

ABSTRACT
Maintenance of genome integrity is crucial to avoid cancer and other genetic diseases. Thus faced with DNA damage, cells mount a DNA damage response to avoid genome instability. The DNA damage response is partially inhibited during mitosis presumably to avoid erroneous processing of the segregating chromosomes. Yet our recent study shows that TopBP1-mediated DNA processing during mitosis is highly important to reduce transmission of DNA damage to daughter cells.1 Here we provide an overview of the DNA damage response and DNA repair during mitosis. One role of TopBP1 during mitosis is to stimulate unscheduled DNA synthesis at underreplicated regions. We speculated that such genomic regions are likely to hold stalled replication forks or post-replicative gaps, which become the substrate for DNA synthesis upon entry into mitosis. Thus, we addressed whether the translesion pathways for fork restart or post-replicative gap filling are required for unscheduled DNA synthesis in mitosis. Using genetics in the avian DT40 cell line, we provide evidence that unscheduled DNA synthesis in mitosis does not require the translesion synthesis scaffold factor Rev1 or PCNA ubiquitylation at K164, which serve to recruit translesion polymerases to stalled forks. In line with this finding, translesion polymerase η foci do not colocalize with TopBP1 or FANCD2 in mitosis. Taken together, we conclude that TopBP1 promotes unscheduled DNA synthesis in mitosis independently of the examined translesion polymerases.
KEYWORDS: fork restart, mitosis, PCNA ubiquitylation, polymerase η, post-replicative gaps, replication stress, REV1, TopBP1, translesion synthesis
DNA double-strand breaks trigger a DNA damage response
DNA damage elicits a cellular response that involves checkpoint arrest, activation of DNA repair mechanisms and in case of severe damage, apoptosis or senescence. This response is in general referred to as the DNA damage response (DDR). However, depending on the type and extent of genomic assaults as well as the stage of the cell cycle, the DDR can engage various cellular pathways.2,3 The canonical DDR to DNA double-strand breaks (DSBs) is initiated by recognition of the break by the MRN complex consisting of MRE11, RAD50 and NBS1 (reviewed in ref. 4). This complex both activates the ATM kinase and processes the DSB for repair.5-8 The DSB also triggers a cascade of events in the surrounding chromatin initiated by the kinases ATM- and DNA-PKcs, which phosphorylate the histone variant H2AX on serine 139. The phosphorylated H2AX is referred to as γ-H2AX (reviewed in refs. 9,10). In brief, γ-H2AX quickly attracts a number of proteins including MDC1,11 which recruits the E3 ubiquitin ligase RNF8 in a phospho-dependent manner.12-14 RNF8 initiates chromatin ubiquitylation to facilitate recruitment of another group of proteins to the flanking chromatin.12-17 The latter group of proteins includes 53BP1, which promotes non-homologous end-joining (NHEJ) 18,19 and BRCA1, which facilitates repair by homologous recombination.20,21
The global checkpoint effect of the canonical DDR is reviewed extensively elsewhere.22 Briefly, checkpoint arrest is a result of ATM activation, which activates the kinase CHK2 by phosphorylation.23 CHK2 phosphorylates and thereby inactivates the CDC25 phosphatases, which are key drivers of the cell cycle.23-25 Moreover, the DDR elicits a transcriptional response via the tumor suppressor p53 that induces expression of a variety of genes involved in DNA repair and apoptosis.26
The DNA damage response to replication stress occurs via ATR activation
Replication stress triggers the DDR by a mechanism that is slightly different from the DSB-mediated DDR.27,28 Replication stress refers to a number of cellular conditions where DNA replication is partially inhibited, thus leading to the formation of stalled replication forks (reviewed in ref. 3). Stalled replication forks generate long tracts of single-stranded DNA (ssDNA), which are bound by the single-strand binding protein RPA.28 ATR is recruited to ssDNA via its interaction partner ATRIP, which binds directly to RPA.29 To gain full kinase activity ATR needs to be activated by the ATR activating domain (AAD) of TopBP1.30 Once activated, the ATR kinase phosphorylates CHK1, which similarly to CHK2 phosphorylates the CDC25 phosphatases and thus the replication stress-induced DDR response converges with the DSB-induced DDR described above. Replication stress-mediated ATR and CHK1 activation also causes stabilization of replication forks and suppression of late origin firing.3
TopBP1 is required for ATR activation
TopBP1 is a multifunctional scaffold protein, which comprises nine BRCT (BRCA1 C-terminus) domains that enable dynamic phospho-dependent protein-protein interactions.31 The BRCT domains are numbered BRCT0 to BRCT8 counting from the N-terminus. Several mechanisms for recruitment of TopBP1 to stalled replication forks are described in the literature. The 9-1-1 clamp consisting of the Rad9, Hus1 and Rad1 proteins, is loaded onto junctions between single- and double-stranded DNA. 9-1-1 is generally considered to be responsible for the recruitment of TopBP1 32 via interaction between the C-terminus of Rad9 and TopBP1 BRCT1+2.33,34 Consistently, interaction between Rad9 and TopBP1 is required for ATR activation.35 Also, the protein RHINO that binds both the 9-1-1 and TopBP1 is required for full activation of ATR.36,37 Somewhat contradictory, TopBP1 has been shown to localize to stalled replication forks independently of the 9-1-1 complex, and rather be responsible for recruitment of 9-1-1.38,39 In line with this, recruitment of TopBP1 to ATR activating DNA structures in Xenopus extracts was shown to rely on the MRN complex. The MRN-mediated recruitment seemed to depend on TopBP1 BRCT3-6.40 In human cells, TopBP1 accumulation at stalled forks induced by the replication inhibitor hydroxyurea is mediated by direct interactions between phosphorylated MDC1 and the BRCT5 domain in TopBP1.41,42
Replication fork restart and post-replicative gap filling mechanisms
As mentioned above, ATR activation in response to replication stress leads to stabilization of stalled forks. Two damage tolerance pathways exist to promote fork restart: template switching and translesion synthesis. Template switching relies on homologous recombination to restart the replication fork. It involves the invasion of nascent DNA on the sister strand, which is used as a template to bypass a potential obstacle on the template strand (reviewed in ref. 43). Translesion synthesis (TLS) applies socalled translesion polymerases to restart the fork (reviewed in ref. 44). Translesion polymerases are low fidelity polymerases capable of replicating across a damaged template to bypass a lesion on the template strand (reviewed in ref. 45). Both pathways are geared to circumvent a damaged template. However, these pathways are also activated in response to the replication inhibitors hydroxyurea or aphidicolin (APH), neither of which directly damage DNA.46,47 As an alternative to fork restart, lagging and leading strand synthesis can be uncoupled leaving a single-stranded gap behind the fork on one strand, which can be filled in post-replicatively.48,49
Translesion polymerases are recruited to stalled forks by monoubiquitylation of PCNA at K164.46,50-52 The E3 ubiquitin ligase and the E2 ubiquitin-conjugating enzyme, Rad18 and Rad6, catalyze this reaction,51,53 while ATR seems to be dispensable for this process.54 Rather RPA-bound ssDNA generated by uncoupling of the replication fork and the replicative helicases recruits Rad18 to stalled forks.54,55 Alternatively, TLS polymerases may be recruited to the stalled fork by the Rev1 scaffold protein.56 In DT40 cells, Rev1 and PCNA ubiquitylation at K164 are central to TLS-mediated restart or gap filling, respectively. Thus, Rev1 recruits TLS polymerases directly to stalled forks to restart replication, whereas PCNA ubiquitylation is required for post-replicative gap filling by TLS polymerases.56
DSB-induced DDR in mitosis
In mitosis the DDR is partially inactivated. An initial response to DSBs including activation of ATM and DNA-PKcs, phosphorylation of H2AX and recruitment of MDC1 is mounted.57 However, the binding of RNF8 to MDC1 is inhibited in mitosis by CDK1-mediated phosphorylation of RNF8 (pT752).58 Therefore the DNA damage-induced ubiquitin cascade is not triggered, hampering the recruitment of downstream factors including BRCA1 and 53BP1. Moreover, in mitosis 53BP1 recruitment to DNA damage is also specifically inhibited by PLK1 and CDK1-mediated phosphorylation of 53BP1.58-60 Transient inhibition of ATM or DNA-PKcs in mitosis disrupting the primary DDR response results in cells becoming hypersensitive to DSB inducing agents.57 This indicates that the initial recognition of DSBs in mitosis is important, and it has been proposed to facilitate the repair of mitotic DNA damage in the subsequent cell cycle.57 However, the repair machineries for DSBs are shut down in mitosis, and this is crucial to avoid telomere fusions.58 Accordingly, RNF8 starts accumulating at the separated chromatids in telophase, which is a late stage of mitosis, when sister telomeres have separated.61
Processing of underreplicated regions and joint molecules in mitosis
When cells are subjected to mild replication stress, some chromosomal loci such as chromosomal fragile sites (CFSs) can escape checkpoint detection and enter mitosis in an underreplicated state.62-64 These underreplicated regions may hold stalled or collapsed replication forks, post-replicative gaps or recombination intermediates. Two proteins belonging to the Fanconi anemia DNA repair pathway, FANCD2 and FANCI, form replication stress-induced foci and a subset of these foci persists into mitosis as pairs of foci on the sister chromatids at CFSs.65,66 Interestingly, the majority of replication stress-induced FANCD2 foci were found to localize to CFSs in mitosis and these sites occasionally undergo unscheduled DNA synthesis at mitosis.67,68 Presumably such unscheduled DNA synthesis could function to complete genome duplication prior to chromosome segregation. Thus, in contrast to DSBs, underreplicated regions may still be processed during mitosis to prevent sister chromatid nondisjunction, which would be accompanied by the formation of DNA bridges in anaphase. To avoid this scenario, joint molecule resolution by Holliday junction resolvases is upregulated during mitosis by several means (reviewed in refs. 69,70). Firstly, the EME1 subunit of the structure-selective endonuclease MUS81-EME1 is phosphorylated by CDK1 in mitosis 71 and this promotes the interaction to another structure-selective endonuclease SLX1-SLX4. As a complex, SLX1-SLX4-MUS81-EME1 efficiently catalyzes joint molecule resolution.72 Moreover, the SLX4-TopBP1 interaction in mitosis is promoted by CDK-mediated phosphorylation.73 Thus, the SLX1-SLX4-MUS81-EME1 Holliday junction resolvase super complex is specifically targeted to chromatin, due to the accumulation of TopBP1 into chromatin-associated foci at mitotic onset.1 Accordingly, TopBP1 depletion around mitosis results in an increase in chromatin bridges in anaphase.74 Secondly, the Holliday junction resolvase GEN1 provides a backup mechanism for joint molecule resolution later in anaphase. GEN1 is cytoplasmic in interphase cells, and thus only gains access to chromatin after nuclear envelope breakdown.75-77
Moreover, MUS81 activity promotes the appearance of “breaks” on metaphase chromosomes at CFSs. At the same time, MUS81 depletion leads to increase in the transmission of DNA damage to daughter cells.67,78 This indicates that MUS81-mediated processing in mitosis, resulting in “breaks” on metaphase chromosomes acts in fact as a mitotic repair mechanism. Similarly, TopBP1 promotes DNA repair during mitosis to avoid the transmission of DNA damage to daughter cells. This seems to involve stimulation of unscheduled DNA synthesis and recruitment of the structure-selective endonuclease SLX4.1
Taken together there is mounting evidence that the final steps of homologous recombination are highly active in mitosis although initiation of homologous recombination, on the other hand, seems to be inhibited. For example, the recombination factor BRCA2 is phosphorylated (pS3291) by CDK2 at mitotic onset.79 This phosphorylation inhibits BRCA2-mediated stabilization of RAD51 nucleofilaments at stalled replication forks.79-82
Regulation of TLS during mitosis is less well characterized, but interestingly studies in yeast revealed that the levels of both Rev1 and the TLS polymerase η (pol η) peak in G2/M.83,84 Similarly the level of Rev3, which is the catalytic subunit of the TLS polymerase zeta peaks in mitosis.85,86 Moreover, Rev3 localizes to chromatin at this stage of the cell cycle and Rev3 has a suggested role for replication of CFS in G2/M in a manner that is independent of the Rev7 subunit of the polymerase zeta complex.86
Replication stress-induced 53BP1 nuclear bodies in G1
In response to replication stress, 53BP1 forms large (2-3 μm) nuclear bodies (NBs) in a symmetrical manner in G1 daughter cells reminiscent of the FANCD2 sister foci in mitosis.87,88 53BP1 NBs are confined to G1, form upon mitotic exit and disassemble again during S-phase. This reveals that they form as a response to replication stress-induced DNA damage inherited from the previous cell cycle. The 53BP1 NBs colocalize with a range of markers that normally assemble in the vicinity of DSBs such as MDC1, γ-H2AX, ATM, RNF8 and BRCA1.88 53BP1 NBs were further found to colocalize with transcriptional regulatory OPT (Oct-1, PTF, transcription) domains, induced by micro-irradiation as well as ionizing radiation.87 Consistently, 53BP1-OPT domains neither colocalize with RNA transcription initiation or elongation factors nor with transcriptionally incorporated 5-fluorouridine, showing that active transcription was not detectable at these sites.87 Inhibition of ATM showed that ATM activity is needed for the efficient formation of 53BP1 NBs in G1 87,88
TopBP1 forms foci in mitosis
TopBP1 forms abundant foci at mitotic onset but only a small fraction of these turns into DNA bridges in anaphase, suggesting that DNA structures other that joint molecules can recruit TopBP1 at mitotic onset. Interestingly, Burgess et al. have shown that chromatin condensation per se can trigger an initial DNA damage response, which results in the recruitment of several DDR factors including MDC1.89 Accordingly, MDC1 forms spontaneous foci on the condensed mitotic chromosomes.57,89 MDC1 is required for TopBP1 recruitment to stalled forks in response to replication stress. Similar to this mechanism, MDC1 accumulation due to condensation could explain the high number of spontaneous TopBP1 foci on mitotic chromatin. It is puzzling, however, that TopBP1 seems to be recruited specifically at the nuclear envelope breakdown, whereas chromatin condensation is initiated in prophase before nuclear envelope breakdown.90 Moreover, the fact that 53BP1 NBs derive from mitotic TopBP1 foci and are increased in number when TopBP1 is depleted, suggests that TopBP1 marks sites in the genome that hold some kind of aberrant DNA structure.1 Intriguingly, BRCT5 of TopBP1 is required for TopBP1 foci formation in mitosis (91 and our unpublished observations). BRCT5 is responsible for TopBP1 interactions to MDC1 and BLM.41,92 However, siRNA knockdown experiments suggest that neither MDC1 nor BLM are responsible for the accumulation of TopBP1 into detectable structures in mitosis.91 Thus, the recruitment mechanism for TopBP1 to mitotic chromatin remains unclear.
The function of TopBP1 in mitosis is independent of ATR
Our recent findings show that the role of TopBP1 in mitosis is not dependent on ATR. In fact, ATR inhibition specifically around mitosis results in fewer or weaker 53BP1 NBs in G1, which is in contrast to TopBP1 depletion.1 Thus, similarly to the role of ATM at DSBs in mitotic cells, ATR could have a role in mitosis by priming aberrant DNA structures for detection by 53BP1 and other repair factors in G1, but apparently this function of ATR does not require TopBP1-mediated activation. Alternatively, the used dose of the inhibitor ATRi 93 might also inhibit ATM in the DT40 cell line.1
REV1 or PCNA K164 ubiquitylation is not required for DNA synthesis in mitosis
One source of TopBP1 foci in mitosis is underreplicated DNA induced by replication stress.1 When cells are subjected to mild replication stress by treatment with the replication inhibitor APH, DNA synthesis can be detected in mitotic cells. Specifically, DNA synthesis is detected by incubating cells with the base analog EdU for 20 min prior to fixation.1 The fact that DNA synthesis in mitosis is induced by replication stress suggests that the sites of DNA synthesis in mitosis are either stalled replication forks or post-replicative gaps. Depletion of TopBP1 leads to decreased EdU incorporation in mitotic cells. Interestingly this phenotype can be complemented with a TopBP1 mutant protein that is cytoplasmic and thus only gains access to DNA after nuclear envelope breakdown.1 This shows that TopBP1-stimulated DNA synthesis takes place in mitosis, but the mechanistic details of this mitotic DNA synthesis are otherwise unknown.
Our recent studies of TopBP1 in mitosis show that the majority of TopBP1 foci do not mark joint molecules between sister chromatids (at least not in anaphase).1 Given that template switching would generate a joint molecule, whereas TLS would not, we speculated whether this class of TopBP1 foci marks post-replicative gaps or stalled replication forks that are destined for TLS-mediated repair pathways. Therefore we have investigated whether TLS pathways involving Rev1 or PCNA ubiquitylation are required for DNA synthesis in mitosis. For this purpose we took advantage of a REV1−/− cell line 94 and of DT40 cell lines lacking both endogenous alleles of PCNA but expressing either wild-type human PCNA (hPCNA) or human PCNA with lysine 164 substituted for alanine.95 This latter cell line, which we refer to as hPCNA K164R, is not capable of PCNA ubiquitylation.95
To address whether REV1 or PCNA ubiquitylation is required for DNA synthesis in mitosis we pulsed replication stressed REV1−/− cells or hPCNA K164R cells as well as control cell lines with EdU for 20 min, followed by fluorescence labeling (Figs. 1A and B). In both of the mutants, we observed robust EdU incorporation in mitosis, demonstrating that these components of the TLS pathway are not required for DNA synthesis in mitosis. In fact both REV1−/− and hPCNA K164R show slightly elevated levels of DNA synthesis compared to DT40 and hPCNA although this trend is not significant in our dataset (Fig. 1B). In agreement with our previous studies we find that TopBP1 localizes to sites of unscheduled DNA synthesis in mitosis (Fig. 1A). To further address the relationship between TLS and TopBP1 in mitosis, we characterized the localization of pol η and TopBP1 in mitosis (Figs. 1C and D). Both pol η and TopBP1 form foci in mitosis and both types of foci are induced by APH. However, TopBP1 and pol η very rarely colocalize corroborating that pol η is not involved in the TopBP1 pathway for unscheduled DNA synthesis in mitosis detectable by EdU incorporation. In agreement with this we also find that FANCD2 and pol η foci do not colocalize in mitosis (Figs. 1E and F).
Figure 1.
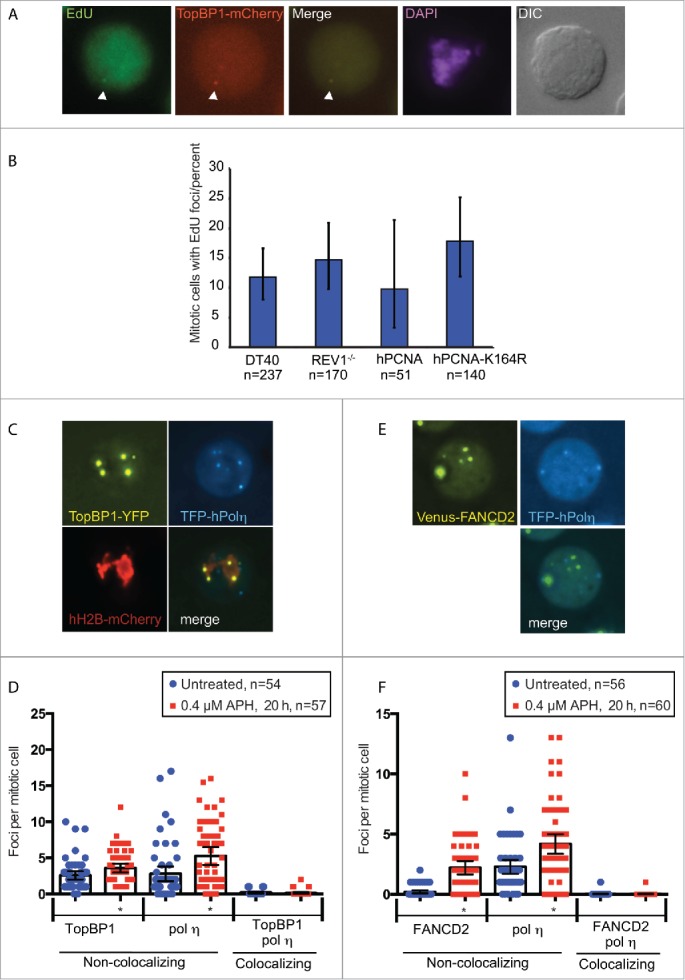
TopBP1 mediates unscheduled DNA synthesis in mitosis independently of TLS pathways. (A) Representative images of fixed DT40 cells (TopBP1mCherry) treated with 0.4 μM APH for 20 h and pulse labeled with 20 μM EdU for 20 min. White arrowheads indicate TopBP1 focus colocalizing with EdU. (B) Quantification of EdU in prometaphase and metaphase cells. (C) Representative images of an APH-treated DT40 cell line (TopBP1YFP-AID/YFP-AID/YFP-AID/ osTIR / TFP-hPol η / hH2B-mCherry). (D) Quantification of TopBP1 and pol η foci. Live cell images were captured of untreated cells or 20 h after addition of 0.4 μM APH. TopBP1 and pol η foci and their colocalization in mitotic cells were quantified. (E) Representative images of an APH-treated DT40 cell line (FANCD2Venus/Venus/ TFP-hPol η). (F) Quantification of FANCD2 and pol η foci. Live cell images were captured of untreated cells or 20 h after addition of 0.4 μM APH. FANCD2 and pol η foci and their colocalization in mitotic cells were quantified. For all graphs in this figure, asterisks indicate significant differences from the untreated (P < 0.05) and error bars represent 95% confidence intervals. Number of cells analyzed is indicated (n).
In summary, our previous work suggested that substantial TopBP1-mediated DNA processing is required during mitosis to avoid transmission of DNA damage to daughter cells.1 One of the functions of TopBP1 during mitosis is to stimulate unscheduled DNA synthesis at underreplicated sites. Here we show that the unscheduled DNA synthesis does not require the bona fide TLS pathways promoted by REV1 or PCNA K164 ubiquitylation.
Methods
All DT40 cell lines and primers used in this study are listed in Tables S1 and S2, respectively
DT40 cell culture
DT40 cells were cultured in RPMI 1640 medium GlutaMAX (Gibco) supplemented with 2% chicken serum (Gibco), 8% fetal bovine serum (Gibco), 2 mM L-glutamine (Gibco), 55 μM β-mercaptoethanol, 50 units/ml penicillin and 50 μg/ml streptomycin at 39°C with 5% CO2.
Generation of TFP-pol η constructs
The construct for expression of TFP-tagged human pol η was generated by PCR amplification using the adapted primers SK3 and SK4 to amplify human pol η cDNA from the plasmid IOH46681 (ultimate). The PCR product was cloned into pcr2.1 by TOPO cloning according to the manufacturers protocol (Thermo Fisher Scientific) and sequenced. TFP encoding cDNA was PCR amplified using NheI and EcoRV adapted primers (SK1 and SK2, respectively) and TOPO-cloned into pcr2.1 for sequencing. Adapted pol η cDNA and TFP fragments were cloned into pExpress containing the chicken β actin promotor. A SpeI fragment containing β actin promotor, TFP and Pol η cDNA was subcloned into either pLox BSR or pLox PURO to obtain pLoxBSR-TFP-pol η or pLoxPURO-TFP-pol η, respectively.
Generation of stable pol η expressing DT40 cell lines
The pol η constructs were transfected into DT40 cells by electroporation (gene pulser Xcell, Biorad). Transfectants harboring the PURO or BSR resistance genes were selected in the presence of 0.5 μg/ml puromycin or 20 μg/ml blasticidin, respectively. Expression of nuclear pol η TFP was confirmed by fluorescence microscopy.
Live cell and immunofluorescence microscopy
For live cell microscopy exponentially growing DT40 cells were imaged at 39°C in growth medium, and mounted on μ-slides prior to imaging (Ibidi, Martinsried, Germany). Where indicated in the figures 0.4 μM aphidicolin (APH; Sigma-Aldrich) was added 20 h prior to imaging. Immunofluorescence microscopy of DT40 cells and metaphase spreads were performed at room temperature.
Fluorophores were visualized on a widefield microscope (DeltaVision Elite; Applied Precision) equipped with a 100× objective lens (U-PLAN S-APO, NA 1.4; Olympus), a cooled EMCCD camera (Evolve 512; Photometrics), and a solid-state illumination source (Insight; Applied Precision, Inc.). Images were acquired using softWoRx (Applied Precision) software. Images were processed with Volocity software (PerkinElmer). Images were pseudocolored according to the approximate emission wavelengths of the fluorophores. Fluorescent proteins used in DT40 were TFP (pmTurquoise2-N1, 96), YFP (enhanced YFP, Takara Bio Inc..), Venus,97 and mCherry.98
Representative images presented in figures were deconvoluted and gamma adjusted using Volocity software.
For EdU incorporation, cells were treated with 20 μM EdU for 20 minutes prior to cell harvest. Cells were washed and resuspended in PBS, and allowed to adhere to poly-L-lysine coated coverslips for 10 minutes. Cell fixation, permeabilization and EdU detection were performed as described in the Click-iT® Plus EdU Alexa Fluor® 594 Imaging Kit manual (Invitrogen, CA, USA). Fixed cells were analyzed on a widefield microscope (AxioImager Z1; Carl Zeiss) equipped with a 100× objective lens (Plan Apochromat, NA 1.4; Carl Zeiss), a cooled CCD camera (Orca-ER; Hamamatsu Photonics), differential interference contrast (DIC), and an illumination source (HXP120C; Carl Zeiss).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank J. Sale for sharing DT40 cell lines.
Funding
This work was supported by The Danish Agency for Science, Technology and Innovation, The Villum Foundation, the Lundbeck Foundation, and the European Research Council (ERC) to M. Lisby and the ERC to V.H. Oestergaard, I. Gallina and R.T. Pedersen.
References
- 1.Pedersen RT, Kruse T, Nilsson J, Oestergaard VH, Lisby M. TopBP1 is required at mitosis to reduce transmission of DNA damage to G1 daughter cells. J Cell Biol 2015; 210:565-82; PMID:26283799; http://dx.doi.org/ 10.1083/jcb.201502107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burgess RC, Misteli T. Not All DDRs Are Created Equal: Non-Canonical DNA Damage Responses. Cell 2015; 162:944-7; PMID:26317463; http://dx.doi.org/ 10.1016/j.cell.2015.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol 2014; 16:2-9; PMID:24366029; http://dx.doi.org/ 10.1038/ncb2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stracker TH, Petrini JH. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol 2011; 12:90-103; PMID:21252998; http://dx.doi.org/ 10.1038/nrm3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 2004; 304:93-6; PMID:15064416; http://dx.doi.org/ 10.1126/science.1091496 [DOI] [PubMed] [Google Scholar]
- 6.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005; 308:551-4; PMID:15790808; http://dx.doi.org/ 10.1126/science.1108297 [DOI] [PubMed] [Google Scholar]
- 7.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature 2007; 450:509-14; PMID:17965729; http://dx.doi.org/ 10.1038/nature06337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 2003; 22:5612-21; PMID:14532133; http://dx.doi.org/ 10.1093/emboj/cdg541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179-204; PMID:20965415; http://dx.doi.org/ 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 2011; 25:409-33; PMID:21363960; http://dx.doi.org/ 10.1101/gad.2021311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005; 123:1213-26; PMID:16377563; http://dx.doi.org/ 10.1016/j.cell.2005.09.038 [DOI] [PubMed] [Google Scholar]
- 12.Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007; 131:901-14; PMID:18001825; http://dx.doi.org/ 10.1016/j.cell.2007.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, et al.. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007; 318:1637-40; PMID:18006705; http://dx.doi.org/ 10.1126/science.1150034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007; 131:887-900; PMID:18001824; http://dx.doi.org/ 10.1016/j.cell.2007.09.040 [DOI] [PubMed] [Google Scholar]
- 15.Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, et al.. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009; 136:435-46; PMID:19203579; http://dx.doi.org/ 10.1016/j.cell.2008.12.041 [DOI] [PubMed] [Google Scholar]
- 16.Stewart GS. Solving the RIDDLE of 53BP1 recruitment to sites of damage. Cell Cycle 2009; 8:1532-8; PMID:19372751; http://dx.doi.org/ 10.4161/cc.8.10.8351 [DOI] [PubMed] [Google Scholar]
- 17.Oestergaard VH, Pentzold C, Pedersen RT, Iosif S, Alpi A, Bekker-Jensen S, Mailand N, Lisby M. RNF8 and RNF168 but not HERC2 are required for DNA damage-induced ubiquitylation in chicken DT40 cells. DNA Repair (Amst) 2012; 11:892-905; PMID:23010445; http://dx.doi.org/ 10.1016/j.dnarep.2012.08.005 [DOI] [PubMed] [Google Scholar]
- 18.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al.. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010; 141:243-54; PMID:20362325; http://dx.doi.org/ 10.1016/j.cell.2010.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakamura K, Sakai W, Kawamoto T, Bree RT, Lowndes NF, Takeda S, Taniguchi Y. Genetic dissection of vertebrate 53BP1: a major role in non-homologous end joining of DNA double strand breaks. DNA Repair (Amst) 2006; 5:741-9; PMID:16644291; http://dx.doi.org/ 10.1016/j.dnarep.2006.03.008 [DOI] [PubMed] [Google Scholar]
- 20.Scully R, Ganesan S, Vlasakova K, Chen J, Socolovsky M, Livingston DM. Genetic analysis of BRCA1 function in a defined tumor cell line. Mol Cell 1999; 4:1093-9; PMID:10635334; http://dx.doi.org/ 10.1016/S1097-2765(00)80238-5 [DOI] [PubMed] [Google Scholar]
- 21.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell 1999; 4:511-8; PMID:10549283; http://dx.doi.org/ 10.1016/S1097-2765(00)80202-6 [DOI] [PubMed] [Google Scholar]
- 22.Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol 2009; 21:245-55; PMID:19230643; http://dx.doi.org/ 10.1016/j.ceb.2009.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998; 282:1893-7; PMID:9836640; http://dx.doi.org/ 10.1126/science.282.5395.1893 [DOI] [PubMed] [Google Scholar]
- 24.Russell P, Nurse P. cdc25+ functions as an inducer in the mitotic control of fission yeast. Cell 1986; 45:145-53; PMID:3955656; http://dx.doi.org/ 10.1016/0092-8674(86)90546-5 [DOI] [PubMed] [Google Scholar]
- 25.Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001; 410:842-7; PMID:11298456; http://dx.doi.org/ 10.1038/35071124 [DOI] [PubMed] [Google Scholar]
- 26.Roos WP, Kaina B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett 2013; 332:237-48; PMID:22261329; http://dx.doi.org/ 10.1016/j.canlet.2012.01.007 [DOI] [PubMed] [Google Scholar]
- 27.Ward IM, Minn K, Chen J. UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J Biol Chem 2004; 279:9677-80; PMID:14742437; http://dx.doi.org/ 10.1074/jbc.C300554200 [DOI] [PubMed] [Google Scholar]
- 28.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev 2005; 19:1040-52; PMID:15833913; http://dx.doi.org/ 10.1101/gad.1301205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003; 300:1542-8; PMID:12791985; http://dx.doi.org/ 10.1126/science.1083430 [DOI] [PubMed] [Google Scholar]
- 30.Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell 2006; 124:943-55; PMID:16530042; http://dx.doi.org/ 10.1016/j.cell.2005.12.041 [DOI] [PubMed] [Google Scholar]
- 31.Wardlaw CP, Carr AM, Oliver AW. TopBP1: A BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair (Amst) 2014; 22:165-74; PMID:25087188; http://dx.doi.org/ 10.1016/j.dnarep.2014.06.004 [DOI] [PubMed] [Google Scholar]
- 32.Helt CE, Wang W, Keng PC, Bambara RA. Evidence that DNA damage detection machinery participates in DNA repair. Cell Cycle 2005; 4:529-32; PMID:15876866; http://dx.doi.org/ 10.4161/cc.4.4.1598 [DOI] [PubMed] [Google Scholar]
- 33.Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev 2007; 21:1472-7; PMID:17575048; http://dx.doi.org/ 10.1101/gad.1547007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee J, Kumagai A, Dunphy WG. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J Biol Chem 2007; 282:28036-44; PMID:17636252; http://dx.doi.org/ 10.1074/jbc.M704635200 [DOI] [PubMed] [Google Scholar]
- 35.Rappas M, Oliver AW, Pearl LH. Structure and function of the Rad9-binding region of the DNA-damage checkpoint adaptor TopBP1. Nucleic Acids Res 2011; 39:313-24; PMID:20724438; http://dx.doi.org/ 10.1093/nar/gkq743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindsey-Boltz LA, Kemp MG, Capp C, Sancar A. RHINO forms a stoichiometric complex with the 9-1-1 checkpoint clamp and mediates ATR-Chk1 signaling. Cell Cycle 2015; 14:99-108; PMID:25602520; http://dx.doi.org/ 10.4161/15384101.2014.967076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cotta-Ramusino C, McDonald ER 3rd, Hurov K, Sowa ME, Harper JW, Elledge SJ. A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science 2011; 332:1313-7; PMID:21659603; http://dx.doi.org/ 10.1126/science.1203430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan S, Michael WM. TopBP1 and DNA polymerase alpha-mediated recruitment of the 9-1-1 complex to stalled replication forks: implications for a replication restart-based mechanism for ATR checkpoint activation. Cell Cycle 2009; 8:2877-84; PMID:19652550; http://dx.doi.org/ 10.4161/cc.8.18.9485 [DOI] [PubMed] [Google Scholar]
- 39.Yan S, Michael WM. TopBP1 and DNA polymerase-alpha directly recruit the 9-1-1 complex to stalled DNA replication forks. J Cell Biol 2009; 184:793-804; PMID:19289795; http://dx.doi.org/ 10.1083/jcb.200810185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duursma AM, Driscoll R, Elias JE, Cimprich KA. A role for the MRN complex in ATR activation via TOPBP1 recruitment. Mol Cell 2013; 50:116-22; PMID:23582259; http://dx.doi.org/ 10.1016/j.molcel.2013.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Gong Z, Chen J. MDC1 collaborates with TopBP1 in DNA replication checkpoint control. J Cell Biol 2011; 193:267-73; PMID:21482717; http://dx.doi.org/ 10.1083/jcb.201010026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leung CC, Sun L, Gong Z, Burkat M, Edwards R, Assmus M, Chen J, Glover JN. Structural insights into recognition of MDC1 by TopBP1 in DNA replication checkpoint control. Structure 2013; 21:1450-9; PMID:23891287; http://dx.doi.org/ 10.1016/j.str.2013.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Princz LN, Gritenaite D, Pfander B. The Slx4-Dpb11 scaffold complex: -coordinating the response to replication fork stalling in S-phase and the subsequent mitosis. Cell Cycle 2015; 14(4):488-494; http://dx.doi.org/10.4161/15384101.2014.989126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Friedberg EC, Lehmann AR, Fuchs RP. Trading places: how do DNA polymerases switch during translesion DNA synthesis? Mol Cell 2005; 18:499-505; PMID:15916957; http://dx.doi.org/ 10.1016/j.molcel.2005.03.032 [DOI] [PubMed] [Google Scholar]
- 45.Goodman MF, Woodgate R. Translesion DNA polymerases. Cold Spring Harb Perspect Biol 2013; 5:a010363; PMID:23838442; http://dx.doi.org/ 10.1101/cshperspect.a010363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kannouche PL, Wing J, Lehmann AR. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell 2004; 14:491-500; PMID:15149598; http://dx.doi.org/ 10.1016/S1097-2765(04)00259-X [DOI] [PubMed] [Google Scholar]
- 47.Leach CA, Michael WM. Ubiquitin/SUMO modification of PCNA promotes replication fork progression in Xenopus laevis egg extracts. J Cell Biol 2005; 171:947-54; PMID:16344309; http://dx.doi.org/ 10.1083/jcb.200508100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prakash L. Characterization of postreplication repair in Saccharomyces cerevisiae and effects of rad6, rad18, rev3 and rad52 mutations. Mol Gen Genet 1981; 184:471-8; PMID:7038396; http://dx.doi.org/ 10.1007/BF00352525 [DOI] [PubMed] [Google Scholar]
- 49.Rupp WD, Wilde CE 3rd, Reno DL, Howard-Flanders P. Exchanges between DNA strands in ultraviolet-irradiated Escherichia coli. J Mol Biol 1971; 61:25-44; PMID:4947693; http://dx.doi.org/ 10.1016/0022-2836(71)90204-X [DOI] [PubMed] [Google Scholar]
- 50.Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, Kannouche P, Wider G, Peter M, Lehmann AR, et al.. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 2005; 310:1821-4; PMID:16357261; http://dx.doi.org/ 10.1126/science.1120615 [DOI] [PubMed] [Google Scholar]
- 51.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002; 419:135-41; PMID:12226657; http://dx.doi.org/ 10.1038/nature00991 [DOI] [PubMed] [Google Scholar]
- 52.Bi X, Barkley LR, Slater DM, Tateishi S, Yamaizumi M, Ohmori H, Vaziri C. Rad18 regulates DNA polymerase kappa and is required for recovery from S-phase checkpoint-mediated arrest. Mol Cell Biol 2006; 26:3527-40; PMID:16611994; http://dx.doi.org/ 10.1128/MCB.26.9.3527-3540.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J 2004; 23:3886-96; PMID:15359278; http://dx.doi.org/ 10.1038/sj.emboj.7600383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chang DJ, Lupardus PJ, Cimprich KA. Monoubiquitination of proliferating cell nuclear antigen induced by stalled replication requires uncoupling of DNA polymerase and mini-chromosome maintenance helicase activities. J Biol Chem 2006; 281:32081-8; PMID:16959771; http://dx.doi.org/ 10.1074/jbc.M606799200 [DOI] [PubMed] [Google Scholar]
- 55.Davies AA, Huttner D, Daigaku Y, Chen S, Ulrich HD. Activation of ubiquitin-dependent DNA damage bypass is mediated by replication protein a. Mol Cell 2008; 29:625-36; PMID:18342608; http://dx.doi.org/ 10.1016/j.molcel.2007.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Edmunds CE, Simpson LJ, Sale JE. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell 2008; 30:519-29; PMID:18498753; http://dx.doi.org/ 10.1016/j.molcel.2008.03.024 [DOI] [PubMed] [Google Scholar]
- 57.Giunta S, Belotserkovskaya R, Jackson SP. DNA damage signaling in response to double-strand breaks during mitosis. J Cell Biol 2010; 190:197-207; PMID:20660628; http://dx.doi.org/ 10.1083/jcb.200911156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Orthwein A, Fradet-Turcotte A, Noordermeer SM, Canny MD, Brun CM, Strecker J, Escribano-Diaz C, Durocher D. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 2014; 344:189-93; PMID:24652939; http://dx.doi.org/ 10.1126/science.1248024 [DOI] [PubMed] [Google Scholar]
- 59.Benada J, Burdova K, Lidak T, von Morgen P, Macurek L. Polo-like kinase 1 inhibits DNA damage response during mitosis. Cell Cycle 2015; 14:219-31; PMID:25607646; http://dx.doi.org/ 10.4161/15384101.2014.977067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee DH, Acharya SS, Kwon M, Drane P, Guan Y, Adelmant G, Kalev P, Shah J, Pellman D, Marto JA, et al.. Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Mol Cell 2014; 54:512-25; PMID:24703952; http://dx.doi.org/ 10.1016/j.molcel.2014.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Giunta S, Jackson SP. Give me a break, but not in mitosis: the mitotic DNA damage response marks DNA double-strand breaks with early signaling events. Cell Cycle 2011; 10:1215-21; PMID:21412056; http://dx.doi.org/ 10.4161/cc.10.8.15334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Palakodeti A, Han Y, Jiang Y, Le Beau MM. The role of late/slow replication of the FRA16D in common fragile site induction. Genes Chromosomes Cancer 2004; 39:71-6; PMID:14603443; http://dx.doi.org/ 10.1002/gcc.10290 [DOI] [PubMed] [Google Scholar]
- 63.Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell 2002; 111:779-89; PMID:12526805; http://dx.doi.org/ 10.1016/S0092-8674(02)01113-3 [DOI] [PubMed] [Google Scholar]
- 64.Letessier A, Millot GA, Koundrioukoff S, Lachages AM, Vogt N, Hansen RS, Malfoy B, Brison O, Debatisse M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011; 470:120-3; PMID:21258320; http://dx.doi.org/ 10.1038/nature09745 [DOI] [PubMed] [Google Scholar]
- 65.Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 2009; 11:753-60; PMID:19465922; http://dx.doi.org/ 10.1038/ncb1882 [DOI] [PubMed] [Google Scholar]
- 66.Naim V, Rosselli F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat Cell Biol 2009; 11:761-8; PMID:19465921; http://dx.doi.org/ 10.1038/ncb1883 [DOI] [PubMed] [Google Scholar]
- 67.Naim V, Wilhelm T, Debatisse M, Rosselli F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat Cell Biol 2013; 15:1008-15; PMID:23811686; http://dx.doi.org/ 10.1038/ncb2793 [DOI] [PubMed] [Google Scholar]
- 68.Bergoglio V, Boyer AS, Walsh E, Naim V, Legube G, Lee MY, Rey L, Rosselli F, Cazaux C, Eckert KA, et al.. DNA synthesis by Pol eta promotes fragile site stability by preventing under-replicated DNA in mitosis. J Cell Biol 2013; 201:395-408; PMID:23609533; http://dx.doi.org/ 10.1083/jcb.201207066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wyatt HD, West SC. Holliday junction resolvases. Cold Spring Harb Perspect Biol 2014; 6:a023192; PMID:25183833; http://dx.doi.org/ 10.1101/cshperspect.a023192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matos J, West SC. Holliday junction resolution: regulation in space and time. DNA Repair (Amst) 2014; 19:176-81; PMID:24767945; http://dx.doi.org/ 10.1016/j.dnarep.2014.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matos J, Blanco MG, Maslen S, Skehel JM, West SC. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 2011; 147:158-72; PMID:21962513; http://dx.doi.org/ 10.1016/j.cell.2011.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wyatt HD, Sarbajna S, Matos J, West SC. Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol Cell 2013; 52:234-47; PMID:24076221; http://dx.doi.org/ 10.1016/j.molcel.2013.08.035 [DOI] [PubMed] [Google Scholar]
- 73.Gritenaite D, Princz LN, Szakal B, Bantele SC, Wendeler L, Schilbach S, Habermann BH, Matos J, Lisby M, Branzei D, et al.. A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev 2014; 28:1604-19; PMID:25030699; http://dx.doi.org/ 10.1101/gad.240515.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Germann SM, Schramke V, Pedersen RT, Gallina I, Eckert-Boulet N, Oestergaard VH, Lisby M. TopBP1/Dpb11 binds DNA anaphase bridges to prevent genome instability. J Cell Biol 2014; 204:45-59; PMID:24379413; http://dx.doi.org/ 10.1083/jcb.201305157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chan YW, West SC. Spatial control of the GEN1 Holliday junction resolvase ensures genome stability. Nat Commun 2014; 5:4844; PMID:25209024; http://dx.doi.org/ 10.1038/ncomms5844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sarbajna S, Davies D, West SC. Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev 2014; 28:1124-36; PMID:24831703; http://dx.doi.org/ 10.1101/gad.238303.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wechsler T, Newman S, West SC. Aberrant chromosome morphology in human cells defective for Holliday junction resolution. Nature 2011; 471:642-6; PMID:21399624; http://dx.doi.org/ 10.1038/nature09790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ying S, Minocherhomji S, Chan KL, Palmai-Pallag T, Chu WK, Wass T, Mankouri HW, Liu Y, Hickson ID. MUS81 promotes common fragile site expression. Nat Cell Biol 2013; 15:1001-7; PMID:23811685; http://dx.doi.org/ 10.1038/ncb2773 [DOI] [PubMed] [Google Scholar]
- 79.Ayoub N, Rajendra E, Su X, Jeyasekharan AD, Mahen R, Venkitaraman AR. The carboxyl terminus of Brca2 links the disassembly of Rad51 complexes to mitotic entry. Curr Biol 2009; 19:1075-85; PMID:19540122; http://dx.doi.org/ 10.1016/j.cub.2009.05.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pefani DE, Latusek R, Pires I, Grawenda AM, Yee KS, Hamilton G, van der Weyden L, Esashi F, Hammond EM, O'Neill E. RASSF1A-LATS1 signalling stabilizes replication forks by restricting CDK2-mediated phosphorylation of BRCA2. Nat Cell Biol 2014; 16:962-71, 1–8; http://dx.doi.org/ 10.1038/ncb3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011; 145:529-42; PMID:21565612; http://dx.doi.org/ 10.1016/j.cell.2011.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 2005; 434:598-604; PMID:15800615; http://dx.doi.org/ 10.1038/nature03404 [DOI] [PubMed] [Google Scholar]
- 83.Waters LS, Walker GC. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)/M phase rather than S phase. Proc Natl Acad Sci U S A 2006; 103:8971-6; PMID:16751278; http://dx.doi.org/ 10.1073/pnas.0510167103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Plachta M, Halas A, McIntyre J, Sledziewska-Gojska E. The steady-state level and stability of TLS polymerase eta are cell cycle dependent in the yeast S. cerevisiae. DNA Repair (Amst) 2015; 29:147-53; PMID:25766643 [DOI] [PubMed] [Google Scholar]
- 85.Nelson JR, Lawrence CW, Hinkle DC. Thymine-thymine dimer bypass by yeast DNA polymerase zeta. Science 1996; 272:1646-9; PMID:8658138; http://dx.doi.org/ 10.1126/science.272.5268.1646 [DOI] [PubMed] [Google Scholar]
- 86.Bhat A, Andersen PL, Qin Z, Xiao W. Rev3, the catalytic subunit of Polzeta, is required for maintaining fragile site stability in human cells. Nucleic Acids Res 2013; 41:2328-39; PMID:23303771; http://dx.doi.org/ 10.1093/nar/gks1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP. Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol 2011; 193:97-108; PMID:21444690; http://dx.doi.org/ 10.1083/jcb.201011083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, et al.. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 2011; 13:243-53; PMID:21317883; http://dx.doi.org/ 10.1038/ncb2201 [DOI] [PubMed] [Google Scholar]
- 89.Burgess RC, Burman B, Kruhlak MJ, Misteli T. Activation of DNA damage response signaling by condensed chromatin. Cell Rep 2014; 9:1703-17; PMID:25464843; http://dx.doi.org/ 10.1016/j.celrep.2014.10.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Swedlow JR, Hirano T. The making of the mitotic chromosome: modern insights into classical questions. Mol Cell 2003; 11:557-69; PMID:12667441; http://dx.doi.org/ 10.1016/S1097-2765(03)00103-5 [DOI] [PubMed] [Google Scholar]
- 91.Broderick R, Nieminuszczy J, Blackford AN, Winczura A, Niedzwiedz W. TOPBP1 recruits TOP2A to ultra-fine anaphase bridges to aid in their resolution. Nat Commun 2015; 6:6572; PMID:25762097; http://dx.doi.org/ 10.1038/ncomms7572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang J, Chen J, Gong Z. TopBP1 controls BLM protein level to maintain genome stability. Mol Cell 2013; 52:667-78; PMID:24239288; http://dx.doi.org/ 10.1016/j.molcel.2013.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, Oyarzabal J, Pastor J, Bischoff JR, Fernandez-Capetillo O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol 2011; 18:721-7; PMID:21552262; http://dx.doi.org/ 10.1038/nsmb.2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Simpson LJ, Sale JE. Rev1 is essential for DNA damage tolerance and non-templated immunoglobulin gene mutation in a vertebrate cell line. EMBO J 2003; 22:1654-64; PMID:12660171; http://dx.doi.org/ 10.1093/emboj/cdg161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Simpson LJ, Ross AL, Szuts D, Alviani CA, Oestergaard VH, Patel KJ, Sale JE. RAD18-independent ubiquitination of proliferating-cell nuclear antigen in the avian cell line DT40. EMBO Rep 2006; 7:927-32; PMID:16888649; http://dx.doi.org/ 10.1038/sj.embor.7400777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Goedhart J, van Weeren L, Hink MA, Vischer NO, Jalink K, Gadella TW Jr. Bright cyan fluorescent protein variants identified by fluorescence lifetime screening. Nat Methods 2010; 7:137-9; PMID:20081836; http://dx.doi.org/ 10.1038/nmeth.1415 [DOI] [PubMed] [Google Scholar]
- 97.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol 2002; 20:87-90; PMID:11753368; http://dx.doi.org/ 10.1038/nbt0102-87 [DOI] [PubMed] [Google Scholar]
- 98.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 2004; 22:1567-72; PMID:15558047; http://dx.doi.org/ 10.1038/nbt1037 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.