

Abstract
Naive CD4+ T cells undergo massive proliferation and differentiation into at least four distinct helper T cell subsets after recognition of foreign antigen–derived peptides presented by dendritic cells. Each helper T cell subset expresses a distinct set of genes that encode unique transcription factor(s), as well as hallmark cytokines. The cytokine environment created by activated CD4+ T cells, dendritic cells and/or other cell types during the course of differentiation is a major determinant for the helper T cell fate. This Review focuses on the role of cytokines of the common γ-chain (γc) family in the determination of the effector helper T cell phenotype that naive CD4+ T cells adopt after being activated and in the function of these helper T cells.
Cytokines regulate a variety of cellular responses, including proliferation, differentiation and survival. Among the several different classes of cytokines, type I cytokines have a particularly important role. Type I cytokines have a four α-helix bundle structure and bind transmembrane proteins whose extracellular regions contain a hematopoietin receptor domain. This evolutionarily conserved, 200–amino acid region derived from a tandem of two ancestral fibronectin-like domains has four conserved cysteine residues in the amino-terminal segment and a tryptophan-serine doublet near the carboxy-terminal end1. Although they were first defined as cytokines, the type I cytokines also include hematopoietic factors and endocrine hormones. Their receptors belong to the type I cytokine receptor superfamily.
An important subfamily of the type I cytokines are those that use the common γ-chain (γc) to generate signaling receptor complexes. These include interleukin 2 (IL-2), IL-4, IL-7, IL-9, IL-15 and IL-21 (ref. 2). In addition, the cytokines IL-13 and TSLP are closely related to IL-4 and IL-7, respectively, and, although they do not use γc, they use an alternative chain (IL-13Rα1 for IL-13, and TSLPR for TSLP) that may have comparable features. In general, the receptor chain that binds the cytokine with high affinity is designated the α-chain (for example, IL-4Rα); the IL-2 receptor (IL-2R) and IL-15R complexes are exceptions to this rule. IL-2R consists of three subunits: the IL-2 receptor β-chain (IL-2Rβ (CD122)), which is the analog of the α-chains in other receptor complexes; the common γ-chain (γc (CD132)); and a third chain (IL-2Rα (CD25)) that is not a member of the structural family of type I cytokine receptors. IL-2Rα alone binds IL-2, although it does so with low affinity and no signaling ability. An intermediate-affinity IL-2R complex is composed of IL-2Rβ and γc, and all three subunits form the high-affinity IL-2R complex, which has both a rapid on rate and a slow off rate3. The IL-15R complex is similar to the IL-2R complex, with IL-2Rβ and γc but with the IL-15Rαhomolog of IL-2Rα. The IL-15R system, although structurally homologous to the IL-2R system, is unique in that not all the receptor subunits are necessarily expressed on the same cells. IL-15 may be captured by cells that express an IL-15Rα chain and then may be presented in trans (that is, as an IL-15-IL-15Rα complex) to neighboring cells that express IL-2Rβ and γc chains, which leads to IL-15-mediated immune responses4.
Each of the receptors for the γc family of cytokines transduces signals through the kinases Jak1 and Jak3, but different members of the family activate different transcription factors of the STAT family2. IL-2, IL-7, IL-9 and IL-15 activate mainly STAT5 (both STAT5A and STAT5B); IL-4 activates STAT6 and, to a lesser extent, STAT5; and IL-21 activates mainly STAT3. In this Review, we will discuss the role of cytokines of the γc family in the fate of peripheral CD4+ T cells during their differentiation into effector helper T cells after exposure to their cognate antigens and in their function. We emphasize results from mouse systems, recognizing that although the principles of helper T cell differentiation in mice and humans are similar, there are some differences in detail5.
T cells originate in the thymus and undergo a process of selection in which cells able to bind complexes of self peptide and major histocompatibility complex (MHC) with some threshold affinity are rescued from apoptosis (positive selection), whereas cells with high affinity for self peptide–MHC complexes are eliminated (negative selection). Cytokines of the γc family have a crucial role in this selection process; excellent reviews of this topic have been published elsewhere6–8.
When naive CD4+ T cells recognize foreign antigen–derived peptides presented in the context of MHC class II on dendritic cells (DCs) in the periphery, these cells undergo a process that includes massive proliferation and differentiation into distinct helper T cell subsets. There is still considerable uncertainty about the number of these subsets, the precursor-product relationships among them and their ability to convert from one to another. At least four different subsets of helper T cells (TH1, TH2, TH17 and regulatory T cells (Treg cells)) have been studied in great detail. Each expresses a distinct set of regulatory transcription factors, including what is sometimes designated a ‘master regulator’, as well as hallmark cytokines. Indeed, the cytokine environment created by activated CD4+ T cells themselves, by ‘partner’ DCs and/or by other cell types during the course of differentiation is one of the key determinants for the differentiation into distinct helper T cell subsets9.
TH1 differentiation: the role of IL-2
The master regulatory factor T-bet has a central role in TH1 differentiation. In developing TH1 cells, IL-2-driven activation of STAT5 controls the binding of T-bet to the conserved noncoding sequence CNS-1 in the promoter of the gene (Ifng) encoding interferon-γ (IFN-γ) and thereby regulates IFN-γ production and the positive feedback process through which TH1 differentiation proceeds10. In addition, IL-2 regulates induction of the IL-12Rβ2 subunit of the IL-12 receptor, an event critical to the completion of TH1 differentiation because of the central role of IL-12 in both the induction of T-bet and the production of IFN-γ11. This IL-2 effect also depends on STAT5 activation12. There is a still unresolved controversy about whether the induction of T-bet expression requires the IL-2–STAT5 pathway10,12. The key cytokines involved in TH1 differentiation are IFN-γ and IL-12, with the latter being a type I cytokine that does not use γc.
TH2 differentiation: the role of IL-2 and IL-4
In vitro TH2 differentiation requires stimulation via the T cell antigen receptor (TCR) and IL-4-mediated activation of STAT6, which jointly induce the expression of GATA-3, a TH2 master regulatory transcription factor13. However, the very first report on in vitro TH2 differentiation pointed out the requirement for both IL-4 and IL-2 for optimal TH2 development14. Although the role of IL-2 in TH2 differentiation was unclear at that time, subsequent studies have indicated a central role for IL-2 in TH2 differentiation. Neutralization of endogenous IL-2 results in the failure of naive CD4+ T cells to undergo TH2 development without affecting their proliferation. In such experiments, the ‘differentiation’ and ‘proliferation and survival’ functions of IL-2 can be distinguished because IL-4, a key component of in vitro TH2 differentiation, can provide the second two functions in place of IL-2 but cannot provide its differentiation function15. Indeed, ectopic expression of constitutively active STAT5A in activated CD4+ T cells leads to robust TH2 differentiation even when IL-4 activity is blocked and exogenous IL-12 is provided16. STAT5 binds to DNase I–hypersensitivity site II in the second intron of Il4 (Fig. 1), although the importance of this second intron site relative to that of other potential STAT5-binding sites in the larger Il4 genetic region has not been established. Thus, the IL-2–STAT5 pathway may function to maintain the Il4 locus in an open configuration so that transcription factors required for TH2 differentiation gain access to their binding sites.
Figure 1.
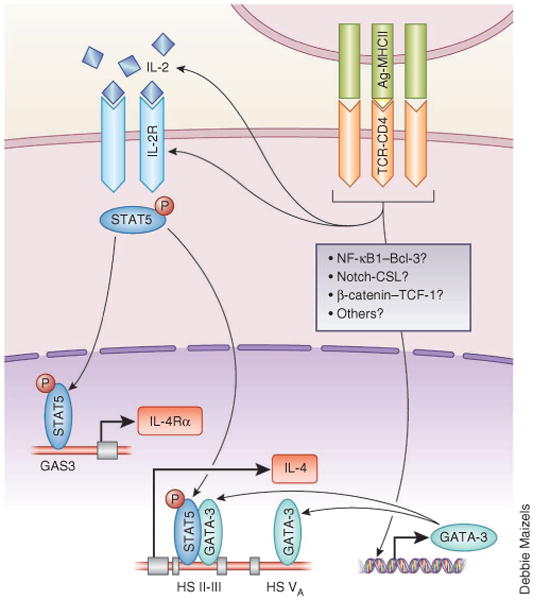
A model for early TH2 differentiation. The recognition of an antigen–MHC class II complex (Ag-MHCII) by the TCR on a naive CD4+ T cell leads to the upregulation of GATA-3 expression independently of IL-4 during the early stages of TH2 differentiation. The NF-ºB1-Bcl-3 complex, the Notch-CSL pathway and the Wnt–β-catenin–TCF-1 pathway have been each proposed to have a critical role in TCR-driven GATA-3 expression. GATA-3 binds to several sites on loci encoding TH2 cytokines, ncluding DNase I–hypersensitivity sites II and III (HS II-III) and site V (HS VA) in the Il4 locus. Stimulation via the TCR also induces the production of IL-2 and expression of the IL-2R complex, which results in the activation of STAT5. Activated STAT5 binds to DNase I–hypersensitivity sites II and III, which act together with GATA-3 to induce small amounts of early IL-4 production, and also binds to the IFN-γ-activated GAS3 motif in the Il4ra locus to upregulate IL-4Rα expression.
IL-2 also upregulates IL-4RαC expression on activated CD4+ T cells in a STAT5-dependent manner, which enhances the capacity of IL-4 to induce signals in developing TH2 cells. The activation of STAT5 by IL-2 results in the binding of STAT5 to the IFN-γ-activated motif GAS3 in the Il4ra locus during the early stage of TH2 differentiation17 (Fig. 1). It should be noted that IL-4 itself also upregulates IL-4Rα expression, so the relative importance of IL-2 and IL-4 in enhancing the sensitivity of the developing CD4+ T cells to IL-4 almost certainly depends on the timing of the availability of these cytokines during the TH2 differentiation process.
In vivo, differentiation of CD4+ T cells down the TH2 pathway may proceed through either IL-4-dependent routes or IL-4-independent routes. Identification of the sources of IL-4 that initiate IL-4-dependent TH2 differentiation in vivo has been of great interest for many years; among the cell types that have been considered are memory TH2 cells, natural killer T cells, mast cells, basophils, eosinophils and the naive CD4+ T cells themselves18. It has been reported that basophils serve as TH2-inducing professional antigen-presenting cells in vivo, given their ability to express functional MHC class II molecules and to produce a large amount of IL-4 (refs. 19–21). However, the methodology used to establish such a function for basophils involves depletion of these cells through the use of a monoclonal antibody to the FcεRI receptor for immunoglobulin E. It has been shown that this antibody results in the depletion of not only basophils but also a population of ‘inflammatory’ DCs expressing Fc∈RI that induce mainly TH2 differentiation22. Furthermore, in another mouse model of depletion of basophils selectively in vivo in the offspring of the crossing of mice that express Cre recombinase in basophils (Basoph8 mice) with mice expressing diphtheria toxin α-chain from the ubiquitous Rosa26 locus (Rosa-DTα mice), the absence of basophils does not affect the ability of CD4+ T cells to produce IL-4 after infection with Schistosoma mansoni23. Whether basophils are dispensable for all aspects of TH2 differentiation remains to be established.
A second main determinant of TH2 differentiation is the strength of signals generated by engagement of the TCR. In vivo immunization with a low concentration of antigen favors antibody production over delayed-type hypersensitivity24. TH1 differentiation is favored when agonist peptides are used for immunization, whereas altered peptide ligands, which interact with the TCR with lower affinity, favor TH2 differentiation25. Indeed, in a wide variety of mutant mice in which TCR signals are partially impaired, TH2 differentiation is favored26–28. In vitro differentiation of cells stimulated with low and high concentrations of peptide has shown that naive CD4+ T cells that receive weak TCR signals increase their expression of GATA-3 in an IL-4-independent manner at ∼14–24 hours after activation, whereas cells that receive strong TCR-mediated signals do not29 (Fig. 2). The NF-κB1–Bcl-3 complex, the Notch-CSL pathway and the Wnt–β-catenin–TCF-1 pathway have each been proposed to serve a critical role in TCR-driven GATA-3 expression30–34. However, gain or loss of function of these pathways, which are the modalities used to study the role of these molecules in this process, may lead to either abnormal thymic development or poor T cell proliferation and survival, even if T cell development seems phenotypically normal35–37. Therefore, it is conceivable that the impairment of both GATA-3 expression and subsequent TH2 differentiation may not be due to the direct regulation of GATA-3 expression by these pathways but may instead be secondary to either abnormal development or insufficient activation of CD4+ T cells derived from donors in which the genes encoding these molecules have been manipulated.
Figure 2.
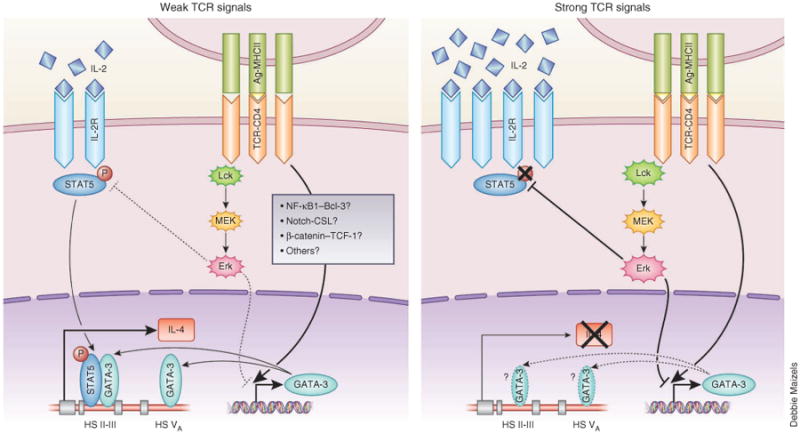
A model for the regulation of early TH2 differentiation by TCR signal strength. When naive CD4+ T cells receive strong TCR signals (right), prolonged and intense activation of the Erk pathway results in not only the failure to upregulate TCR-driven early expression of GATA-3 but also transient inhibition of IL-2-mediated activation of STAT5, at least during the first 24 hours after stimulation, despite the abundant IL-2 production and IL-2R expression. This defect in both GATA-3 upregulation and STAT5 activation leads to a lack of both early production of IL-4 and subsequent TH2 differentiation. In contrast, when naive CD4+ T cells receive weak TCR signals (left), the degree of activation of the Erk pathway is not strong enough to suppress TCR-driven early expression of GATA-3 or to block STAT5 activation in response to small amounts of IL-2, which allows T cells to generate early production of IL-4 and to undergo subsequent TH2 differentiation.
Naive CD4+ T cells that have received strong TCR signals not only fail to upregulate TCR-driven expression of GATA-3 but also do not activate STAT5 in response to IL-2 endogenously produced by activated CD4+ T cells themselves, at least during the first 24 hours after stimulation. This defect in both GATA-3 induction and STAT5 activation through the action of the kinase Erk pathway results in a failure to produce IL-4 or to undergo subsequent TH2 differentiation29 (Fig. 2). The physiological relevance of the TCR signal strength–mediated regulation of in vivo TH1 differentiation versus TH2 differentiation has been an open question. However, two studies have shown that omega-1, a T2 RNase derived from S. mansoni egg antigen, acts on DCs to suppress IL-12 production and to diminish the intensity of TCR-mediated signals that naive CD4+ T cells receive, which indicates that manipulation of DC function may result in weak TCR signals even if the antigen amount is not limiting and thus favor in vivo TH2 differentiation38,39.
Indeed, although IL-4 is essential for the differentiation of naive CD4+ T cells into TH2 cells in vitro14,40, its requirement for in vivo TH2 differentiation has been a matter of intense debate, given the fact that mice deficient in either IL-4 or STAT6 show normal TH2 differentiation in vivo in response to infection with Nippostrongylus brasilienis or Trichuris muris41,42. In contrast, the deletion of Gata3 specifically in activated T cells results in the failure to undergo TH2 differentiation and instead results in the appearance of IFN-γ-producing CD4+ T cells after infection with N. brasilienis43, which indicates an indispensable role for GATA-3 in TH2 development in vivo. Future investigations should focus on clarifying the mechanisms by which helminth infection regulates the induction of GATA-3 and suppression of transcription factors for differentiation into other helper T cell subsets during the early stages of the activation of naive CD4+ T cells. It will also be of particular interest to identify helminth-derived substances similar to S. mansoni egg antigen–derived omega-1 that may be responsible for lowering TCR-mediated signals.
Treg cell development and function: role of IL-2, IL-7 and IL-15
Most thymocytes that express TCRs that recognize self peptide in the context of MHC molecules with very low or high affinity are eliminated by failure of positive selection or by negative selection in the thymus, respectively. However, some cells with high-affinity TCRs escape the negative selection process and thus are potentially able to mediate destructive autoimmunity. To prevent such autoreactive T cells from being activated in the periphery, the immune system has evolved several means, one of which involves Treg cells, a specialized subset of CD4+ T cells with high expression of CD25 and the master regulatory transcription factor Foxp3 (ref. 44).
Similar to autoreactive T cells, thymus-derived Treg cells, often called ‘natural Treg cells’ (nTreg cells), express a TCR with higher affinity for self peptide–MHC than that of TCRs on the bulk of conventional CD4+ T cells, which indicates that nTreg cells escape clonal deletion by negative selection during thymic development45. It has been proposed that there are two steps in the development of nTreg cells, as follows: a fraction of immature CD4+ single-positive thymocytes with self-reactive TCRs of sufficiently high affinity gives rise to Foxp3−CD25+ Treg precursor cells that then give rise to cells that express Foxp3 in response to cytokines of the γc family without further activation through the TCR46. Indeed, mice deficient in IL-2, CD25 or IL-2Rβ have 50% fewer Foxp3+ thymocytes as wild-type mice have47–49. Although the loss of IL-7 or IL-15 alone does not perturb the generation of thymic Foxp3+ cells, the combined elimination of IL-2, IL-7 and IL-15 leads to complete abrogation of Foxp3+ thymocytes, which indicates a substantial role for IL-2 and compensatory roles for IL-7 and IL-15 in nTreg cell development50,51.
Whether transforming growth factor (TGF-β) is required for the thymic development of nTreg cells remains controversial52,53. However, TGF-β is essential for the differentiation of peripheral naive CD4+ T cells into Foxp3+ cells with regulatory ability, called ‘induced Treg cells’ (iTreg cells)54. IL-2 is required for TGF-β-mediated iTreg cell differentiation in vitro55. Interestingly, the vitamin A metabolite retinoic acid promotes TGF-β-mediated induction of Foxp3 expression in CD4+ T cells in vitro independently of the IL-2–STAT5 pathway56. Moreover, retinoic acid derived from CD103+ DCs in the mesenteric lymph nodes and the lamina propria of the small intestine has an essential role in the conversion of naive CD4+ T cells into Foxp3+ T cells with suppressive activity57–59. However, the proposal of a role for retinoic acid in generating iTreg cells in the gut has been challenged by the observation that retinoic acid does not act as a cofactor for TGF-β-mediated induction of Foxp3 but is required for optimal activation of the TCR-proximal signaling cascades in CD4+ T cells60.
How nTreg cells suppress the immune responses of conventional T cells has been of great interest. It has been shown that nTreg cells abrogate the induction of Il2 mRNA expression in CD4+CD25− responder cells in vitro without affecting the initial activation of responder T cells, and that nTreg cells require IL-2 for their ability to block Il2 transcription in the target T cells61. Given that nTreg cells do not produce IL-2 in response to stimulation via the TCR, the source of IL-2 required for nTreg cells to demonstrate suppressor activity may well be the conventional CD4+ T cells that will be the eventual targets of the activated Treg cells. Interestingly, detailed kinetic analysis by IL-2-capture assay has shown that nTreg cells do not begin to suppress IL-2 production by CD4+CD25− responder cells until 6 hours of coculture, so nTreg cells can receive IL-2 signals until then to activate their suppressor function62. However, that model has been challenged by the observation that under certain circumstances, nTreg cells do not abolish IL-2 production by CD4+CD25− responder cells but instead compete for IL-2 and for other cytokines that are essential for T cell survival in vivo, which leads to apoptosis of the responders owing to cytokine deprivation63. Overexpression of the antiapoptotic protein Bcl-2 or loss of the proapoptotic protein Bim in CD4+CD25− responder cells renders these cells resistant to nTreg cell–mediated suppression61. Further investigation of these contradictory findings in terms of ‘abrogated IL-2 production’ versus ‘cytokine deprivation’ will provide better understanding of the mechanism through which nTreg cells suppress the immunological responses of CD4+CD25− T cells.
Generation of TH17 and follicular helper T cells: role of IL-21
TH17 cells are important in protection from bacterial and fungal infection and in the development of autoimmunity. It has been proposed that the following three steps control the differentiation of naive CD4+ T cells into TH17 cells: differentiation induced by TGF-β and IL-6; IL-21-driven amplification; and IL-23-mediated stabilization64,65. During the differentiation step, TGF-β and IL-6 in combination with stimulation via the TCR cause naive CD4+ T cells to express IL-23R and to induce the TH 17 master regulatory transcription factor RORγt, and to produce IL-17A, IL-17F and IL-21 (refs. 66–69). Induction of IL-21 production depends on IL-6-driven activation of STAT3 and stimulation via the inducible costimulator ICOS70,71. During the amplification step, IL-21 acts together with TGF-β to further upregulate IL-17 production and IL-23R expression in a STAT3-dependent manner. Genetic loss of either IL-21 or its receptor IL-21R results in less TH17 differentiation both in vitro and in vivo70,72,73. If mice are depleted of Treg cells by treatment with antibody to CD25 before immunization, IL-21 is able to induce TH17 responses in vivo even in the absence of IL-6, although to a lesser degree than if IL-6 is available. The cellular source of IL-21 in this setting remains unclear72. Thus, IL-21 seems to have an important role in the positive feedback regulation of TH17 differentiation through its activation of STAT3. However, that conclusion has been challenged by a study reporting that mice deficient in either IL-21 or IL-21R are still able to mount TH17 responses in vivo, which indicates a dispensable role for IL-21 in TH17 differentiation when proinflammatory cytokines such as IL-6, IL-1 and TNF are abundantly available74.
Follicular helper T cells (TFH cells) promote T cell–dependent humoral immune responses by providing T cell help to B cells and thereby promote the formation of germinal centers, affinity maturation of antibody-secreting B cells and long-lived antibody responses75. TFH cells express the transcriptional repressor Bcl-6 as their master regulator76–78 and the CXC chemokine receptor CXCR5 (refs. 79,80) and have high expression of the costimulatory molecules ICOS79,80, PD-1 (ref. 81) and BTLA82, as well as the signaling adaptor molecule SAP83, but downregulate their expression of the transcription factor Blimp-1 (ref. 77). It is still a point of considerable controversy whether TFH cells originate directly from naive CD4+ T cells as a distinct subset, similar to TH1, TH2, TH17 and iTreg cells, or whether TFH cells emerge from CD4+ T cells that have adopted a TH1, TH2 or TH17 cell fate. One key issue about the acquisition of TFH identity is clarification of the timing and mechanism by which responding CD4+ T cells acquire expression of Bcl-6 and CXCR5. CXCR5 expression is essential for the migration of activated CD4+ T cells to the follicles, where they interact with B cells that express the same cognate peptide presented by DCs to undergo maturation into functional TFH cells.
Mice with T cell-specific deletion of Stat3 have a much lower frequency of CD4+CXCR5+ cells that arise in vivo in response to immunization with keyhole limpet hemocyanin in complete Freund's adjuvant, a phenotype that resembles that seen in deficiency in IL-6 or IL-21 (ref. 84). In contrast, mice deficient in IL-6 or IL-21 generate TFH cells normally after infection with lymphocytic choriomeningitis virus (LCMV), which indicates a redundant role for these cytokines in the development of TFH cells85. Although the combined absence of IL-6 and IL-21 results in a failure to secrete antigen-specific immunoglobulin G after infection with LCMV, such mice have only slightly less generation of TFH cells than that of their wild-type counterparts86. As the dependence of LCMV-generated TFH cells on STAT3 has not been determined, it is uncertain whether IL-6 and IL-21 may be replaced by another activator of STAT3 in this case or whether a STAT3-independent pathway exists for the generation of TFH cells.
TH17 and TFH differentiation: role of IL-2
The IL-2–STAT5 pathway has been demonstrated to block TH17 differentiation. When IL-2 is exogenously provided to TH17-polarizing cultures, the generation of IL-17-producing cells is impaired, whereas there is an increase in the frequency of Foxp3+ cells87. Naive CD4+ T cells from mice of the OT-II strain (with transgenic expression of an MHC class II–restricted, ovalbumin-specific TCR) that are deficient in recombination-activating gene 1 and are on the scurfy background, which lack functional Foxp3, still fail to undergo TH17 differentiation under TH17-polarizing conditions in the presence of exogenous IL-2 (ref. 88). STAT5 competes with STAT3 for binding to multiple sites in the Il17a-Il17f locus; binding of STAT5 to these sites is associated with repressive epigenetic marks across the Il17a promoter region and enhancer elements88, which suggests a mechanism through which IL-2 directly represses TH17 differentiation (Fig. 3).
Figure 3.

A model for early determination of iTreg fate versus TH17 fate controlled by TCR signal strength. During early TH17 differentiation phase, naive CD4+ T cells require a combination of the cytokines IL-6 and TGF-β and costimulation (including ICOS), as well as strong TCR signals, to induce the expression of RORγt and TH17 cytokines (right). When naive CD4+ T cells receive weak TCR signals under TH17-polarizing conditions, the differentiation of Foxp3-expressing iTreg cells is favored (left). Although weak TCR signals induce only small amounts of IL-2 production and IL-2R expression, IL-2-mediated STAT5 activation blocks IL-17A production and induces Foxp3 expression, which suppresses the induction of RORγt and thereby favors iTreg differentiation. In contrast, despite abundant IL-2 production and IL-2R expression, transient inhibition of STAT5 activation by strong TCR signals leads to the failure to induce Foxp3 but allows expression of genes encoding RORγt and TH17 cytokines and thereby favors TH17 differentiation. Smad, signal-transducer protein(s) downstream of the receptor for TGF-β (TGF-βR).
Two days after infection with LCMV, T cells from SMARTA mice (which have transgenic expression of a TCR specific for LCMV glycoprotein) CD4+ develop into the following two subpopulations: CD25hi cells, which have high expression of Blimp-1 and undergo a program of differentiation into effector T cells (Teff cells); and CD25int cells, which express Bcl-6 and CXCR5 and undergo differentiation into TFH cells89. The failure of CD25hi cells to become TFH cells suggests an inhibitory role for in vivo IL-2-generated signals in the development of TFH cells. In addition, the IL-2–STAT5 pathway blocks the generation of TFH cells by inducing Blimp-1, which results in the suppression of Bcl-6 expression90–93 (Fig. 4). Interestingly, the crucial role of the IL-2–STAT5 pathway in the fate ‘decisions’ to develop into either Teff cells or TFH cells through regulation of the expression of the two mutually exclusive transcriptional regulators Blimp-1 and Bcl-6 resembles that observed for the differentiation of iTreg cells and TH17 cells, in which Foxp3 and RORγt reciprocally regulate each other (Figs. 3 and 4).
Figure 4.

A model for early determination of Teff cell fate versus TFH cell fate controlled by TCR signal strength. During the early phase of differentiation into the TFH cell subset, naive CD4+ T cells require the cytokine IL-6 and costimulation (including ICOS), as well as strong TCR signals, to induce the expression of Bcl-6 (right). Although strong TCR signals induce abundant production of IL-2 and expression of the IL-2R complex, transient inhibition of STAT5 activation, presumably through the action of the Erk pathway, leads to the failure to express Blimp-1 and allows Bcl-6 expression induced by IL-6 and stimulation via ICOS and thereby results in differentiation into the TFH cell subset. In contrast, T cells that have received weak TCR signals under TFH cell–polarizing conditions can activate STAT5 in response to small amounts of IL-2, which induces Blimp-1 expression and suppresses Bcl-6 expression and thereby results in ‘preferential’ differentiation into the Teff cell subset (left).
In addition to the mutual regulation of helper T cell differentiation as a result of the induction of distinctive transcription factors, TCR signal strength is also a key element in determining fate ‘decisions’ to develop into iTreg cells or TH17 cells, as well as Teff cells or TFH cells. If naive CD4+ T cells receive weak TCR signals, they do not differentiate into TH17 cells and express Foxp3 instead, even though they are exposed to TH17-inducing cytokines94–96. Furthermore, the generation and function of TFH cells depends on the strength of the binding of the TCR to a foreign peptide–MHC class II complex97. Notably, strong TCR signals transiently inhibit IL-2-driven activation of STAT5, despite abundant production of IL-2 by activated CD4+ T cells, which thereby blocks TH2 development29,98. Thus, it is conceivable that the generation of TH17 cells and TFH cells may require strong TCR signals to block the IL-2–STAT5 pathway so that the inhibitory effects of IL-2 can be abrogated (Figs. 3 and 4). Clarifying the molecular basis that underlies TCR signal strength–mediated control of the IL-2–STAT5 pathway may provide better understanding of the delicate balance between helper T cell subset fates that are reciprocally regulated.
Concluding remarks
Cytokines of the γc family have crucial roles in the fate ‘decisions’ of naive CD4+ T cells to differentiate into distinct helper T cell subsets and in the function of these effector helper T cells. We have emphasized here the roles of cytokines of the γc family in this process through the activation of STAT proteins and the genes targeted by activated STAT proteins, particularly the so-called master regulatory transcription factors. Cytokines of the γc family signal through other signaling pathways as well, including activation of the metabolic checkpoint kinase mTOR. The two mTOR pathways, mTORC1 and mTORC2, have a unique function in influencing the fate of cells developing into distinct helper T cell subsets99,100. Whether these differences can be accounted for by the action of the cytokines that determine the ‘choice’ of helper T cell phenotype is not yet clear but is an important area for continued study.
Comprehensive analysis of the cytokine dependence of in vivo helper T cell differentiation as well as the regulation of the change in phenotype of differentiated cells (‘plasticity’) is of great potential importance because this may provide opportunities for the development of drugs that can ensure that appropriate responses are mounted to given challenges and, possibly, to alter ‘inappropriate’ responses.
Acknowledgments
Supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases (US National Institutes of Health).
Footnotes
Competing Financial Interests: The authors declare no competing financial interests.
References
- 1.Bazan JF. Structural design and molecular evolution of a cytokine receptor superfamily. Proc Natl Acad Sci USA. 1990;87:6934–6938. doi: 10.1073/pnas.87.18.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by γc family cytokines. Nat Rev Immunol. 2009;9:480–490. doi: 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X, Lupardus P, Laporte SL, Garcia KC. Structural biology of shared cytokine receptors. Annu Rev Immunol. 2009;27:29–60. doi: 10.1146/annurev.immunol.24.021605.090616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dubois S, Mariner J, Waldmann TA, Tagaya Y. IL-15Rα recycles and presents IL-15 in trans to neighboring cells. Immunity. 2002;17:537–547. doi: 10.1016/s1074-7613(02)00429-6. [DOI] [PubMed] [Google Scholar]
- 5.Rautajoki KJ, Kylaniemi MK, Raghav SK, Rao K, Lahesmaa R. An insight into molecular mechanisms of human T helper cell differentiation. Ann Med. 2008;40:322–335. doi: 10.1080/07853890802068582. [DOI] [PubMed] [Google Scholar]
- 6.Fry TJ, Mackall CL. The many faces of IL-7: from lymphopoiesis to peripheral T cell maintenance. J Immunol. 2005;174:6571–6576. doi: 10.4049/jimmunol.174.11.6571. [DOI] [PubMed] [Google Scholar]
- 7.Singer A, Adoro S, Park JH. Lineage fate and intense debate: myths, models and mechanisms of CD4- versus CD8-lineage choice. Nat Rev Immunol. 2008;8:788–801. doi: 10.1038/nri2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vicente R, et al. Molecular and cellular basis of T cell lineage commitment. Semin Immunol. 2010;22:270–275. doi: 10.1016/j.smim.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi M, Lin TH, Appell KC, Berg LJ. Janus-kinase-3-dependent signals induce chromatin remodeling at the Ifng locus during T helper 1 cell differentiation. Immunity. 2008;28:763–773. doi: 10.1016/j.immuni.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang JT, Segal BM, Shevach EM. Role of costimulation in the induction of the IL-12/IL-12 receptor pathway and the development of autoimmunity. J Immunol. 2000;164:100–106. doi: 10.4049/jimmunol.164.1.100. [DOI] [PubMed] [Google Scholar]
- 12.Liao W, Lin JX, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat Immunol. 2011;12:551–559. doi: 10.1038/ni.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 14.Le Gros G, Ben-Sasson SZ, Seder R, Finkelman FD, Paul WE. Generation of interleukin 4 (IL-4)-producing cells in vivo and in vitro: IL-2 and IL-4 are required for in vitro generation of IL-4-producing cells. J Exp Med. 1990;172:921–929. doi: 10.1084/jem.172.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cote-Sierra J, et al. Interleukin 2 plays a central role in Th2 differentiation. Proc Natl Acad Sci USA. 2004;101:3880–3885. doi: 10.1073/pnas.0400339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 activation plays a critical role in Th2 differentiation. Immunity. 2003;19:739–748. doi: 10.1016/s1074-7613(03)00292-9. [DOI] [PubMed] [Google Scholar]
- 17.Liao W, et al. Priming for T helper type 2 differentiation by interleukin 2-mediated induction of interleukin 4 receptor α-chain expression. Nat Immunol. 2008;11:1288–1296. doi: 10.1038/ni.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamane H, Paul WE. Memory CD4+ T cells: fate determination, positive feedback and plasticity. Cell Mol Life Sci. 2012;69:1577–1583. doi: 10.1007/s00018-012-0966-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perrigoue JG, et al. MHC class II-dependent basophil-CD4+ T cell interactions promote TH2 cytokine-dependent immunity. Nat Immunol. 2009;10:697–705. doi: 10.1038/ni.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshimoto T, et al. Basophils contribute to TH2-IgE responses in vivo via IL-4 production and presentation of peptide-MHC class II complexes to CD4+ T cells. Nat Immunol. 2009;10:706–712. doi: 10.1038/ni.1737. [DOI] [PubMed] [Google Scholar]
- 21.Sokol CL, et al. Basophils function as antigen-presenting cells for an allergen-induced T helper type 2 response. Nat Immunol. 2009;10:713–720. doi: 10.1038/ni.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hammad H, et al. Inflammatory dendritic cells–not basophils–are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J Exp Med. 2010;207:2097–2111. doi: 10.1084/jem.20101563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sullivan BM, et al. Genetic analysis of basophil function in vivo. Nat Immunol. 2011;12:527–535. doi: 10.1038/ni.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parish CR, Liew FY. Immune response to chemically modified flagellin. 3. Enhanced cell-mediated immunity during high and low zone antibody tolerance to flagellin. J Exp Med. 1972;135:298–311. doi: 10.1084/jem.135.2.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Constant SL, Bottomly K. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu Rev Immunol. 1997;15:297–322. doi: 10.1146/annurev.immunol.15.1.297. [DOI] [PubMed] [Google Scholar]
- 26.Aguado E, et al. Induction of T helper type 2 immunity by a point mutation in the LAT adaptor. Science. 2002;296:2036–2040. doi: 10.1126/science.1069057. [DOI] [PubMed] [Google Scholar]
- 27.Sommers CL, et al. A LAT mutation that inhibits T cell development yet induces lymphoproliferation. Science. 2002;296:2040–2043. doi: 10.1126/science.1069066. [DOI] [PubMed] [Google Scholar]
- 28.Altin JA, et al. Decreased T-cell receptor signaling through CARD11 differentially compromises forkhead box protein 3-positive regulatory versus TH2 effector cells to cause allergy. J Allergy Clin Immunol. 2011;127:1277–1285. doi: 10.1016/j.jaci.2010.12.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamane H, Zhu J, Paul WE. Independent roles for IL-2 and GATA-3 in stimulating naive CD4+ T cells to generate a Th2-inducing cytokine environment. J Exp Med. 2005;202:793–804. doi: 10.1084/jem.20051304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das J, et al. A critical role for NF-κB in Gata3 expression and TH2 differentiation in allergic airway infammation. Nat Immunol. 2001;2:45–50. doi: 10.1038/83158. [DOI] [PubMed] [Google Scholar]
- 31.Corn RA, Hunter C, Liou HC, Siebenlist U, Boothby MR. Opposing roles for RelB and Bcl-3 in regulation of T-box expressed in T cells, GATA-3, and Th effector differentiation. J Immunol. 2005;175:2102–2010. doi: 10.4049/jimmunol.175.4.2102. [DOI] [PubMed] [Google Scholar]
- 32.Amsen D, et al. Direct regulation of Gata3 expression determines the T helper differentiation potential of Notch. Immunity. 2007;27:89–99. doi: 10.1016/j.immuni.2007.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fang TC, et al. Notch directly regulates Gata3 expression during T helper 2 cell differentiation. Immunity. 2007;27:100–110. doi: 10.1016/j.immuni.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu Q, et al. T cell factor 1 initiates the T helper type 2 fate by inducing the transcription factor GATA-3 and repressing interferon-γ. Nat Immunol. 2009;10:992–999. doi: 10.1038/ni.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schulze-Luehrmann J, Ghosh S. Antigen-receptor signaling to nuclear factor κB. Immunity. 2006;25:701–715. doi: 10.1016/j.immuni.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 36.Ioannidis V, Beermann F, Clevers H, Held W. The β-catenin-TCF-1 pathway ensures CD4+CD8+ thymocyte survival. Nat Immunol. 2001;2:691–697. doi: 10.1038/90623. [DOI] [PubMed] [Google Scholar]
- 37.Ong CT, Sedy JR, Murphy KM, Kopan R. Notch and presenilin regulate cellular expansion and cytokine secretion but cannot instruct Th1/Th2 fate acquisition. PLoS ONE. 2008;3:e2823. doi: 10.1371/journal.pone.0002823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Everts B, et al. Omega-1, a glycoprotein secreted by Schistosoma mansoni eggs, drives Th2 responses. J Exp Med. 2009;206:1673–1680. doi: 10.1084/jem.20082460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steinfelder S, et al. The major component in schistosome eggs responsible for conditioning dendritic cells for Th2 polarization is a T2 ribonuclease (omega-1) J Exp Med. 2009;206:1681–1690. doi: 10.1084/jem.20082462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swain SL, Weinberg AD, English M, Huston G. IL-4 directs the development of Th2-like helper effectors. J Immunol. 1990;145:3796–3806. [PubMed] [Google Scholar]
- 41.van Panhuys N, et al. In vivo studies fail to reveal a role for IL-4 or STAT6 signaling in Th2 lymphocyte differentiation. Proc Natl Acad Sci USA. 2008;105:12423–12428. doi: 10.1073/pnas.0806372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paul WE, Zhu J. How are TH2-type immune responses initiated and amplifed? Nat Rev Immunol. 2010;10:225–235. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu J, et al. Conditional deletion of Gata3 shows its essential function in TH1-TH2 responses. Nat Immunol. 2004;5:1157–1165. doi: 10.1038/ni1128. [DOI] [PubMed] [Google Scholar]
- 44.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 45.Hinterberger M, et al. Autonomous role of medullary thymic epithelial cells in central CD4+ T cell tolerance. Nat Immunol. 2010;6:512–519. doi: 10.1038/ni.1874. [DOI] [PubMed] [Google Scholar]
- 46.Lio CW, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity. 2008;28:100–111. doi: 10.1016/j.immuni.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burchill MA, et al. Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity. 2008;28:112–121. doi: 10.1016/j.immuni.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 49.Yu A, Zhu L, Altman NH, Malek TR. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity. 2009;30:204–217. doi: 10.1016/j.immuni.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor β-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178:280–290. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- 51.Vang KB, et al. IL-2, -7, and -15, but not thymic stromal lymphopoeitin, redundantly govern CD4+Foxp3+ regulatory T cell development. J Immunol. 2007;181:3285–3290. doi: 10.4049/jimmunol.181.5.3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Y, et al. A critical function for TGF-β signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. 2008;9:632–640. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- 53.Ouyang W, Beckett O, Ma Q, Li MO. Transforming growth factor-β signaling curbs thymic negative selection promoting regulatory T cell development. Immunity. 2010;32:642–653. doi: 10.1016/j.immuni.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen W, et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting Edge: IL-2 is essential for TGF-β-mediated induction of Foxp3+ T regulatory cells. J Immunol. 2007;178:4022–4026. doi: 10.4049/jimmunol.178.7.4022. [DOI] [PubMed] [Google Scholar]
- 56.Elias KM, et al. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111:1013–1020. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Coombes JL, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced Treg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007;204:1765–1774. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun CM, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hall JA, et al. Essential role for retinoic acid in the promotion of CD4+ T cell effector responses via retinoic acid receptor α. Immunity. 2011;34:435–447. doi: 10.1016/j.immuni.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thornton AM, Donovan EE, Piccirillo CA, Shevach EM. Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J Immunol. 2004;172:6519–6523. doi: 10.4049/jimmunol.172.11.6519. [DOI] [PubMed] [Google Scholar]
- 62.Sojka DK, Hughson A, Sukiennicki TL, Fowell DJ. Early kinetic window of target T cell susceptibility to CD25+ regulatory T cell activity. J Immunol. 2005;175:7274–7280. doi: 10.4049/jimmunol.175.11.7274. [DOI] [PubMed] [Google Scholar]
- 63.Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8:1353–1362. doi: 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- 64.Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of TH17 cells. Nature. 2008;453:1051–1057. doi: 10.1038/nature07036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–348. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 66.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 67.Mangan PR, et al. Transforming growth factor-β induces development of the TH17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 68.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 69.Ivanov II, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 70.Nurieva R, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 71.Bauquet AT, et al. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat Immunol. 2009;10:167–175. doi: 10.1038/ni.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Korn T, et al. IL-21 initiates an alternative pathway to induce proinflammatory TH17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou L, et al. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2008;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 74.Sonderegger I, Kisielow J, Meier R, King C, Kopf M. IL-21 and IL-21R are not required for development of Th17 cells and autoimmunity in vivo. Eur J Immunol. 2008;38:1833–1838. doi: 10.1002/eji.200838511. [DOI] [PubMed] [Google Scholar]
- 75.Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 76.Nurieva RI, et al. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325:1001–1005. doi: 10.1126/science.1176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Johnston RJ, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cells. Science. 2009;325:1006–1010. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu D, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity. 2009;31:457–468. doi: 10.1016/j.immuni.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 79.Breitfeld D, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med. 2000;192:1545–1552. doi: 10.1084/jem.192.11.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schaerli P, et al. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. 2000;192:1553–1562. doi: 10.1084/jem.192.11.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Haynes NM, et al. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol. 2007;179:5099–5108. doi: 10.4049/jimmunol.179.8.5099. [DOI] [PubMed] [Google Scholar]
- 82.M'Hidi H, et al. High expression of the inhibitory receptor BTLA in T-follicular helper cells and in B-cell small lymphocytic lymphoma/chronic lymphocytic leukemia. Am J Clin Pathol. 2009;132:589–596. doi: 10.1309/AJCPPHKGYYGGL39C. [DOI] [PubMed] [Google Scholar]
- 83.Yusuf I, et al. Germinal center T follicular helper cell IL-4 production is dependent on signaling lymphocytic activation molecule receptor (CD150) J Immunol. 2010;185:190–202. doi: 10.4049/jimmunol.0903505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nurieva RI, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29:138–149. doi: 10.1016/j.immuni.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Poholek AC, et al. In vivo regulation of Bcl6 and T follicular helper cell development. J Immunol. 2010;185:313–326. doi: 10.4049/jimmunol.0904023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Eto D, et al. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PLoS ONE. 2011;6:e17739. doi: 10.1371/journal.pone.0017739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Laurence A, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 88.Yang XP, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Choi YS, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34:932–946. doi: 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Oestreich KJ, Mohn SE, Weinmann AS. Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profle. Nat Immunol. 2012;13:405–411. doi: 10.1038/ni.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ballesteros-Tato A, et al. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity. 2012;36:847–856. doi: 10.1016/j.immuni.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med. 2012;209:243–250. doi: 10.1084/jem.20111174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nurieva RI, et al. STAT5 protein negatively regulates T follicular hlper (Tfh) cell generation and function. J Biol Chem. 2012;287:11234–11239. doi: 10.1074/jbc.M111.324046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Turner MS, Kane LP, Morel PA. Dominant role of antigen dose in CD4+Foxp3+ regulatory T cell induction and expansion. J Immunol. 2009;183:4895–4903. doi: 10.4049/jimmunol.0901459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Iezzi G, et al. CD40–CD40L cross-talk integrates strong antigenic signals and microbial stimuli to induce development of IL-17-producing CD4+ T cells. Proc Natl Acad Sci USA. 2009;106:876–881. doi: 10.1073/pnas.0810769106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Molinero LL, Miller ML, Evaristo C, Alegre ML. High TCR stimuli prevent induced regulatory T cell differentiation in a NF-κB-dependent manner. J Immunol. 2011;186:4609–4617. doi: 10.4049/jimmunol.1002361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol. 2009;10:375–384. doi: 10.1038/ni.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee IH, Li WP, Hisert KB, Ivashkiv LB. Inhibition of interleukin 2 signaling and signal transducer and activator of transcription (STAT)5 activation during T cell receptor-mediated feedback inhibition of T cell expansion. J Exp Med. 1999;190:1263–1274. doi: 10.1084/jem.190.9.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol. 2012;30:39–68. doi: 10.1146/annurev-immunol-020711-075024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. 2012;12:325–338. doi: 10.1038/nri3198. [DOI] [PMC free article] [PubMed] [Google Scholar]