

Protein modification by ubiquitin plays a critical role in multiple cellular processes and is counteracted by the activity of ∼90 deubiquitinating enzymes (DUBs) encoded by the human genome. Beside the catalytic core domain most DUBs contain various protein-protein-interaction domains, which determine substrate specificity and recruitment to multi-molecular complexes. However, the physiological relevance of most mammalian DUBs in particular cell types remains elusive.1
Several DUBs like A20, Cylindromatosis (CYLD), USP9X, and USP15 were shown to be essential for proper T cell function.2 We now identified ubiquitin-specific protease 8 (USP8) as a novel component of the T cell receptor (TCR) signalosome that interacts with the adapter molecule Gads and the regulatory protein 14–3–3β.3 Gads is highly abundant in T cells and modulates signal diversification.4 As typical for 14–3–3 proteins, the interaction with 14–3–3β was dependent on a serine phosphorylation motif within the 14–3–3 binding motif (14–3–3BM) of USP8 (Fig. 1). In several cell types including hepatocytes, fibroblasts and adenoma cells USP8 was shown to control endosomal sorting of transmembrane proteins.5,6 By mediating deubiquitination of cargo substrates and components of the endosomal sorting complex (ESCRT) USP8 counteracts lysosomal degradation and thus stabilizes receptor tyrosine kinases such as the EGFR.5-7 Remarkably, in contrast to MEFs and hepatocytes, lack of USP8 had no influence on the stability of components of the endosomal sorting machinery in T cells.3,5 The adaptor molecule STAM2 for example, which interacts with USP8 via SH3 binding (Fig. 1) was not destabilized in T cells. The observation that USP8 has distinct functions in different cells depending on varying interacting proteins or substrates supports a unifying concept for a wide range of other DUBs encompassing multiple protein interaction modules. With respect to USP8, T cell-specific function might arise from the high affinity of the N-terminal SH3 binding motif (SH3BM) of USP8 to Gads, which is predominantly expressed in T cells.
Figure 1.
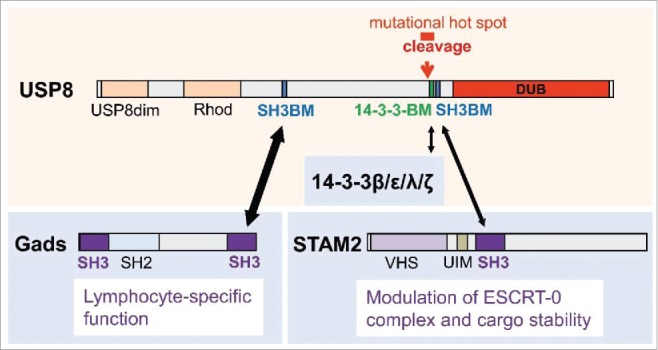
USP8 structure-function relationships. Functional domains, interacting proteins and modules, the region affected by CD-causing mutations and the site of proteolytic processing are depicted. USP8dim: USP8 dimerization domain; Rhod: Rhodanese-like domain; VHS: Domain present in VPS-27, Hrs and STAM; UIM: Ubiquitin-interaction-motif.
T cell-specific deletion in mice (Usp8fl/flCd4-Cre) revealed that USP8 is essential for thymocyte transition to the CD4+ and CD8+ single positive (SP) stages and efficient positive selection.3 USP8 was not required for CD3/CD28-induced canonical signal transduction or negative selection, but critical for Foxo1-mediated IL7Rα and C-C chemokine receptor type 7 (CCR7) expression. In vivo reconstitution showed that enzymatic activity and SH3-binding are essential for USP8 function in T cells. Of note, Usp8fl/flCd4-Cre mice developed lethal colitis that was associated with disturbed T cell homeostasis, predominance of CD8+ γδ-T cells in the intestine and compromised regulatory T cell function.3
Recently, USP8 has received much attention as somatic mutations of the gene encoding USP8 that lead to the accumulation of the EGFR on the surface of corticotroph cell pituitary adenomas represent the primary cause of Cushing's disease (CD).6 Because these cells produce excessive adrenocorticotropic hormone (ACTH), adrenal glands are stimulated to produce elevated levels of cortisol. The resulting symptoms are referred to as “Cushing's Syndrome” and include: central obesity, myopathy, skin and mood changes, impaired reproductive function, hypertension, cardiovascular disease and metabolic syndrome. All Usp8 mutations found in CD are located in or around the sequence encoding the 14–3–3 binding motif (Fig. 1). Intriguingly, disruption of 14–3–3 binding by most of these mutations is associated with enhanced proteolytic processing of USP8 yielding a truncated, highly active protein, which deubiquitinates EGFR and ultimately leads to enhanced recycling and expression of activated EGFR at the plasma membrane of corticotrophs.6 Sustained EGFR signaling is in turn responsible for the forced expression of the ACTH precursor proopiomelanocorticotropin (POMC).6
Similar to the USP8 variants found in CD patients, we identified rapid processing of USP8 in murine T cells upon activation of the TCR.3 This processing was caspase-dependent and resulted in the generation of mouse USP8 fragments that were homologous to the fragments of the human USP8 variants found in corticotropinomas. These findings confirmed that murine and human USP8 are both proteolytically cleaved. Our results also clearly show that USP8 processing can occur even when the 14–3–3 binding site is not mutated demonstrating that USP8 cleavage is not only a pathological process, but rather an endogenous regulatory mechanism. Moreover, we showed for the first time that USP8 is processed under physiological conditions by a receptor-driven stimulus in a caspase dependent manner. As binding of USP8 to 14–3–3 strictly depends on phosphorylation, USP8 cleavage and activity may be regulated by kinases and phosphatases. Furthermore processing might be affected by other posttranslational modifications or interacting proteins like Gads.
SP thymocyte maturation requires low-affinity TCR interactions with self-peptide-MHC to trigger a signaling cascade that allows for positive selection.4 Our studies did not reveal a defect in TCR-induced canonical signaling cascades. However, whether USP8 is involved in the discrimination of low- and high-affinity signals in specific thymocyte populations remains to be tested. Likewise, USP8 may control TCR-stabilization/upregulation during development. It will be interesting to see whether similar to CD somatic USP8 mutations might underlie human T cell defects or malignancies, or autoimmune disorders.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Komander D, et al. Nat Rev Mol Cell Biol 2009; 10:550-63; PMID:19626045; http://dx.doi.org/ 10.1038/nrm2731 [DOI] [PubMed] [Google Scholar]
- 2.Sun SC. Nat Rev Immunol 2008; 8:501-11; PMID:18535581; http://dx.doi.org/ 10.1038/nri2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dufner A, et al. Nat Immunol 2015; 16(9):950-60; PMID:26214742 [DOI] [PubMed] [Google Scholar]
- 4.Brownlie RJ, Zamoyska R. Nat Rev Immunol 2013; 13:257-69; PMID:23524462; http://dx.doi.org/ 10.1038/nri3403 [DOI] [PubMed] [Google Scholar]
- 5.Niendorf S, et al. Mol Cell Biol 2007; 27:5029-39; PMID:17452457; http://dx.doi.org/ 10.1128/MCB.01566-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Theodoropoulou M, et al. Eur J Endocrinol 2015; 173(4):M73-83; PMID:26012588 http://dx.doi.org/ 10.1530/EJE-15-0320 [DOI] [PubMed] [Google Scholar]
- 7.Wright MH, et al. Cell Biochem Biophys 2011; 60:39-46; PMID:21448666; http://dx.doi.org/ 10.1007/s12013-011-9181-9 [DOI] [PubMed] [Google Scholar]