

Abstract
The cyclin-dependent kinase (CDK) inhibitor p27Kip1 has been shown to regulate cellular proliferation via inhibition of CDK activities. It is now recognized that p27Kip1 can regulate cellular processes through non-canonical, CDK-independent mechanisms. We have developed an inducible p27Kip1 model in cultured cells to explore CDK-independent p27Kip1 regulation of biological processes. We present evidence that p27Kip1 can function in a CDK-independent manner to inhibit entry and/or progression of S phase. Even though this p27Kip1 mechanism is non-canonical it does requires the intact cyclin-binding motif in p27Kip1. We suggest a mechanism similar to that proposed in post-mitotic neural cells whereby p27Kip1 functions to coordinate growth arrest and apoptosis. Our hypothesis supports the concept that p27Kip1 is a gatekeeper for the entry and progression of S phase through interaction with specific protein(s) or via binding to specific DNA sequences in a CDK-independent manner.
Keywords: cell cycle, p27Kip1, non-canonical, cyclin-dependent kinases, cyclin F
Introduction
The cyclin-dependent kinase (CDK) activities have been shown to play a major role in the regulation of the cell cycle and p27Kip1 (hereafter p27) can regulate CDK activities.1-3 The p27 protein was originally recognized as an inhibitor of CDK activities for complexes containing CDK2 and shown to inhibit cyclin E and cyclin A activities which regulate G1 and S phase traverse.4-6 In addition to CDK inhibition, p27 putatively has other multifarious interactions with cyclin D/cdk4 complexes.7 Since cellular levels of p27 are elevated in response to high cell density, serum deprivation, and TGFβ, it was hypothesized p27 brought cells into quiescence and held them in G0 through the inhibition of CDK activities.8 Numerous reports have characterized the regulation of p27 including the control of its transcription,9,10 translation,11,12 post-translational modifications.7,13,14 cellular localization15-19 and stability.20-23 The regulation of its stability plays a major role in adjusting cellular level of p27; the ubiquitin-proteasome system has been shown to be a major regulator of p27 cellular abundance.21,22 Importantly, the level of p27 is low in many aggressive tumors and it is thought that protein degradation primarily accounts for this low abundance in most cancers.23 However, multiple studies have now documented a gain of cytoplasmic p27 localization along with loss of the nuclear localization of p27 in some cancers.19,24,25 For example, the phosphorylation of p27 on serine 10 marks it for transport to the cytoplasm and the phosphorylation of threonine 157 by activated AKT in breast cancers retains human p27 in the cytoplasm and thus reducing its ability to inhibit the nuclear CDK activities responsible for cell cycle traverse and cellular division.7,13,25 Multiple mechanisms for cytoplasmic location of p27 have been implicated in various aggressive cancers. A decrease in nuclear p27 sanctions the CDK activities required to ensure the initiation of the cell cycle, DNA synthesis and the completion of the S phase.
More recently, p27 has been implicated in cancers through the regulation of cellular processes by CDK-independent mechanisms. For example, p27 was shown to stimulate cellular migration through direct binding to RhoA.26 The C-terminal of p27 protein interacts with RhoA and blocks the GEF-mediated activation of RhoA; however, the effects of this interaction remain controversial.27 Cytosolic compartmentalized p27 also interacts with Rac, stathmin, Grb2 and 14–3–3 through its C-terminus.28 The interactions of p27 with RhoA, Rac and stathmin individually affect cell movement and migration. The physiological significance of the interactions of p27 with Grb2 and 14–3–3 are not well understood, but AKT phosphorylation of p27 allows its binding to 14–3–3 which helps limit the nuclear compartmentalization of p27.25 Another non-canonical process of cellular regulation by p27 was suggested by Besson et al, who demonstrated that p27, independently of its CDK inhibitory activity, functioned as a dominant oncogene in vivo, promoting stem-cell expansion and spontaneous multi-organ tumorigenesis.29 In addition, other non-canonical cell control mechanisms have been described for nuclear localized p27. Nallamshetty et al.30 reported that p27 binds MCM7 to inhibit S phase entry and DNA synthesis independent of CDK inhibition. p27 has been hypothesized to directly regulate the gene expression of Twist1 and Brachyury via non-CDK mechanisms and thus affect self-renewal and pluripotency of human stem cells, suggesting a role for p27 on epithelial to mesenchymal transition (EMT).31 Moreover, p27 associates with the SRR2 enhancer of Sox2 gene in association with p130-E2F4-SIN3A.32 p27 has also been shown to promote neuronal differentiation by stabilizing Neurogenin2 protein through interactions with the N-terminal of p27.33 Taken together these and other published reports point out that p27 exerts regulatory growth control via CDK-independent mechanisms. In our investigations using a p27 construct that cannot bind or inhibit CDKs, but harbors an intact cyclin-binding region, we demonstrate a unique cell cycle control event. We now show that p27 can function as the S phase gatekeeper through a non-canonical process.
Results
Growing U2OS cells expressing p27K incorporate Brdu
We have shown that a p27 mutant (p27K) constructed such that it cannot bind and inhibit CDKs, when overexpressed under the control of the TET-ON promoter, brought about a decrease in the growth rate of U2OS cells in culture.34 The induced expression of p27K was shown to cause nuclear alterations commonly seen in mitotic catastrophe. We demonstrated this phenotype was dependent upon the interaction of p27K with cyclin F such that the sequestration of cyclin F altered growth rates, in part, through mitotic abnormalities. The mitotic alterations stemmed from elevated CP110 due to its increased stability caused by a lack of cyclin F to target CP110 for destruction.34,35 To continue and extend our evaluation of the effect of the overexpression of p27K on U2OS cells progressing through their cell cycle, we compared Brdu incorporation into DNA of growing parental U2OS cells to that of U2OS cells expressing p27K. Cultures of U2OS cells with p27K under the TET-responsive promoter were grown in media with and without the addition of doxycycline (Dox). As an additional control we also employed non-induced U2OS cell with a wild type p27 under a TET-responsive promoter (p27WT). After 24 and 48 h of Dox stimulation, Brdu was added to each culture condition for 1 and 24 h to allow the evaluation of DNA synthesis through Brdu incorporation during cell cycle progression. Culturing for 24 and 48 h in the presence of Dox allowed cells adequate time to proliferate and adjust to the p27K before the addition of Brdu. After incubation with Brdu, the cells were collected and flow cytometric analysis was performed. In agreement with and extending previous findings,34 Figure 1A shows the Brdu incorporation after 60 min of incubation in U2OS cells with overexpressed p27K (middle panel) as compared to parental cells (left panel) and non-induced p27K cells (right panel). It was noted that more cells in the induced p27K remained in the unlabeled S phase (see R2 gate in Fig 1A). When cells were labeled for 60 min after the 48 h incubation in Dox, the cellular Brdu distribution was similar to that seen at 60 min after 24 h in Dox (data not shown); 10% fewer cells expressing p27K incorporated Brdu than in control cells, which is similar to the difference in labeled cells after 24 h of incorporation of Brdu (Fig. 1C). However, as can be seen in Figure 1B, 10% of the cells displayed Annexin V on their surface in conditions of p27K expression. The incorporation of Brdu was also distributed through the cell cycle equally during 24 h incubation with Brdu (Fig. 1C). These data point out the p27K expressing cells incorporate Brdu in a similar fashion to the parental cells and thus indicate comparable growth in the parental cells and those expressing p27K. Although, once again, more p27K expressing cells remained in the unlabeled S phase fraction (see R2 gate). After 72 h in culture with Dox, fewer cells were seen in the confluent cultures of U2OS cells expressing p27K (Fig. 2A), however, as can be seen in Figure 2B the incorporation of Brdu was similar in parental cells, non-induced U2OS-p2WT cells and those expressing p27K. We had previously observed a slower growth rate for cultures of U2OS cells expressing p27K, even though cell cycle traverse apparently occurred normally, which accounts for the fewer total cells in culture. It was observed that the percent of cells which did not incorporate Brdu were equal in all cultures excluding possible apoptotic cells (Figs. 1, 2). These results and our published data34 concerning the effects of p27K expression and its sequestration of cyclin F on cell proliferation made us keenly interested in comparing the growth properties of p27K-expressing U2OS cells to that reported for embryonic fibroblastic cells prepared from cyclin F−/− mice.36
Figure 1.

Parental U2OS, U20S-p27WT, and U2OS-p27K cellswere grown in culture. After 24 h, DOX (1µg/ml) was added to one set of cultures of U2OS-p27K cells. After another 48 h, BrdU (30 µM) was added to all cell cultures including induced and non-induced p27K, parental U2OS cells and U2OS-p27WT cells. Cultures were incubated with BrdU for 60 min (Panel A) and 24 h (Panel C). Cells in each condition and at the indicated times were removed from plates and processed for cell cycle analysis and BrdU incorporation. In Panel B annexin V staining (Materials and Methods) for cells isolated at each time and condition are shown.
Figure 2.

Parental, U2OS-p27WT, and U2OS-p27K cells were grown and treated as described in Figure 1 with one set of U2OS –p27K cells receiving DOX (1μg/ml) and then cells were incubated for 72 h. BrdU (30 μM) was added at this time for 1 h. Panel A shows picture of the cell populations at 72 h and Panel B shows the BrdU incorporation into the cells in all populations.
p27K inhibits late G1/S phase traverse
Since cyclin F−/− MEFs derived from a cyclin F knockout mouse grew in culture, although at a reduced rate, and since the cyclin F−/− fibroblasts did not initiate growth properly when serum was added to serum starved cyclin F−/− MEFs,36 it could be hypothesized that the re-stimulation of growth from G0 required cyclin F for G1 and S phase traverse. Since we had previously shown the overexpression of p27K sequestered cyclin F which in turn could limit availability of cyclin F, we sought to compare U2OS cells expressing p27K to cyclin F−/− MEF cells. Because it is not possible to use serum starvation of U2OS cells to investigate their reentry into the G1 phase as was done with cyclin F−/− fibroblasts, we used release from a nocodazole-induced mitotic block to determine the effect of p27K expression and possible cyclin F sequestration on G1 and S phase traverse in U2OS cells. Figure 3A demonstrates p27K expressing cells released from nocodazole inhibition move from 4N to 2N and this progression was similar to uninduced p27K and parental cells. However, the induction of p27K in U2OS cells before release from the mitotic inhibition brought about the inhibition of the cells in the progression of the cell cycle after the completion of cell division and during the traverse of late G1 phase or early S phase (Fig. 2B). Figure 3C shows the induction of p27K in response to Dox. Parental cells had moved through S phase and entered G2/M within this time frame as did non-induced U2OS-p27WT used as a control and looked like parental U2OS cells. Of note, that induction of p27WT does not inhibit the transition from 4N to 2N (Fig. 3 panel A). We noted that cells overexpressing p27CK (a p27 construct that cannot bind cyclins or CDKs34) traversed the cell cycle as did parental cells, and on the other extreme, cells expressing p27WT arrested after mitosis in G1 (data not shown) indicating p27WT expression which inhibited CDK activities inhibited G1 cell cycle traverse. In order to examine the extent of G1 and S phase traverse, the parental and p27K cells were cultured under identical conditions as described for Figure 3A and B and then cultured in the presence of Brdu following release from nocodazole inhibition. Figure 3D shows that cells in either condition were able to leave mitosis and enter into G1. Most of the parental cells traverse S phase (Fig. 3). On the other hand, a portion of the cells expressing p27K entered S phase, but displayed a lack of complete S phase traverse which was reminiscent of the lack of S phase traverse of cyclin F−/− fibroblasts that had been serum starved and then re-stimulated with serum addition.36 Serum starved cyclin F−/− fibroblasts induced cyclin D, cyclin E and a limited amount of cyclin A after serum addition, although they displayed a paucity of S and G2 phase traverse.36 Likewise, when U2OS cells were blocked with nocodazole and then released from mitotic inhibition under conditions for expression of p27K, several G1 phase genes, including cyclin D, cyclin E, and limited cyclin A were induced (Fig. 4) and the cells did not traverse S phase like cyclin F−/− MEFs. However, in parental cells and non-induced U2OS-p27WT cells traverse of G1 and entry into S phase was normal. The inhibition of cell cycle traverse by the expression of p27K was not cell density dependent. Cultures of U2OS cells at various densities were placed in nocodazole-induced mitotic inhibition. The expression of p27K inhibited cell cycle traverse when cultures were released from the nocodazole inhibition regardless of culture density (Fig. 5). The U2OS cells expressing p27K were able to complete cell division after release from the mitotic inhibition, but were not able to progress through S phase (Fig. 5).
Figure 3.

U2OS cells, U2OS-p27WT cells and U2OS-p27K cells were placed in nocodazole inhibition as described in Material and Methods. Some cultures of U2OS-p27K and U2OS-p27WT cells had Dox (1µg/ml) added for 6 h. Cultures of parental, non-induced U2OS-p27WT, non-induced U2OS-p27K, and induced U2OS-p27K cells were released from the nocodazole block. Panel A shows propidium iodide straining of these cells, as described in Material and Methods, before release and 15 h after release from the mitotic inhibition. Data show 2N and 4N staining. Panel B shows the cell cycle distribution with propidium iodide staining of parental cells, U2OS-p27WT, and U2OS-p27K induced 6 h before mitotic release. Panel C illustrates the induction of p27K by 1 µg/ml of Dox. Panel D shows the analysis of BrdU incorporation in cells treated as in Panel B when released into fresh medium containing 30 μM BrdU.
Figure 4.
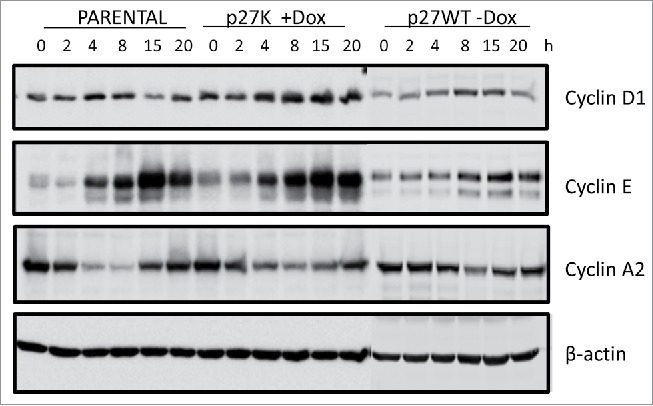
Parental, U2OS-p27WT and U2OS-p27K cells where placed in a nocodazole-induced mitotic block as described in Figure 3 and Material and Methods. Dox (1µg/ml) was added to cultures of U2OS-p27K cells 6 h before mitotic release. Cells were washed and placed in fresh medium with Dox. At indicated times cells were collected and extracts prepared. Extracts were analyzed by western blot analysis for cyclin D1, Cyclin E and cyclin A. β-Actin was used as a loading control.
Figure 5.

Parental cells were plated at 1million cells/dish (upper panel) and U2OS-p27K cells were plated at various densities as indicated (lower panel). All cells were put into nocodazole-induced mitotic inhibition as described in Figure 4 and Materials and Methods. Cells were released from inhibition and placed in fresh medium. Cell cycle analysis was performed on cells in mitotic inhibition and 14 h post mitotic release. Pictures show cell density.
Cyclin F decreases after release from nocodazole inhibition and p27K sequesters cyclin F
Parental cells and uninduced U2OS-p27WT cells mitotically blocked according to the procedure described for experiments shown in Figure 3 have elevated level of cyclin F (Fig. 6A). Upon release from the nocodazole inhibition, the level of cyclin F decreases dramatically and this decrease can be inhibited by the proteasome inhibitor MG132 (Fig. 6B). We noticed that majority of cyclin F was detected as a slower-migrating form in the presence of MG132. From the blot of cyclin F in cells incubated in the presence of cycloheximide, the half-life of cyclin F was determined to be approximately 1 h. Our data shows that cyclin F diminished to a non-detectable level during G1 phase and began a return to peak levels in G2 phase (8–20 h in panel A, Fig. 6). We sought to determine the increase in cyclin F during S phase. Fig. 6 (panel C) shows that parental cells released from a double thymidine block begin an increase in cyclin F immediately in S phase as has been shown for HeLa cells.35 Taken together, our data indicate a decrease in cyclin F following the release from mitotic inhibition and then followed by an increase in cyclin F during S phase; however, we found cells held in the nocodazole-induced mitotic block for extended time (>16 h) also showed a decrease in cyclin F (Fig. 6D). Since cyclin F increased after release from the double thymidine block we could not rule out a requirement for cyclin F during entry and/or traverse of S phase. Finally, Figure 6E presents data showing that p27WT and p27K binds and coimmunoprecipitates cyclin F while the p27C and p27CK mutants with altered cyclin-binding regions did not. The expression of p27K, although allowing CDK activities, binds and sequesters cyclin F. Our data on cell cycle traverse indicated that the U2OS cells overexpressing p27K mimicked the cyclin F knockout cells in failure to traverse the cell cycle while being able to induce the expression of the cell cycle G1 phase-specific genes that are normally expressed during G1 phase traverse. From these data it was thought that sequestration of cyclin F by p27K rendered U2OS cells unable to complete cell cycle traverse in a manner similar to MEFs derived from cyclin F knockout mice. Thus, we hypothesized that p27K expression was regulating the cell cycle through the non-canonical process of binding cyclin F and implied cyclin F function is absolutely required in late G1 or S phase.
Figure 6.

Panel A shows the western blot analysis for cyclin F in parental U2OS cells, U2OS-p27WT cells and in U2OS-p27K cells with induced p27K that were released from a nocodazole-induced mitotic block. The p27K was induced 6 h before release from mitotic inhibition. Cells were collected at the indicated times. Panel B shows cyclin F at indicated times. CTRL is parental cells released from nocodazole inhibition. CHX are cells release in presence of 10 µg/ml cycloheximide added 1 h prior to release. MG132 show cyclin F when cells were released in the presence of 10µM MG132 added 2 h prior to release. Panel C shows cyclin F at the indicated times after parental cells were released from double thymidine block using 2 mM thymidine. Panel D shows cyclin F at the indicated times after parental cells were released from thymidine block (as in Panel C) into the fresh medium with or without nocodazole inhibition. Panel E shows the co-immunoprecipitation of cyclin F with various p27 constructs: wild-type (p27WT), altered cyclin-binding site (p27C), altered CDK-binding site (p27K) and altered cyclin- and CDK- binding sites (p27CK). U2OS-derived p27-inducible cells were grown in medium containing Dox (1µg/ml) for 24 h. Cells were harvested, extracts prepared and HA-tagged p27 proteins were immunoprecipitated and separated by SDS-PAGE gel. Immunoblots were analyzed for cyclin F.
Cyclin F is not required for G1 and S phase traverse
In order to test this hypothesis, siRNA was used to knockdown cyclin F in U2OS cells to determine the progression through G1 and into S phase in the absence of cyclin F after release from mitotic inhibition. U2OS cells were placed in a nocodazole-induced mitotic inhibition. At 12 h before release, siRNA targeting cyclin F was introduced to the nocodazole-inhibited cells. Figure 7A shows the knockdown of cyclin F was essentially complete. When cells with silenced cyclin F were released from the mitotic inhibition, they underwent traverse of the cell cycle as well as the untransfected U2OS cells and those cells treated in an identical manner with non-targeting siRNA (Fig. 7B). We repeated the knockdown of cyclin F at different times, including, before nocodazole inhibition (data not shown) and results indicated that cyclin F was not required for progression through G1 and S phase (Fig. 7C). Thus, cyclin F was not necessary for U2OS cells to leave the mitotic block and traverse through S phase, indicating our hypothesis that cyclin F was required for G1 and/or S phase was not substantiated. In order to have a more direct comparison to cells that had been in low serum, we repeated the siRNA knockdown of cyclin F in nocodazole-inhibited U2OS cells which had been incubated and released in the presence of limited serum (0.1%). The limited serum did not produce a requirement for cyclin F (data not shown). It is not known why U2OS cells without cyclin F traverse the cell cycle whereas the growth arrested cyclin F—/— fibroblasts after serum stimulation did not.
Figure 7.

U2OS cells were placed in a nocodazole-induced mitotic arrest as described in Materials and Methods. Some cultures received cyclin F siRNA (Cyclin F-siRNA), others received a non-targeting siRNA (NT-siRNA) and cells not treated with siRNA (untransfected) were also used (On-TARGETplus SMARTpool siRNAs from Dharmacon were used and knockdown was performed as per manufacturer's instruction using Dharmafect-1). After the cyclin F knockdown with siRNAs,cells were placed in a nocodazole-induced mitotic arrest as described in Materials and Methods. Cells were then rinsed and released into fresh medium. Panel A shows cyclin F at the indicated times following release from the nocodazole arrest. Panel B shows the cell cycle distribution at 0,16, 20, and 24 h after release from the mitotic arrest. Panel C shows the incorporation of BrdU at various times after the release from mitotic arrest for untransfected, cyclin F-siRNA and NT-siRNA treated cells.
p27K expression in mid to late G1 inhibits cellular traverse of G1/S phase
Since the absence of cyclin F did not inhibit the traverse of G1/S phase, we asked whether expression of p27K was required before or after the release from the mitotic block in order to inhibit S phase traverse. p27K cells were inhibited with nocodazole to establish a mitotic block. After release from the mitotic block the expression of p27K was induced at various times by the addition of Dox as indicated in Figure 8. Induction of p27K began rapidly after addition of Dox (Fig. 3C) and was substantially elevated within 60 min. Figure 8 presents data illustrating that p27K must be present before 3–4 h after release from the mitotic inhibition in order to completely inhibit all cells from progressing through S phase. We noted that the expression of p27K before release from the mitotic inhibition inhibited 60% of the cells from labeling with Brdu (Fig. 3). Even though some cells labeled with Brdu, our data point out, as observed earlier, that only few of the cells completed S phase. Thus, the execution point for the step inhibited by p27K is between 3–4 h post-release from the mitotic inhibition.
Figure 8.
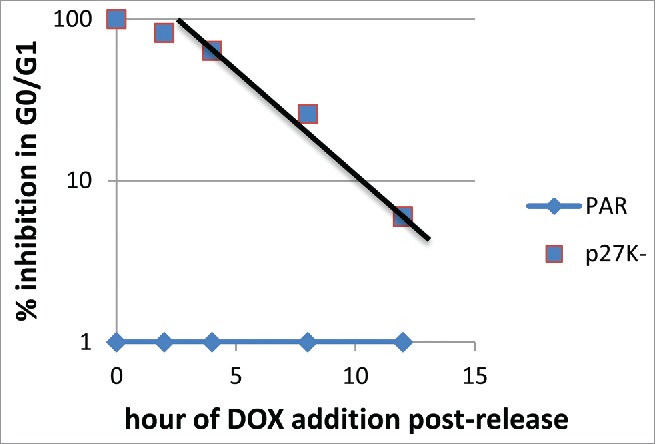
Parental and U2OS-p27K were placed in nocodazole-induced mitotic arrest. Cells were released from the mitotic arrest, p27K had been induced 6 h before release for one culture and at 0, 2, 4, 8, 12 h after release for other cultures. Cells were incubated in medium with BrdU. The percent of cells not entering S phase (DNA synthesis) was determined.
p27K inhibits cell cycle progression in G1/S and alters CDT1 and ORC1 expression
Our data clearly indicate that the expression of p27K does not allow cells to traverse S phase; however, some Brdu incorporation is observed in the initiation of DNA synthesis. In order to determine if the initiation of DNA synthesis is altered by the presence of p27K, we allowed U2OS cells expressing p27K to leave a mitotic block and followed the expression of CDT1 and ORC1. As seen earlier (Fig. 3), cells traversed G1 and induced cyclin D1, cyclin E and a small amount of cyclin A. The induction of the cyclin D and E appeared normal, but with lack of entry and/or traverse of S phase, cyclin A was not substantially induced. Figure 9 shows the levels of CDT1 and ORC1 found in extracts of cells at various times after the cells were released from mitotic inhibition and proceeded through G1 phase to S phase. Even though CDT1 and ORC1 are induced in cells expressing p27K as in parental cells, the eventual decrease in the expressed amount of the proteins was delayed from that seen in the parental cells. These results showing the decrease in CDT1 level suggest that ORIs are being licensed and some DNA synthesis is initiated but not completed as was evident with Brdu incorporation.
Figure 9.
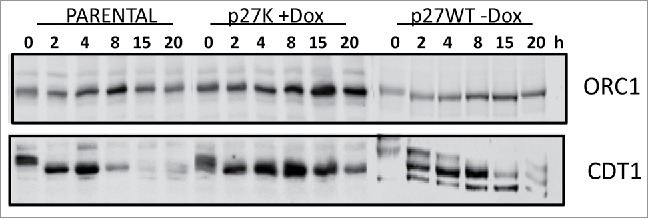
Parental, U2OS-p27WT, and U2OS-p27K cells were inhibited in a nocodazole-induced mitotic arrest. Dox was added as indicated for 6 h and then cells were washed and placed in fresh medium with or without Dox. At the indicated times cells were harvested and immunoblotted to determine comparative amounts of ORC1 and CDT1.
Discussion
The CDK inhibitor p27, increases in response to high cell density, serum starvation, and TGFβ, all of which cause growth arrest. It is thought that p27 causes cells to enter G0/G1 and remain in growth arrest through the inhibition of the CDK activities. Our data and data from many others indicate that p27 exerts regulatory control over numerous cell cycle CDK-controlled events and serves as a tumor suppressor. On the other hand, p27 has also been shown to control cellular events through non-CDK dependent regulation. Non-canonical regulation by p27 includes regulation of cellular migration, stem cell pool size, transcription, and some of these activities are prooncogenic in nature. We had previously shown that a p27 construct (termed p27K) which had been constructed with alanine substituted at residue 62 and 64 did not bind cdk2 or cdk4, and did not inhibit the CDK activities when expressed in U2OS cells.34 Even though the p27K did not inhibit CDK activity, it did alter growth rates of cultured U2OS cells. In order to study the effect of p27K on cell cycle control and to examine its control of specific cell cycle phases in U2OS cells we employed release from nocodazole-induced mitotic inhibition. Our data shows that U2OS cells expressing p27K did not enter and/or complete S phase when released from nocodazole-induced mitotic inhibition (Fig. 3). Cells overexpressing p27K released from nocodazole inhibition did undergo cell division and traversed G1 phase showing the same expression of cyclin D and cyclin E as parental cells (Fig. 3). The experiments illustrated in Figure 3 were performed using p27K clone #2 (ref 34), and in order to insure the results were not clone specific the experiments were repeated using separate p27K-inducible clones with the same results.
Our experiments presented above, set up an interesting condition in which we had the overexpression of p27K without inhibition of CDK activities allowing us to observe an unusual situation. While the cells expressing an excess of p27 mutant that is deficient for CDK binding (due to the substitution of only 2 critical amino acids within its CDK-binding domain) could traverse the cell cycle, these cells could not complete the cell cycle after release from nocodazole-induced mitotic inhibition. We point out that this function of p27 requires an intact cyclin-binding sequence since the p27CK construct did not exhibit the same effect. We had anticipated that p27CK would exhibit regulatory control on the cell cycle because for most of the non-canonical biological regulatory effects which have been published, the p27 did not require cyclin- or CDK-binding sites and are dependent on the C-terminus. But this was not the case. We have previously shown the overexpression of p27WT or p27C (p27 construct with mutated cyclin-binding site) inhibited CDK activities and the progression through G1.34 Since we knew that the p27K protein bound cyclin F34, we hypothesized that the cyclin F was being sequestered and the U2OS cells were acting similar to serum-starved quiescent MEFs derived from cyclin F−/− mice in their significantly delayed capacity to reenter the cell cycle upon serum stimulation. This hypothesis put forth the idea that cyclin F is required during cell cycle reentry and/or G1/S phase traverse. However, the data in Figure 7 shows that cyclin F was not required for U2OS cells to proceed from mitotic inhibition to the next G2 phase. The required role that had been demonstrated for cyclin F in the cell cycle reentry upon serum stimulation of cyclin F −/− MEFs is not clear at this time. It is possible that these MEFs have a target protein of cyclin F that must be altered to allow cell cycle traverse. Since the MEFs are derived from the cyclin F−/− mouse the difference could stem from mouse embryonic cells verses human tumor derived cells. Our data shows that the overexpression of p27K does not inhibit mitosis and cell division. However, the p27K altered an event necessary for S phase entry or S phase traverse. If it inhibited the G1 cyclin-dependent kinase activities these cell would behave like p27WT expressing cells and not traverse the cell cycle as we previously published.34 In addition, the G1 phase gene expression would not have been observed (Fig. 4 and 9). In many of the non-canonical events that p27 regulates, either cytoplasmic or nuclear, it appears that neither the cyclin- nor the CDK-binding site is required. In our experiments the N-terminus appears to be responsible for the described regulatory event. Our data clearly showed that the p27CK did not bring about growth inhibition; therefore, the cyclin- binding site is required and CDK-binding site is not required. Since p27CK induced to the same level as that of p27K but did not inhibit the cell cycle, it is not the overexpression of a protein in general that is responsible for the observed inhibition of G1/S phase progression. We were not able to detect the binding of p27K to MCM7 or other proteins of DNA licensing. Figure 9 suggests that CDT1 and ORC1 were not degraded as rapidly in the p27K expressing cell, which is not surprising with the lack of cyclin A expression (Fig. 4) although cyclin E was fully active. Finally, p27 has been suggested to interact with DNA sites that regulate gene expression such as demonstrated for Twist and Brachyury.31 Moreover, p27 associates with the SRR2 enhancer of Sox2 gene in association with p130-E2F4-SIN3A.32 Collectively, these published investigations illuminate cellular events that are, in part, regulated by p27 through non-CDK processes and thus illustrating p27 regulation of cellular proliferation through non-canonical CDK-independent mechanisms. The possible protein or DNA-binding site through which p27K is limiting cell cycle progression in S phase has not been determined; however, it apparently interacts with p27 in a fashion that requires an intact cyclin-binding sequence. Recently, it has been proposed that post-mitotic neurons have a nuclear complex composed of CDK5, p39, E2F1 and p27 which functions as a growth suppressor to inhibit the cell cycle through decreased transcriptional activity.37,38 In this case, p27 is high in the nuclear compartment when post-mitotic free E2F1 might become available for cell growth. These conditions mimic the conditions we have established in our model with active growth yet high levels of p27, and the cells in our model may develop a similar condition to bring about the inhibition of S phase. We have not at present determined possible components in the putative p27K complex, although we have performed mass spectrometry to identify proteins associated with p27K during inhibition of S phase. This putative regulatory event was discovered because the p27K did not inhibit CDK activities which allowed cell cycle progression and the p27K was present in sufficient quantity to reveal the regulatory episode. It is interesting to note that the growth inhibition in response to the overexpression of p27K was most obvious in experimental conditions that synchronized the cells in a G1 phase traverse. The overexpression of wild-type p27, which inhibits CDK activities, did not allow cell cycle progression of G1 to the G1/S border, while the much studied p27CK did not have the necessary cyclin- and CDK-binding sites, and therefore it allowed growth. The nature of the role that p27K, with an intact cyclin-binding site, plays in G1/S phase inhibition in cells released from nocodazole-induced mitotic inhibition is not known, yet it reflects the ability of p27 to inhibit S phase through a non-canonical mechanism. The arrest of cells described by our data may reflect the long established G1/S phase border arrest point.39-41
We suggest that p27 has evolved this regulatory control event to maintain another mechanism to inhibit cell cycle progression using events in late G1 or S phase in which inhibition of CDK activity is not required, such as with Rb deletion. In fact, we propose that p27 could maintain this mechanism even if phosphorylated on tyrosine, as long as the p27 remained localized in the nucleus. It remains to be determined whether p27 provides the gatekeeper control through protein interactions modifying required pathways or via an inhibition by a direct effect on DNA sites of replication.
Materials and Methods
Cell culture
U2OS_TetON cells and U2OS cells, and U2OS_TetON cells stably transfected with pTRE2_Hyg_p27 constructs were cultured as described previously.34,42 p27 was induced with Dox (1µg/ml) to the cultures.
Nocodazole Inhibition
The cells were synchronized in S phase with 2mM thymidine for 16 h. Cells were washed and placed in fresh medium with 100 ng/ml of nocodazole for 16 h. To release from mitotic inhibition, cells were washed with fresh medium and placed in fresh medium.
Propidium iodide staining
Cells were removed from the plates with 0.25% trypsin and 0.5mM EDTA in PBS; an equal volume of medium containing 10% serum was added to neutralize the trypsin. Cells were pelleted and resuspended in PBS (1 ml), and 100% ethanol (3.5 ml) was added slowly. Cells were incubated at 4°C for a minimum of 16 h, pelleted, and resuspended in PBS containing 0.1% Tween-20, 0.5% bovine serum albumin, 100 µg/ml RNase A and 30 mg/ml propidium iodide. After a further incubation at 4°C for at least 4 h, cell cycle distribution was analyzed by BD Accuri-C6. DNA content (FL3: FL4 area) and relative amount of BrdU per cell (FL1:H Anti-BrdU-FITC) are plotted on the x and y axes, respectively, of the scatter plots.
BrdU staining
Cells received 30 µM BrdU for times indicated in the figure legends. Cells were removed from the plates and fixed in PBS and ethanol as described above for propidium iodide staining. Pelleted cells were resuspended in 2 N HCl and incubated for 30 min at 37°C to denature DNA. Sodium borate (final concentration, 0.1 M) was added to neutralize the pH, and cells were washed thoroughly to remove any residual acid. Cells were incubated overnight at 4°C in PBS containing 0.5% bovine serum albumin, 0.1% Tween-20 and a 1/20 dilution of mouse monoclonal anti-BrdU-FITC antibody (eBioscience, San Diego, CA, USA). After washing, cells were incubated in PBS containing 0.1% Tween-20, 0.5% bovine serum albumin, 100 µg/ml RNase A and 30 µg/ml propidium iodide staining for at least 1 h at 4°C. Percent BrdU-labeled cells and DNA content were determined by BD Accuri-C6.
Annexin V binding
Cells were detached from plates with 0.25% trypsin-EDTA and combined with floating cells. Cells were stained with Annexin V-fluorescein isothiocyanate and propidium iodide (BD Pharmingen), and analyzed by BD Accuri-C6.
Western blotting
Cells were rinsed with phosphate-buffered saline (PBS), harvested by scraping, and collected by centrifugation. Cell pellets were suspended in 1X RIPA buffer (Cell Signaling) with protease inhibitor cocktail and phosphatase inhibitor cocktail (Calbiochem) and incubated on ice for 15 min. Insoluble material was removed by centrifugation. Cell extracts normalized for amount of protein were resolved by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked in PBS containing 0.1% Tween-20 and 5% instant milk and incubated with primary antibody in PBS containing 0.1% Tween for 2 h at room temperature or overnight at 4°C. Proteins recognized by the primary antibody were detected by enhanced chemiluminescence using a horseradish peroxidase coupled secondary antibody according to the instructions of the manufacturer (Pierce) or followed with Odyssey Western blot protocol and imaged with the Odessey system LiCOR.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This study was funded by the SRH Foundation and the Gibbs Cancer Center Research Development Fund.
References
- 1.Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev 1995; 9:1149-63; PMID:7758941; http://dx.doi.org/ 10.1101/gad.9.10.1149 [DOI] [PubMed] [Google Scholar]
- 2.Roberts JM, Koff A, Polyak K, Firpo E, Collins S, Ohtsubo M, Massague J. Cyclins, Cdks, and cyclin kinase inhibitors. Cold Spring Harb Symp Quant Biol 1994; 59:31-8; PMID:7587083; http://dx.doi.org/ 10.1101/SQB.1994.059.01.006 [DOI] [PubMed] [Google Scholar]
- 3.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999; 13:1501-12; PMID:10385618; http://dx.doi.org/ 10.1101/gad.13.12.1501 [DOI] [PubMed] [Google Scholar]
- 4.Hengst L, Dulic V, Slingerland JM, Lees E, Reed SI. A cell cycle-regulated inhibitor of cyclin-dependent kinases. Proc Natl Acad Sci U S A 1994; 91:5291-5; PMID:8202483; http://dx.doi.org/ 10.1073/pnas.91.12.5291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-β and contact inhibition to cell cycle arrest. Genes Dev 1994; 8:9-22; PMID:8288131; http://dx.doi.org/ 10.1101/gad.8.1.9 [DOI] [PubMed] [Google Scholar]
- 6.Slingerland JM, Hengst L, Pan CH, Alexander D, Stampfer MR, Reed SI. A novel inhibitor of cyclin-Cdk activity detected in transforming growth factor β-arrested epithelial cells. Mol Cell Biol 1994; 14:3683-94; PMID:8196612; http://dx.doi.org/ 10.1128/MCB.14.6.3683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer 2008; 8:253-67; PMID:18354415; http://dx.doi.org/ 10.1038/nrc2347 [DOI] [PubMed] [Google Scholar]
- 8.Coats S, Flanagan WM, Nourse J, Roberts JM. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science 1996; 272:877-80; PMID:8629023; http://dx.doi.org/ 10.1126/science.272.5263.877 [DOI] [PubMed] [Google Scholar]
- 9.Bagui TK, Cui D, Roy S, Mohapatra S, Shor AC, Ma L, Pledger WJ. Inhibition of p27Kip1 gene transcription by mitogens. Cell Cycle 2009; 8:115-24; PMID:19158484; http://dx.doi.org/ 10.4161/cc.8.1.7527 [DOI] [PubMed] [Google Scholar]
- 10.Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, Medema RH. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol 2002; 168:5024-31; http://dx.doi.org/ 10.4049/jimmunol.168.10.5024 [DOI] [PubMed] [Google Scholar]
- 11.Agrawal D, Hauser P, McPherson F, Dong F, Garcia A, Pledger WJ. Repression of p27kip1 synthesis by platelet-derived growth factor in BALB/c 3T3 cells. Mol Cell Biol 1996; 16:4327-36; PMID:8754833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Millard SS, Vidal A, Markus M, Koff A. A U-rich element in the 5′ untranslated region is necessary for the translation of p27 mRNA. Mol Cell Biol 2000; 20:5947-59; PMID:10913178; http://dx.doi.org/ 10.1128/MCB.20.16.5947-5959.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borriello A, Cucciolla V, Oliva A, Zappia V, Della Ragione F. p27Kip1 metabolism: a fascinating labyrinth. Cell Cycle 2007; 6:1053-61; PMID:17426451; http://dx.doi.org/ 10.4161/cc.6.9.4142 [DOI] [PubMed] [Google Scholar]
- 14.Wander SA, Zhao D, Slingerland JM. p27: a barometer of signaling deregulation and potential predictor of response to targeted therapies. Clin Cancer Res 2011; 17:12-8; PMID:20966355; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-0752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosen DG, Yang G, Cai KQ, Bast RC Jr., Gershenson DM, Silva EG, Liu J. Subcellular localization of p27kip1 expression predicts poor prognosis in human ovarian cancer. Clin Cancer Res 2005; 11:632-7; PMID:15701850 [PubMed] [Google Scholar]
- 16.Wu FY, Wang SE, Sanders ME, Shin I, Rojo F, Baselga J, Arteaga CL. Reduction of cytosolic p27(Kip1) inhibits cancer cell motility, survival, and tumorigenicity. Cancer Res 2006; 66:2162-72; PMID:16489017; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3304 [DOI] [PubMed] [Google Scholar]
- 17.Denicourt C, Saenz CC, Datnow B, Cui XS, Dowdy SF. Relocalized p27Kip1 tumor suppressor functions as a cytoplasmic metastatic oncogene in melanoma. Cancer Res 2007; 67:9238-43; PMID:17909030; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-1375 [DOI] [PubMed] [Google Scholar]
- 18.Wen S, So Y, Singh K, Slingerland JM, Resnick MB, Zhang S, Ruiz V, Moss SF. Promotion of cytoplasmic mislocalization of p27 by Helicobacter pylori in gastric cancer. Oncogene 2012; 31:1771-80; PMID:21841827; http://dx.doi.org/ 10.1038/onc.2011.362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, et al.. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med 2002; 8:1153-60; PMID:12244302; http://dx.doi.org/ 10.1038/nm761 [DOI] [PubMed] [Google Scholar]
- 20.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1999; 1:193-9; PMID:10559916; http://dx.doi.org/ 10.1038/12013 [DOI] [PubMed] [Google Scholar]
- 21.Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol 1999; 9:661-4; PMID:10375532; http://dx.doi.org/ 10.1016/S0960-9822(99)80290-5 [DOI] [PubMed] [Google Scholar]
- 22.Hara T, Kamura T, Nakayama K, Oshikawa K, Hatakeyama S, Nakayama K. Degradation of p27(Kip1) at the G(0)-G(1) transition mediated by a Skp2-independent ubiquitination pathway. J Biol Chem 2001; 276:48937-43; PMID:11682478; http://dx.doi.org/ 10.1074/jbc.M107274200 [DOI] [PubMed] [Google Scholar]
- 23.Gstaiger M, Jordan R, Lim M, Catzavelos C, Mestan J, Slingerland J, Krek W. Skp2 is oncogenic and overexpressed in human cancers. Proc Natl Acad Sci U S A 2001; 98:5043-8; PMID:11309491; http://dx.doi.org/ 10.1073/pnas.081474898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, Arteaga CL. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med 2002; 8:1145-52; PMID:12244301; http://dx.doi.org/ 10.1038/nm759 [DOI] [PubMed] [Google Scholar]
- 25.Viglietto G, Motti ML, Bruni P, Melillo RM, D'Alessio A, Califano D, Vinci F, Chiappetta G, Tsichlis P, Bellacosa A, et al.. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med 2002; 8:1136-44; PMID:12244303; http://dx.doi.org/ 10.1038/nm762 [DOI] [PubMed] [Google Scholar]
- 26.Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev 2004; 18:862-76; PMID:15078817; http://dx.doi.org/ 10.1101/gad.1185504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang XQ, Lui EL, Cai Q, Ching WY, Liu KS, Poon RT, Fan ST. p27Kip1 promotes migration of metastatic hepatocellular carcinoma cells. Tumour Biol 2008; 29:217-23; PMID:18781093; http://dx.doi.org/ 10.1159/000152939 [DOI] [PubMed] [Google Scholar]
- 28.Denicourt C, Dowdy SF. Cip/Kip proteins: more than just CDKs inhibitors. Genes Dev 2004; 18:851-5; PMID:15107401; http://dx.doi.org/ 10.1101/gad.1205304 [DOI] [PubMed] [Google Scholar]
- 29.Besson A, Hwang HC, Cicero S, Donovan SL, Gurian-West M, Johnson D, Clurman BE, Dyer MA, Roberts JM. Discovery of an oncogenic activity in p27Kip1 that causes stem cell expansion and a multiple tumor phenotype. Genes Dev 2007; 21:1731-46; PMID:17626791; http://dx.doi.org/ 10.1101/gad.1556607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nallamshetty S, Crook M, Boehm M, Yoshimoto T, Olive M, Nabel EG. The cell cycle regulator p27Kip1 interacts with MCM7, a DNA replication licensing factor, to inhibit initiation of DNA replication. FEBS Lett 2005; 579:6529-36; PMID:16289477; http://dx.doi.org/ 10.1016/j.febslet.2005.10.028 [DOI] [PubMed] [Google Scholar]
- 31.Menchon C, Edel MJ, Izpisua Belmonte JC. The cell cycle inhibitor p27Kip(1) controls self-renewal and pluripotency of human embryonic stem cells by regulating the cell cycle, Brachyury and Twist. Cell Cycle 2011; 10:1435-47; PMID:21478681; http://dx.doi.org/ 10.4161/cc.10.9.15421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pippa R, Espinosa L, Gundem G, Garcia-Escudero R, Dominguez A, Orlando S, Gallastegui E, Saiz C, Besson A, Pujol MJ, et al.. p27Kip1 represses transcription by direct interaction with p130/E2F4 at the promoters of target genes. Oncogene 2012; 31:4207-20; PMID:22179826; http://dx.doi.org/ 10.1038/onc.2011.582 [DOI] [PubMed] [Google Scholar]
- 33.Nguyen L, Besson A, Roberts JM, Guillemot F. Coupling cell cycle exit, neuronal differentiation and migration in cortical neurogenesis. Cell Cycle 2006:2314-8; http://dx.doi.org/ 10.4161/cc.5.20.3381 [DOI] [PubMed] [Google Scholar]
- 34.Sharma SS, Ma L, Bagui TK, Forinash KD, Pledger WJ. A p27Kip1 mutant that does not inhibit CDK activity promotes centrosome amplification and micronucleation. Oncogene 2012; 31:3989-98; PMID:22158041; http://dx.doi.org/ 10.1038/onc.2011.550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.D'Angiolella V, Donato V, Vijayakumar S, Saraf A, Florens L, Washburn MP, Dynlacht B, Pagano M. SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature 2010; 466:138-42; PMID:20596027; http://dx.doi.org/ 10.1038/nature09140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tetzlaff MT, Bai C, Finegold M, Wilson J, Harper JW, Mahon KA, Elledge SJ. Cyclin F disruption compromises placental development and affects normal cell cycle execution. Mol Cell Biol 2004; 24:2487-98; PMID:14993286; http://dx.doi.org/ 10.1128/MCB.24.6.2487-2498.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, Li H, Herrup K. Cdk5 nuclear localization is p27-dependent in nerve cells: implications for cell cycle suppression and caspase-3 activation. J Biol Chem 2010; 285:14052-61; PMID:20189989; http://dx.doi.org/ 10.1074/jbc.M109.068262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J, Herrup K. Nucleocytoplasmic Cdk5 is involved in neuronal cell cycle and death in post-mitotic neurons. Cell Cycle 2011; 10:1208-14; PMID:21415596; http://dx.doi.org/ 10.4161/cc.10.8.15328 [DOI] [PubMed] [Google Scholar]
- 39.Pledger WJ, Stiles CD, Antoniades HN, Scher CD. An ordered sequence of events is required before BALB/c-3T3 cells become committed to DNA synthesis. Proc Natl Acad Sci U S A 1978; 75:2839-43; PMID:275855; http://dx.doi.org/ 10.1073/pnas.75.6.2839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leof EB, Van Wyk JJ, O'Keefe EJ, Pledger WJ. Epidermal growth factor (EGF) is required only during the traverse of early G1 in PDGF stimulated density-arrested BALB/c-3T3 cells. Exp Cell Res 1983; 147:202-8; PMID:6604639; http://dx.doi.org/ 10.1016/0014-4827(83)90285-9 [DOI] [PubMed] [Google Scholar]
- 41.Leof EB, Wharton W, van Wyk JJ, Pledger WJ. Epidermal growth factor (EGF) and somatomedin C regulate G1 progression in competent BALB/c-3T3 cells. Exp Cell Res 1982; 141:107-15; PMID:6749535; http://dx.doi.org/ 10.1016/0014-4827(82)90073-8 [DOI] [PubMed] [Google Scholar]
- 42.Fu W, Ma L, Chu B, Wang X, Bui MM, Gemmer J, Altiok S, Pledger WJ. The cyclin-dependent kinase inhibitor SCH 727965 (dinacliclib) induces the apoptosis of osteosarcoma cells. Mol Cancer Ther 2011; 10:1018-27; PMID:21490307; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0167 [DOI] [PMC free article] [PubMed] [Google Scholar]