent kinase-mediated caspase-9 activation. Cancer Res 2006; 66:11254-62; PMID:17145870; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-1569 [DOI] [PubMed] [Google Scholar]
- 10.Ahmed W, Rahmani M, Dent P, Grant S. The cyclin-dependent kinase inhibitor p21(CIP1/WAF1) blocks paclitaxel-induced G2M arrest and attenuates mitochondrial injury and apoptosis in p53-null human leukemia cells. Cell Cycle 2004; 3:1305-11; PMID:15467449; http://dx.doi.org/ 10.4161/cc.3.10.1161 [DOI] [PubMed] [Google Scholar]
- 11.Wang Z, Van Tuyle G, Conrad D, Fisher PB, Dent P, Grant S. Dysregulation of the cyclin-dependent kinase inhibitor p21WAF1/CIP1/MDA6 increases the susceptibility of human leukemia cells (U937) to 1-β-D-arabinofuranosylcytosine-mediated mitochondrial dysfunction and apoptosis. Cancer Res 1999; 59:1259-67; PMID:10096557 [PubMed] [Google Scholar]
- 12.Wuerzberger-Davis SM, Chang PY, Berchtold C, Miyamoto S. Enhanced G2-M arrest by nuclear factor-{kappa}B-dependent p21waf1/cip1 induction. Mol Cancer Res 2005; 3:345-53; PMID:15972853; http://dx.doi.org/ 10.1158/1541-7786.MCR-05-0028 [DOI] [PubMed] [Google Scholar]
- 13.Rommer A, Steinmetz B, Herbst F, Hackl H, Heffeter P, Heilos D, Filipits M, Steinleitner K, Hemmati S, Herbacek I, et al. EVI1 inhibits apoptosis induced by antileukemic drugs via upregulation of CDKN1A/p21/WAF in human myeloid cells. PLoS One 2013; 8:e56308; PMID:23457546; http://dx.doi.org/ 10.1371/journal.pone.0056308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newbold A, Salmon JM, Martin BP, Stanley K, Johnstone RW. The role of p21 and p27 in HDACi-mediated tumor cell death and cell cycle arrest in the Emu-myc model of B-cell lymphoma. Oncogene 2013; 33(47):5415-23; PMID: 24292681; http://dx.doi:16413492 10.1038/onc.2013.482 [DOI] [PubMed] [Google Scholar]
- 15.Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al.. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006; 124:207-19; PMID:16413492; http://dx.doi.org/ 10.1016/j.cell.2005.10.043 [DOI] [PubMed] [Google Scholar]
- 16.Hill R, Madureira PA, Waisman DM, Lee PW. DNA-PKcs binding to p53 on the p21WAF1/CIP1 promoter blocks transcription resulting in cell death. Oncotarget 2011; 2:1094-108; PMID:22190353; http://dx.doi.org/ 10.18632/oncotarget.378 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.El-Deiry WS. The role of p53 in chemosensitivity and radiosensitivity. Oncogene 2003; 22:7486-95; PMID:14576853; http://dx.doi.org/ 10.1038/sj.onc.1206949 [DOI] [PubMed] [Google Scholar]
- 18.Wada M, Bartram CR, Nakamura H, Hachiya M, Chen DL, Borenstein J, Miller CW, Ludwig L, Hansen-Hagge TE, Ludwig WD, et al. Analysis of p53 mutations in a large series of lymphoid hematologic malignancies of childhood. Blood 1993; 82:3163-9; PMID:8219205 [PubMed] [Google Scholar]
- 19.Blau O, Avigad S, Stark B, Kodman Y, Luria D, Cohen IJ, Zaizov R. Exon 5 mutations in the p53 gene in relapsed childhood acute lymphoblastic leukemia. Leuk Res 1997; 21:721-9; PMID:9379679; http://dx.doi.org/ 10.1016/S0145-2126(97)80032-X [DOI] [PubMed] [Google Scholar]
- 20.Pettitt AR, Sherrington PD, Stewart G, Cawley JC, Taylor AM, Stankovic T. p53 dysfunction in B-cell chronic lymphocytic leukemia: inactivation of ATM as an alternative to TP53 mutation. Blood 2001; 98:814-22; PMID:11468183; http://dx.doi.org/ 10.1182/blood.V98.3.814 [DOI] [PubMed] [Google Scholar]
- 21.Marks DI, Kurz BW, Link MP, Ng E, Shuster JJ, Lauer SJ, Brodsky I, Haines DS. High incidence of potential p53 inactivation in poor outcome childhood acute lymphoblastic leukemia at diagnosis. Blood 1996; 87:1155-61; PMID:8562942 [PubMed] [Google Scholar]
- 22.Hogarth LA, Hall AG. Increased BAX expression is associated with an increased risk of relapse in childhood acute lymphocytic leukemia. Blood 1999; 93:2671-8; PMID:10194447 [PubMed] [Google Scholar]
- 23.Davies C, Hogarth LA, Dietrich PA, Bachmann PS, Mackenzie KL, Hall AG, Lock RB. p53-independent epigenetic repression of the p21(WAF1) gene in T-cell acute lymphoblastic leukemia. J Biol Chem 2011; 286:37639-50; PMID:21903579; http://dx.doi.org/ 10.1074/jbc.M111.272336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Groves MJ, Maccallum SF, Boylan MT, Haydock S, Cunningham J, Gelly K, Gowans D, Kerr R, Coates PJ, Tauro S. Heterogeneity of p53-pathway Protein Expression in Chemosensitive Chronic Lymphocytic Leukemia: A Pilot Study. J Cancer 2012; 3:354-61; PMID:22962562; http://dx.doi.org/ 10.7150/jca.4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zenz T, Habe S, Denzel T, Mohr J, Winkler D, Buhler A, Sarno A, Groner S, Mertens D, Busch R, et al.. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial. Blood 2009; 114:2589-97; PMID:19643983; http://dx.doi.org/ 10.1182/blood-2009-05-224071 [DOI] [PubMed] [Google Scholar]
- 26.Canman CE, Gilmer TM, Coutts SB, Kastan MB. Growth factor modulation of p53-mediated growth arrest vs. apoptosis. Genes Dev 1995; 9:600-11; PMID:7698649; http://dx.doi.org/ 10.1101/gad.9.5.600 [DOI] [PubMed] [Google Scholar]
- 27.Bastin-Coyette L, Cardoen S, Smal C, de Viron E, Arts A, Amsailale R, Van Den Neste E, Bontemps F. Nucleoside analogs induce proteasomal down-regulation of p21 in chronic lymphocytic leukemia cell lines. Biochem Pharmacol 2011; 81:586-93; PMID:21168391; http://dx.doi.org/ 10.1016/j.bcp.2010.12.009 [DOI] [PubMed] [Google Scholar]
- 28.Wang Z, Wang S, Fisher PB, Dent P, Grant S. Evidence of a functional role for the cyclin-dependent kinase inhibitor p21CIP1 in leukemic cell (U937) differentiation induced by low concentrations of 1-β-D-arabinofuranosylcytosine. Differentiation 2000; 66:1-13; PMID:10997587; http://dx.doi.org/ 10.1046/j.1432-0436.2000.066001001.x [DOI] [PubMed] [Google Scholar]
- 29.Lazzarini R, Moretti S, Orecchia S, Betta PG, Procopio A, Catalano A. Enhanced antitumor therapy by inhibition of p21WAF1 in human malignant mesothelioma. Clin Cancer Res 2008; 14:5099-107; PMID:18698027; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-0255 [DOI] [PubMed] [Google Scholar]
- 30.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 2009; 9:400-14; PMID:19440234; http://dx.doi.org/ 10.1038/nrc2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W, Kornblau SM, Kobayashi T, Gambel A, Claxton D, Deisseroth AB. High levels of constitutive WAF1/Cip1 protein are associated with chemoresistance in acute myelogenous leukemia. Clin Cancer Res 1995; 1:1051-7; PMID:9816079 [PubMed] [Google Scholar]
- 32.Roman-Gomez J, Castillejo JA, Jimenez A, Gonzalez MG, Moreno F, Rodriguez Mdel C, Barrios M, Maldonado J, Torres A. 5' CpG island hypermethylation is associated with transcriptional silencing of the p21(CIP1/WAF1/SDI1) gene and confers poor prognosis in acute lymphoblastic leukemia. Blood 2002; 99:2291-6; PMID:11895758; http://dx.doi.org/ 10.1182/blood.V99.7.2291 [DOI] [PubMed] [Google Scholar]
- 33.Jänicke RU, Sohn D, Essmann F, Schulze-Osthoff K. The multiple battles fought by anti-apoptotic p21. Cell Cycle 2007; 6:407-13; http://dx.doi.org/ 10.4161/cc.6.4.3855 [DOI] [PubMed] [Google Scholar]
- 34.Harvey KJ, Lukovic D, Ucker DS. Caspase-dependent Cdk activity is a requisite effector of apoptotic death events. J Cell Biol 2000; 148:59-72; PMID:10629218; http://dx.doi.org/ 10.1083/jcb.148.1.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin SJ, Finucane DM, Amarante-Mendes GP, O'Brien GA, Green DR. Phosphatidylserine externalization during CD95-induced apoptosis of cells and cytoplasts requires ICE/CED-3 protease activity. J Biol Chem 1996; 271:28753-6; PMID:8910516; http://dx.doi.org/ 10.1074/jbc.271.46.28753 [DOI] [PubMed] [Google Scholar]
- 36.Balasubramanian K, Mirnikjoo B, Schroit AJ. Regulated externalization of phosphatidylserine at the cell surface: implications for apoptosis. J Biol Chem 2007; 282:18357-64; PMID:17470427; http://dx.doi.org/ 10.1074/jbc.M700202200 [DOI] [PubMed] [Google Scholar]
- 37.Verhoven B, Krahling S, Schlegel RA, Williamson P. Regulation of phosphatidylserine exposure and phagocytosis of apoptotic T lymphocytes. Cell Death Differ 1999; 6:262-70; PMID:10200577; http://dx.doi.org/ 10.1038/sj.cdd.4400491 [DOI] [PubMed] [Google Scholar]
- 38.Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, O'Sullivan B, He Z, Peng Y, Tan AC, et al.. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med 2011; 17:860-6; PMID:21725296; http://dx.doi.org/ 10.1038/nm.2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gregory CD, Pound JD. Cell death in the neighbourhood: direct microenvironmental effects of apoptosis in normal and neoplastic tissues. J Pathol 2011; 223:177-94; PMID:21125674; http://dx.doi.org/ 10.1002/path.2792 [DOI] [PubMed] [Google Scholar]
- 40.Weiss RH. p21Waf1/Cip1 as a therapeutic target in breast and other cancers. Cancer Cell 2003; 4:425-9; PMID:14706334; http://dx.doi.org/ 10.1016/S1535-6108(03)00308-8 [DOI] [PubMed] [Google Scholar]
- 41.Chang CC, Heller JD, Kuo J, Huang RC. Tetra-O-methyl nordihydroguaiaretic acid induces growth arrest and cellular apoptosis by inhibiting Cdc2 and survivin expression. Proc Natl Acad Sci U S A 2004; 101:13239-44; PMID:15329416; http://dx.doi.org/ 10.1073/pnas.0405407101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forster K, Obermeier A, Mitina O, Simon N, Warmuth M, Krause G, Hallek M. Role of p21(WAF1/CIP1) as an attenuator of both proliferative and drug-induced apoptotic signals in BCR-ABL-transformed hematopoietic cells. Ann Hematol 2008; 87:183-93; PMID:17960378; http://dx.doi.org/ 10.1007/s00277-007-0400-9 [DOI] [PubMed] [Google Scholar]
- 43.Fulda S. Inhibitor of apoptosis proteins in pediatric leukemia: molecular pathways and novel approaches to therapy. Front Oncol 2014; 4:3; PMID:24478984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suzuki A, Kawano H, Hayashida M, Hayasaki Y, Tsutomi Y, Akahane K. Procaspase 3/p21 complex formation to resist fas-mediated cell death is initiated as a result of the phosphorylation of p21 by protein kinase A. Cell Death Differ 2000; 7:721-8; PMID:10918446; http://dx.doi.org/ 10.1038/sj.cdd.4400706 [DOI] [PubMed] [Google Scholar]
- 45.Choi J, Hwang YK, Sung KW, Lee SH, Yoo KH, Jung HL, Koo HH, Kim HJ, Kang HJ, Shin HY, et al. Expression of Livin, an antiapoptotic protein, is an independent favorable prognostic factor in childhood acute lymphoblastic leukemia. Blood 2007; 109:471-7; PMID:16990595; http://dx.doi.org/ 10.1182/blood-2006-07-032557 [DOI] [PubMed] [Google Scholar]
- 46.Mahyar-Roemer M, Roemer K. p21 Waf1/Cip1 can protect human colon carcinoma cells against p53-dependent and p53-independent apoptosis induced by natural chemopreventive and therapeutic agents. Oncogene 2001; 20:3387-98; PMID:11423989; http://dx.doi.org/ 10.1038/sj.onc.1204440 [DOI] [PubMed] [Google Scholar]
- 47.Waldman T, Lengauer C, Kinzler KW, Vogelstein B. Uncoupling of S phase and mitosis induced by anticancer agents in cells lacking p21. Nature 1996; 381:713-6; PMID:8649519; http://dx.doi.org/ 10.1038/381713a0 [DOI] [PubMed] [Google Scholar]
- 48.Freemerman AJ, Vrana JA, Tombes RM, Jiang H, Chellappan SP, Fisher PB, Grant S. Effects of antisense p21 (WAF1/CIP1/MDA6) expression on the induction of differentiation and drug-mediated apoptosis in human myeloid leukemia cells (HL-60). Leukemia 1997; 11:504-13; PMID:9096690; http://dx.doi.org/ 10.1038/sj.leu.2400625 [DOI] [PubMed] [Google Scholar]
- 49.Ruan S, Okcu MF, Pong RC, Andreeff M, Levin V, Hsieh JT, Zhang W. Attenuation of WAF1/Cip1 expression by an antisense adenovirus expression vector sensitizes glioblastoma cells to apoptosis induced by chemotherapeutic agents 1,3-bis(2-chloroethyl)-1-nitrosourea and cisplatin. Clin Cancer Res 1999; 5:197-202; PMID:9918219 [PubMed] [Google Scholar]
- 50.Baptiste-Okoh N, Barsotti AM, Prives C. Caspase 2 is both required for p53-mediated apoptosis and downregulated by p53 in a p21-dependent manner. Cell Cycle 2008; 7:1133-8; PMID:18418048; http://dx.doi.org/ 10.4161/cc.7.9.5805 [DOI] [PubMed] [Google Scholar]
- 51.Ocker M, Schneider-Stock R. Histone deacetylase inhibitors: signalling towards p21CIP1/WAF1. Int J Biochem Cell Biol 2007; 39:1367-74; PMID:17412634; http://dx.doi.org/ 10.1016/j.biocel.2007.03.001 [DOI] [PubMed] [Google Scholar]
- 52.Smolewski P. Terameprocol, a novel site-specific transcription inhibitor with anticancer activity. IDrugs 2008; 11:204-14; PMID:18311658 [PubMed] [Google Scholar]
- 53.Suryani S, Carol H, Chonghaile TN, Frismantas V, Sarmah C, High L, Bornhauser B, Cowley MJ, Szymanska B, Evans K, et al. Cell and molecular determinants of in vivo efficacy of the BH3 mimetic ABT-263 against pediatric acute lymphoblastic leukemia xenografts. Clin Cancer Res 2014; 20:4520-31; PMID:25013123; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-0259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keshelava N, Houghton PJ, Morton CL, Lock RB, Carol H, Keir ST, Maris JM, Reynolds CP, Gorlick R, Kolb EA, et al. Initial testing (stage 1) of vorinostat (SAHA) by the pediatric preclinical testing program. Pediatr Blood Cancer 2009; 53:505-8; PMID:19418547; http://dx.doi.org/ 10.1002/pbc.21988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carol H, Gorlick R, Kolb EA, Morton CL, Manesh DM, Keir ST, Reynolds CP, Kang MH, Maris JM, Wozniak A, et al. Initial testing (stage 1) of the histone deacetylase inhibitor, quisinostat (JNJ-26481585), by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 2014; 61:245-52; PMID:24038993; http://dx.doi.org/ 10.1002/pbc.24724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bachmann PS, Gorman R, Papa RA, Bardell JE, Ford J, Kees UR, Marshall GM, Lock RB. Divergent mechanisms of glucocorticoid resistance in experimental models of pediatric acute lymphoblastic leukemia. Cancer Res 2007; 67:4482-90; PMID:17483364; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-4244 [DOI] [PubMed] [Google Scholar]
- 57.Lock RB, Liem N, Farnsworth ML, Milross CG, Xue C, Tajbakhsh M, Haber M, Norris MD, Marshall GM, Rice AM. The nonobese diabetic severe combined immunodeficient (NOD/SCID) mouse model of childhood acute lymphoblastic leukemia reveals intrinsic differences in biologic characteristics at diagnosis and relapse. Blood 2002; 99:4100-8; PMID:12010813; http://dx.doi.org/ 10.1182/blood.V99.11.4100 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.