

Abstract
Purpose
Measurement of autophagy in cancer and correlation with histopathologic grading or clinical outcomes has been limited. Accordingly, we investigated LC3B as an autophagosome marker by analyzing nearly 1400 tumors from 20 types of cancer, focussing on correlations with clinical outcomes in melanoma and breast cancer.
Experimental Design
Staining protocols were developed for automated quantitative analysis (AQUA) using antibodies vs LC3 isoform B (LC3B) and Ki-67. Clinically annotated breast and melanoma TMA’s and a multitumor array were employed. An AQUA program was developed to quantitate LC3B distribution in punctate and diffuse compartments of the cell.
Results
LC3B staining was moderate to high in the large majority of tumors. The % area occupied by punctate LC3B was elevated 3–5 fold at high LC3B intensities. In breast cancer and melanoma TMAs, LC3B and Ki-67 showed strong correlations (p < 0.0001) and in multitumor TMAs mitotic figures were most often seen in tumors with highest LC3B expression (p < 0.002). In breast cancer, LC3B expression was elevated in node-positive vs node-negative primaries and associated with increased nuclear grade and shortened survival. In a melanoma TMA with no survival data, LC3B levels were highest in nodal, visceral and cutaneous metastases.
Conclusions
The results reveal a common expression of LC3B in malignancy and support emerging evidence that autophagy plays a significant role in cancer progression. High LC3B was associated proliferation, invasion and metastasis, high nuclear grade and worse outcome. Thus autophagy presents a key target of therapeutic vulnerability in solid tumors.
INTRODUCTION
Preclinical studies have established autophagy as a therapeutic target in cancer and several clinical trials are underway combining autophagy inhibitors with standard cytotoxic and targeted anticancer therapies (1). Autophagy is part of a conserved integrated stress response whereby in times of nutrient deprivation, hypoxia and DNA damage, or endoplasmic reticulum stress, a cell’s own cytoplasmic components --damaged organelles, misfolded proteins--are engulfed into autophagosomes and degraded following fusion of the autophagosomes with lysosomes to form autophagolysosomes. Recycling of degraded cellular components from autophagolysosomes provides a source of amino acids, nucleotides, and lipids for ATP production and macromolecular synthesis (2). Autophagy plays dual roles in cancer. On the one hand it acts as a suppressor of tumor development (3), and can induce senescence (4). On the other hand, once a tumor is established, autophagy is a cancer cell survival factor (5,6). For example, autophagy can confer resistance to cytotoxic therapeutics (7–8). Indeed, malignant cells bearing activating mutations in Ras are autophagy-addicted, requiring active autophagy for survival (9). Numerous preclinical studies have demonstarted that genetic or pharmacological autophagy inhibition can enhance the antitumor efficacy of variety of anticancer therapies (7–14).
While there is growing interest in clinical trials targeting autophagy in cancer therapy, the clinical implications of elevated or suppresssed autophagy at different stages and in different malignancies is as yet unknown. A critical shortcoming in the field is the lack of immunohistochemical tools that can be applied to traditional FFPE tissue. Here, we analyzed autophagy in nearly 1400 tumor specimens from 20 different types of cancer with a major focus on melanoma and breast cancer. As a marker, we used punctate and diffuse staining of a key autophagosomal modulating protein, LC3 (microtubule-associated protein 1, light chain 3). We employed automated quantitative analyses (AQUA) and direct observer scoring to assess LC3 expression, cellular distribution between punctate and diffuse compartments, and associations with tumor progression. AQUA is linked to a fluorescent microscope system that detects the expression of biomarker proteins by measuring the intensity of antibody-conjugated fluorophores within a specified subcellular compartment (typically including the nucleus, cytoplasm, and plasma membrane) within the tumor region of each tissue microarray spot. The result is a quantitative score of immunofluorescence intensity for the tumor (15–17).
LC3 is first synthesized as pro-LC3 that is cleaved by cysteine protease Atg4B to its cytosolic form, LC3-I. During active autophagy, LC3-I is conjugated to phosphatidylethanolamine where it resides in the lipid membrane of nascent autophagosomes as LC3-II (1). LC3-II migrates separately from LC3-I on gel electrophoresis (18). LC3-II is localized to the cytosolic and luminal surfaces of mature, double-membraned autophagic vessicles (AV’s) and modulates AV growth and cargo recruitment. Some proteins recruited by LC3-II are themselves cargo adaptor proteins (autophagy receptors) and include p62, Nbr1, and NIX (1). During autophagic flux, autophagosomes fuse with lysosomes to form autolysosomes and the contents are rapidly degraded with lysosomal hydrolases. As this proceeds, cytoplasmic surface LC3-II is delipidated to LC3-I and recycled into the cytosol while luminal LC3-II is degraded by hydrolases. Here we used an antibody that was generated against the N-terminus of full length human LC3B sequence to investigate LC3 expression and cellular distribution in FFPE specimens of human cancers by immunohistochemistry and AQUA. In mammals, LC3 is expressed as three isoforms, A, B, and C. We used the B isoform, LC3B, due to its broad tissue specificities and previous use as an autophagosome marker in cancer (19–21). By a number of approaches we show herein that LC3B expression is remarkably common in a variety of metastatic human cancers. We chose to investigate the correlation between LC3B staining and lymph node metastases and clinical outcome in melanoma a cancer reported to have high levels of autophagy (22,23) and breast cancer, a cancer that is commonly referred to as autophagy deficient due monoallelic loss of the autophagy gene beclin (24). Here we find that LC3B is strongly associated with proliferation, metastasis and at least in breast cancer, worse patient outcome.
MATERIALS AND METHODS
Immunohistochemistry
LC3B (microtuble-associated protein light chain 3, isoform B) was detected using rabbit anti-LC3B polyclonal antibody ab48394 (Abcam, Inc., Santa Cruz, CA) generated versus a synthetic peptide corresponding to an N-terminal portion of the human LC3B protein sequence. The LC3B antibody recognizes human LC3B I and II as confirmed by Western blotting. Ki-67 was detected using mouse anti-human Ki-67 antibody 556026 (BD Biosciences, San Jose, CA). Sections from formalin-fixed, paraffin-embedded tissue were deparaffinized and rehydrated by standard procedures. For direct observer scoring via light microscopy, slides were stained with anti-LC3B (1:500, without citric acid/antigen retrieval) by standard immunoperoxidase techniques using the brown chromogen generated from diaminobenzidine, and counter-stained with hematoxylin.
AQUA (automated quantitative analysis)
Fluorescence-conjugated antibodies were employed as previously described (15–17). For detection of LC3B, slides were incubated with anti-LC3B (1:5000, overnight at 4° C) with no citric acid/antigen retrieval. Similar procedures were used to detect Ki-67 through immunofluorescence except that citric acid antigen retrieval was used prior to staining (22). The antibodies were visualized using a species-specific horseradish-peroxidase-conjugated secondary antibody (rabbit Envision, Dako) followed by Cy5 tyramide. The LC3B or Ki-67 signals were read within a tumor mask created using mouse-anti-cytokeratin (1:100 AE1/AE3, Dako, Carpinteria, CA) for breast cancer, or a cocktail of anti-S100 (1:100, Biogenex, Fremont CA) and anti-HMB45 for melanoma (1:25, Abcam, Cambridge, MA). The tumor mask antibodies were incubated along with the anti-LC3B and visualized with Cy3-goat-anti-mouse IgG (1:100, Jackson Immunoresearch, West Grove, PA).
Light microscope scoring of microarrays
Multitumor microarray YTMA96 from the Yale Critical Technologies Group were stained by immunohistochemistry for LC3B using the immunoperoxidase reaction coupled with a brown chromogen. Individual sections were evaluated under the light microscope by two independent observers (RL and JP). Staining intensities were scored and pooled into two groups of 0/1+ (none-weak) and 2+/3+ (moderate to strong).
Nuclear Grade
Nuclear grade information for the breast carcinoma TMA was obtained from the pathology reports of the individual cases. Nuclear grade information was not available for the melanoma or multitumor TMAs.
Assessment of LC3B cellular distribution through AQUA
The relative distribution of LC3B in punctate (granular/vesicular) and diffuse (cytosolic) compartments of the cytoplasm were assessed within antibody masks as described above (S100 for melanoma, anti-cytokeratin for breast cancer). The local LC3B immunofluorescence intensity was averaged for each pixel based on the surrounding 15-pixels (~2μ2). Pixels with an LC3B expression level above the local mean were designated as “punctate” and the remaining pixels as “diffuse”. Over-saturated pixels were excluded from the analysis. The expression of LC3B in each designation was then calculated, as was the percentage of pixels assigned to the punctate category. Using small tissue microarrays (TMAs) of melanoma and breast carcinoma with about 40 cases each (Yale Cancer Center Critical Technologies group) the program was validated through color overlays demonstrating coincidence between the algorithm-generated patterns and the actual fluorescence emmisions of LC3B.
Multitumor tissue microarray YTMA96
A multiple tumor microarray, YTMA96, was provided by the Yale Cancer Center Critical Technologies group and used for observer evaluation of immunohistochemical staining with the light microscope. The array consists of the following primary tumors: Invasive ductal carcinoma of the breast (n=20), invasive colon carcinoma (n=20), clear cell carcinoma of the kidney (n=20), invasive and metastatic malignant melanoma (n=20), papillary thyroid carcinoma (n=20), glioblastoma multiforme (n=11), astrocytoma (n=11), non-small cell carcinoma of the lung (n=20), mixed types of ovarian carcinoma (n=20), hepatocellular carcinoma (n=20), endometrial carcinoma of the uterus (n=20), Hodgkin lymphoma (n=10), mixed non-Hodgkin lymphoma (n=10), mixed sarcomas (n=20), pancreatic adenocarcinoma (n=20), germ cell neoplasms of the testis (n=20), prostatic adenocarcinoma (n=20), gastric adenocarcinoma (n=20), transitional cell carcinoma of the bladder (n=20), squamous cell carcinoma of the skin (n=20). In addition, unmatched normal specimens from the following normal tissues were included (n=10 of each type): breast, colon, skin, kidney, brain, thyroid, lung, ovary, liver, endometrium, lymph node, muscle, pancreas, testis, prostate, stomach, bladder. All specimens were taken from deidentified blocks of specimens submitted to Yale surgical pathology. Tumors were of mixed stage and grade.
Breast carcinoma microarray YTMA49
A microarray of human primary breast carcinomas, YTMA49, was provided by the Yale Cancer Center Critical Technologies Group for use in the AQUA analyses of fluorescent staining intensities. The microarray consists of 640 cases of node-negative (n = 321) and node-positive (n = 319) primary tumors. FFPE specimens of breast carcinoma were identified from the archives of the Department of Pathology at Yale University between 1962 and 1980. Complete treatment information was unavailable; however, most of the node-positive patients were treated with local radiation and approximately 15% were given chemotherapy consisting primarily of adriamycin, cytoxan, and 5-fluorouricil. The node-negative patients were routinely treated with surgery and/or local radiation alone. About one-fourth of the patients subsequently received tamoxifen (post-1978). Representative regions of invasive carcinoma were selected for coring by a pathologist (RC). All patients were followed until death or for a minimum of 20 years. Patients were censored if they died of causes other than breast cancer or survived more than 20 years after their initial date of diagnosis. The prognostic utility of YTMA49 was assessed using standard clinicopathologic variables and are presented in the supplemental materials. Correlations between these variables and LC3B expression are also provided.
Melanoma tissue microarray YTMA20
Melanoma array YTMA20, was provided by the Yale Cancer Center Critical Technologies Group for use in the AQUA analyses. This array consists 446 cases of primary (n = 285) and unmatched metastases (n = 161) from the following sites: lymph node (n=86), skin (n=43), visceral organs (n=32). This array was constructed from FFPE specimens taken from the archives of the Yale University, Department of Pathology as available from 1959–1994. A dermatopathologist reviewed slides from all blocks to select representative areas of invasive tumor to be cored. No adjudicated survival data exists for this array, however pathology criteria for the primaries (Breslow depth, Clark levels, tumor infiltrating lymphocytes, and ulceration) and metastases (site) are provided in the supplemental materials.
Statistical analyses
Statistical analyses for AQUA were performed using Statview 5.0.1 (SAS Institute Inc, Cary, NC). Prognostic significance was assessed using multivariate Cox proportional hazards model with 30-year follow-up data. Survival curves were calculated using the Kaplan-Meier method, with significance evaluated using the log-rank test. For the distribution of mitotic figures the p-value was calculated through Pearson’s chi-squared test with Yates’ continuity correction (Supplemental Table S5).
RESULTS
Normal Tissues
While autophagy has been long known to be operational in many disease states in addition to malignancies, less is known about autophagy in healthy tissues and cells. Previous studies in primary cutaneous melanomas from our group found that while most if not all invasive melanomas were positive for LC3B, normal epidermal keratinocytes and melanocytes proximal to the tumor as well as dermal fibroblasts and lymphocytes were negative for LC3B staining. On the other hand, melanoma-associated macrophages showed strong expression of LC3B with abundant autophagosomes (22). In the multitumor array YTMA96, there were also a number of normal tissues included that showed a variety of staining LC3B staining patterns (Supplemental Fig. S1a–f). Many tissues were negative for LC3B as seen in a section of normal brain tissue (Fig. S1a). However renal (Fig. S1b) and pancreatic tissues showed strong LC3B cytoplasmic expression with punctate and diffuse staining in the epithelial lining of tubules and glands. This is consistent with roles for autophagy in the physiology of these organs (25, 26). Stromal fibroblasts and tumor-infiltrating lymphocytes were LCB3-negative (Fig. S1c–e), while tumor-associated macrophages were strongly positive with punctate and diffuse staining recently reported (Fig S1e, f) (22). Thus at least in some cases LC3B expression alone cannot be used to distinguish between normal and malignant tissues, however to avoid this problem tumor-specific antibody masks were used in the breast carcinoma and melanoma AQUA analyses herein (Methods).
Multitumor microarray YTMA96
The multitumor microarray YTMA96, containing invasive or metastatic specimens from 20 different types of human cancers was stained with the anti-LC3B antibody and scored for staining intensity by two independent observers (Methods). Staining generally, but not always, approached homogeneity in the sections. Examples of different cases with strong and weak LC3B staining are shown (Fig. 1). For all tumors, 84 ± 11% (245/292) showed moderate to strong LC3B staining with a range between the different tumor types from 60% to 97% (Supplemental Table S1). The brown LC3B staining was localized predominantly to the cytoplasm while nuclei stained blue from the hematoxylin counterstain. Cases with moderate to strong staining intensities showed prominent punctate patterns while those with weak intensities showed mostly diffuse patterns. Although the tumors were from different tissues of origin, the staining patterns were similar.
Figure 1.

Multitumor array YTMA96: LC3B immunohistochemical staining with a brown chromogen in 16 cases of human cancers from 8 tissue types. Cancer types are arranged in pairs with high and low staining intensities as follows: a–b, astrocytoma; c–d, breast; e–f, colon; g–h, endometrium; i–j, gastric; k–l, liver; m–n, lung; o–p, pancreas; q–r, prostate; s–t, ovarian. Black arrows denote punctate staining; red asterisks denote mitotic figures. Original images were photophraphed through a Zeiss Axioskop 40 light microscope equipped with a Spot Flex digital camera through a 63x lens (total magnification = 630x). All photographs were cut and assembled together into a single PhotoshopTM document, merged into one layer, and then treated with automatic contrast and brightening tools.
LC3B cytoplasmic distribution
Since punctate LC3B staining is a marker for autophagosomes and a potential sign of active autophagy (though not a proof in itself), one of us (RC) devised an AQUA program to quantitate the amounts of LC3B in the punctate and diffuse compartments of the cytoplasm by combining the parameters of flourescence-conjugated LC3B emission intensity and percent cytoplasmic area (Methods). We assessed this in melanoma and breast carcinoma small TMAs using the appropriate masks to assure staining of tumor cells (Methods). For validation of the program we compared the actual LC3B immunofluoresence patterns to those generated by the algorithm (punctate staining, yellow; diffuse staining, blue) (Fig. 2). Shown is a high LC3B-expressing breast carcinoma (Fig. 2A, B) compared to and a low-expressing one (Fig. 2C, D) with immunofluorescence in the panels on the left (A, C) and the algorithm-generated images on the right (B, D). Arrows point to examples of correspondence between the two within each tumor. The same correspondence was also seen with the melanoma small TMA (not shown). There was little or no nuclear LC3B staining by the AQUA fluorescence analyses, consistent with the IHC analyses (not shown).
Figure 2.

Comparison of fluorescence-conjugated anti-LC3B staining patterns with those generated by LC3B algorithm that generates yellow for punctate and blue for diffuse LC3B staining through an AQUA program (Methods). Representative examples of breast cancers with high (A, B) and low (C, D) LC3B expression are presented. Actual immunofluorescence is shown in the panels on the left (A, C) and the algorithm-generated images on the right (B, D). Arrows show representative areas of correspondence within each tumor.
We thus used the algorithms above to perform quantitative analyses of LC3B distribution in the same 40-case TMAs of melanoma and breast carcinoma (Fig. 3). For both cancers, LC3B total cellular expression/intensity levels varied widely between different tumors (punctate + diffuse; AQUA score). However, when the tumors were ordered from low to high total LC3B intensity, the percent area occupied by the punctate compartment was 3–5 fold higher in tumors with the highest vs lowest total expression levels, occupying 25–30% of the cytoplasm at the highest and 5–10% at the lowest (p = < 0.0001 for both cancers) (Fig. 3A and B). Interestingly, as the percent area occupied by punctate LC3B increased, there was a concommittent reduction in the intensity of LC3B fluorescence intensity in the punctate compartment versus the diffuse compartment (Fig. 3C and D). For melanoma (Fig. 3C), the intensity of LC3B fluoresence in punctate compartment (but not the percent area) was reduced by ~60% in tumors with the highest compared to lowest total LC3B expression levels (p = < 0.0001). For breast carcinoma (Fig. 3D) the punctate LC3B content was reduced by ~20%, although statistical significance for the breast TMA was not reached (p = 0.1218). These results indicate that high total LC3B expression is associated with increased punctate LC3B area. As discussed below, this is apparently due to a higher total number of autophagosomes per cell but with diminished LC3 concentrations, consistent with active LC3 processing during autophagosome-lysosome fusion.
Figure 3.

Distribution of LC3B between punctate and diffuse cytoplasmic compartments at different total cellular LC3B expression levels. A and B: Percent cytoplasm involved with punctate LC3B staining versus total LC3B expression. C and D: Ratio of punctate/diffuse LC3B intensity versus total LC3B expression in melanoma (A, C) and breast cancer (B, D). The best-fit curves were obtained plotting the data as a natural log regression.
Breast cancer microarray YTMA49
Using AQUA, we examined LC3B expression in primary breast carcinomas from patients with or without lymph node metastases in microarray YTMA49. Primary tumors with associated lymph node involvement showed higher LC3B expression than those without (p < 0.0001) (Fig. 4A). Further, breast cancers with highest LC3B expression also showed highest Ki-67 expression, a widely-used marker for cell proliferation and tumor aggressiveness (r = 0.631; p < 0.0001) (Fig. 5A) (17). High LC3B expression was also associated with increased nuclear grade (p < 0.0001), a predictor of tumor agressiveness and survival (Fig. 6A). Likewise, Kaplan-Meier analyses of YTMA49 revealed a direct correlation between increased LC3B expression and disease related survival (Mantel-Cox analysis, p = 0.0016; Cox univariate analysis, p = 0.0013) (Figure 6B). Additional clinicopathologic correlations in microarray YTMA49 are shown in supplemental Table S2. LC3B expression was significantly higher in larger tumors (p = 0.0117), in tumors negative for the estrogen receptor (p = 0.0009) and progesterone receptor (p = 0.0043), and in tumors from women ≤ 50 years vs > 50 years (p = 0.0001). There was no correlation of LC3B with HER2/neu expression (p = 0.2943). Univariant survival statistics regarding the relative risk of various clinicopathologic critera are shown in supplemental Table S3.
Figure 4.
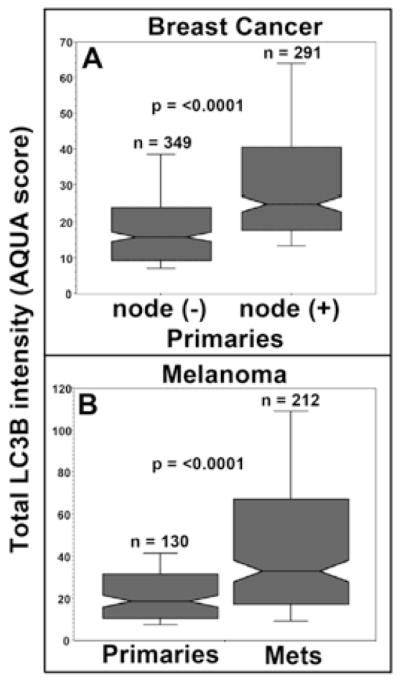
Association of LC3B punctate staining with metastasis. A. Breast carcinoma array YTMA49--box graphs of LCB3 staining intensity in primary tumors comparing lymph node negative vs lymph node positive cases (n = 640). B. Melanoma array YTMA20--box graphs of LCB3 staining intensity in primary melanoma compared to that in lymph node, visceral, and cutaneous metastases (n = 342).
Figure 5.

Associations of LC3B punctate staining with cell proliferation. A. Correlation analyses by AQUA of LC3B cytoplasmic staining and Ki-67 nuclear staining in breast carcinoma microarray YTMA49. B. Mitotic figure (asterix) and punctate LC3B staining (arrow) in a breast carcinoma section. C. Correlation analyses of LC3B cytoplasmic staining and Ki-67 nuclear staining in melanoma microarray YTMA20. D. Mitotic figure and punctate staining in a melanoma section.
Figure 6.

Breast carcinoma tumor array YTMA49--association of LC3B staining intensity, nuclear grade and patient outcome. A. Box graphs of LC3B staining intensity vs nuclear grade. Nuclear grade information obtained from the pathology reports of the individual cases in the TMA. B. Kaplan-Meier analyses of LC3B staining intensity versus patient outcome containing a mixture of node-negative and node-positive primary tumors. To visualize the data, AQUA scores were standardized by converting to z-scores and divided as follows: Blue: <−0.5 (less than 0.5 standard deviations below the mean); Green: −0.5 to 0 (between −0.5 standard deviations below the mean and the mean); Yellow: 0 to 1.5 (between the mean and 1.5 standard deviations above the mean); Red: > 1.5 (greater than 1.5 standard deviations above the mean). Mantel-Cox p-value = 0.0016. When analyzed as a continuous variable in a Cox univariate analysis, the p-value is 0.0013. Dashed line: censored patient.
Melanoma microarray YTMA20
Melanoma microarray YTMA20 was designed for AQUA analyses and consisted of primary tumors (n = 131), and unmatched lymph node, visceral and sub-cutaneous metastases (n = 250). LC3B expression was significantly higher in metastases compared to primary tumors (p < 0.0001) (Figure 4B). This was true for all metastatic locations and there were no correlations with specific sites (Supplemental Table S4). As with the breast cancer microarray YMTA49, there was a strong correlation between LC3B and Ki-67 epression in melanoma YTMA20 (r = 0.694; p < 0.0001) (Fig 4B). In primary melanomas there was a weak but significant correlation of LC3B expression with Breslow depth (p = 0.0479) but not with Clark Level (p = 0.1066), although Clark Level trended in that fashion. LC3B staining intensities were highest in patients ≥ 60 years (p = 0.0085). There were no specific associations with the density of tumor infiltrating lymphocytes, the presence of ulceration, or gender (Supplemental Table S4). There are no data for this TMA regarding nuclear grade or survival times.
Mitotic figures
In a survey of 289 tumor sections on microarray YTMA96, 91 contained one or more mitotic figures. Of these, we had previously scored 89/91 as having moderate to strong LC3B expression levels, while only 2/91 were scored as LC3B negative to weak. Based on random distribution, the expected number of mitotic figures would have been 15/91 since 17% of the tumors in the YTMA96 arrays were LC3B-weak to negative. Thus, the distribution of mitoses was skewed toward moderate to high LC3B-expression (p < 0.002) (Supplemental data-Table S5), consistent with the findings on Ki-67 levels in melanoma and breast cancer above (cf Fig 4). Examples are shown of mitotic figures in close proximity to LC3B-positive punctate structures, presumably autophagosomes, in a melanoma (Fig. 4B) and invasive breast carcinoma (Fig. 4D). Lower magnification photos of mitotic figures associated with LC3B punctate staining are also seen in Fig. 1.
DISCUSSION
From analyses of 1359 tissue specimens from breast carcinoma, melanoma and, on a smaller scale 18 other cancers, LC3B expression was remarkably common regardless of tissue origin. LC3B expression encompassed both the punctate and diffuse compartments of the cytoplasm but not the nucleus. A novel AQUA-based quantitive imaging technique for LC3B staining determined that tumors with high LC3B expression contain more LC3B-postive vesicles, but within these vesicles the intensity of LC3B staining is diminshed compared to that in tumors which have fewer LC3B-positive vesicles. We suggest that the results are consistent with a model in which higher total (punctate + diffuse) LC3B expression levels reflect increased autophagic flux with accelerated AV-lysosome fusion and production of autolysosomes. This would result in decreased LC3B content in the punctate compartment due to increased LC3B-II degradation by lysosomal hydrolases as well as delipidation with release into the cytosol. The results do not seem to be consistent with a block in autophagic flux prior to AV-lysosome fusion, as such a block would likely result in increased rather than decreased LC3B in the punctate compartment. However, since we could not measure the dynamics of these processes in the fixed tissues, further studies are necessary to address this model.
These results are consistent with emerging evidence that while autophagy can suppress the initiation and development of early tumors, it plays a pro-survival role once cancer cells gain metastatic competence (1, 9, 24). Evidence for autophagy serving as a tumor suppressor mechanism was first seen in a mouse model of monoallelic loss of the autophagy gene beclin 1, in which an increased development of spontaneous tumors and elevated rates of tumor proliferation were observed in vivo (3). In humans, 40–75% of carcinomas of the breast, ovary and prostate showed monoallelic deletions of beclin 1 (24). Thus, these findings and those of others demonstrate a role for autophagy as a suppressor of tumor initiation and development. On the other hand, a large volume of literature has emerged demonstrating that once malignancies have become established they rely heavily on autophagy for survival. One explanation for this paradox is that the retained allele of beclin, even in tumors which are haploinsufficient for this gene, is always wild type. Therefore when necessary, autophagy can be induced to high levels as a response to environmental stress even in tumors with “autophagy deficient” phenotypes. Support for a survival role for autophagy came early from mouse models of cancer therapy. Experiments involving AKT-driven tumors expressing GFP-LC3 demonstarted that autophagy was critical for survival of cancer cells within the center of growing tumors (27). Preclinical therapeutic models found that antitumor therapy including chemotherapy and targeted therapies induce autophagy in vivo (7, 10–13), and that blockade of therapy-induced autophagy can augment the efficacy of a variety of cancer therapies. More recently, reports that autophagy promotes survival and aggressiveness of metastatic melanoma (23) and pancreatic cancer (28) have emerged. Many of these early preclinical and clinical investigations studies have been limited to the use of electron microscopy to measure autophagy in tumor tissue. While electron microscopy provides a wealth of ultrastructural data and with trained investigators can accurately measure autophagy in a semi-quantitive manner, it is expensive, time-intensive, and without a molecular marker of autophagy included in the analysis the incorrect scoring of non-autopahosomal structures as autophagosomes may skew the results. In addition, reliance on EM limits autophagy investigations to freshly collected tissue. Reliable, objective methods to measure autophagy in FFPE would allow more thorough investigation of the dozens of genes related to autophagy as potential prognostic or predictive biomarkers for cancer prevention and therapy.
Other groups have previously reported LC3 staining in FFPE. Sato et al. showed that 90% of primary colorectal tumors (72/80) and 100% of lymph node (5/5) and liver (6/6) metastases were positive for LC3 punctate expression (29). Autophagosomes were confirmed by electron microscopy of surgically removed tumor tissue. Normal mucosal epithelium adjacent to the tumors was negative for punctate LC3 staining. When autophagy was induced in colorectal cancer cell lines by nutrient deprivation, agents that interfered with autophagy markedly increased apoptotic death, indicating that autophagy plays a pro-survival role in colorectal cancer cell. Yoshioka et al. showed strong LC3 expression in 53% of esophageal, 58% of gastric and 63% of colorectal cancers. In early esophageal cancers this correlated with high Ki-67 expression (31). It was concluded that LC3 expression is advantageous to tumor development. In pancreatic cancers, Fujii et al observed that 87% (62/71) were LC3B-positive and this correlated with shorter disease-free period and poor patient outcome (31). In primary invasive melanoma, we previously reported that while normal melanocytes adjacent to the tumor and melanoma cells of early in situ melanomas showed little or no LC3B expression, punctate LC3B staining was widespread in nests of florid in situ tumor cells and in 31/31 invasive tumors in the dermis (22; Lazova and Pawelek unpublished). Autophagosomes were confirmed by electron microscopy showing double-membraned structures filled with melanized melanosomes. While these studies laid the groundwork to explore the importance of LC3B staining as a measure of autophagy in cancer, our findings here include a much larger and broader representation of human malignancy, employed objective quantitive immunofluorescence techniques, addressed the issue of LC3B puncta versus total LC3B expression, and established LC3B staining as a potential prognostic marker in multiple malignancies by correlating quantitative staining with survival outcomes.
CONCLUSIONS AND SIGNIFICANCE
Regardless of tissue origin, a large majority of solid tumors contain autophagosomes as assessed by LC3B punctate staining and, in the case of melanoma (22) and colorectal cancer (29) by electron microscopy. Punctate LC3B was associated with tumor cell proliferation, metastasis, high nuclear grade and worse patient outcome. Taken together, it seems most likely that these results, along with those of others, reflect widespread, active autophagy in malignant tumors as a pro-survival mechanism for cancer cells.
There are several potential therapeutic implications to these findings. LC3 staining could be a prognostic marker in many human cancers and could be incorporated into clinical trials of drugs that are known to induce autophagy, or inhibit autophagy. The surprisingly common occurrence of autophagosomes in malignancies points to a key role for autophagy in tumor progression and thus presents a common pathway of therapeutic vulnerability in human malignancies.
Supplementary Material
Statement of Translational Relevance.
Preclinical studies indicate that autophagy can be a suppressor mechanism for the initiation of tumorigenesis but a targetable mechanism of cancer cell survival once a tumor becomes established. Translation of these preclinical findings has lead to clinical trials involving the autophagy inhibitor hydroxychloroquine in combination with existing anticancer therapies that may induce autophagy in tumors. To guide the development of autophagy inhibitors for advanced disease and to understand the prognostic significance of high and low levels of autophagy in early disease, robust biomarkers for autophagy need to be developed that can be assessed in commonly available paraffin-embedded tissue. This study reports novel immunohistochemical protocols for LC3 staining that were applied to hundreds of human cancer pathology specimens through the use of clinically annotated tissue microarrays. The results provide comprehensive evidence that autophagy levels are elevated in most human cancers and are associated with aggressive malignancy and poor outcome. High expression of LC3B was found to be prognostic of high nuclear grade, high mitotic rate, metastases and shortened survival. The staining techniques reported here by light microscopy and quantitive immunofluoresence (AQUA analysis) can now be incorporated into prospective and retrospective clinical studies to establish the clinical utility of LC3 as a potential prognostic and predictive marker in multiple cancers. These techniques can also be applied in the future for other emerging autophagy markers.
Acknowledgments
Funded by a generous gift from the Amway Corporation. We thank James Platt, Yale School of Medicine, for help with statistical analyses regarding the distribution of mitotic figures.
Footnotes
CONFLICTS OF INTEREST
There are no conflicts of interest from any of the authors in this report.
References
- 1.Amaravadi RK, Lippincott-Schwartz J, Yin XM, et al. Principles and Current Strategies for Targeting Autophagy for Cancer Treatment. Clin Cancer Res. 2011;17:654–66. doi: 10.1158/1078-0432.CCR-10-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuma A, Mizushima N. Physiological role of autophagy as an intracellular recycling system: with an emphasis on nutrient metabolism. Semin Cell Dev Biol. 21:683–90. doi: 10.1016/j.semcdb.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Young AR, Narita M, Ferreira M, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amaravadi RK, Thompson CB. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res. 2007;13:7271–9. doi: 10.1158/1078-0432.CCR-07-1595. [DOI] [PubMed] [Google Scholar]
- 6.Amaravadi RK, Yu D, Lum JJ, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–36. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang S, Kimmelman AC. A critical role for autophagy in pancreatic cancer. Autophagy. 2011;7:912–3. doi: 10.4161/auto.7.8.15762. [DOI] [PubMed] [Google Scholar]
- 8.Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–29. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo JY, Chen HY, Mathew R, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–70. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bellodi C, Lidonnici MR, Hamilton A, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest. 2009;119:1109–23. doi: 10.1172/JCI35660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Degtyarev M, De Maziere A, Orr C, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol. 2008;183:101–16. doi: 10.1083/jcb.200801099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carew JS, Nawrocki ST, Kahue CN, et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr- Abl-mediated drug resistance. Blood. 2007 doi: 10.1182/blood-2006-10-050260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding WX, Ni HM, Gao W, et al. Oncogenic transformation confers a selective susceptibility to the combined suppression of the proteasome and autophagy. Mol Cancer Ther. 2009;8:2036–45. doi: 10.1158/1535-7163.MCT-08-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katayama M, Kawaguchi T, Berger MS, Pieper RO. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2007;14:548–58. doi: 10.1038/sj.cdd.4402030. [DOI] [PubMed] [Google Scholar]
- 15.Divito KA, Berger AJ, Camp RL, Dolled-Filhart M, Rimm DL, Kluger HM. Automated quantitative analysis of tissue microarrays reveals an association between high Bcl-2 expression and improved outcome in melanoma. Cancer Res. 2004;64:8773–7. doi: 10.1158/0008-5472.CAN-04-1387. [DOI] [PubMed] [Google Scholar]
- 16.Berger AJ, Camp RL, Divito KA, Kluger HM, Halaban R, Rimm DL. Automated quantitative analysis of HDM2 expression in malignant melanoma shows association with early-stage disease and improved outcome. Cancer Res. 2004;64:8767–72. doi: 10.1158/0008-5472.CAN-04-1384. [DOI] [PubMed] [Google Scholar]
- 17.Camp RL, Chung GG, Rimm DL. Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat Med. 2002;8:1323–7. doi: 10.1038/nm791. [DOI] [PubMed] [Google Scholar]
- 18.Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klionsky DJ, Abeliovich H, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanida I. Autophagy basics. Microbiol Immunol. 2011;55:1–11. doi: 10.1111/j.1348-0421.2010.00271.x. [DOI] [PubMed] [Google Scholar]
- 21.Tanida I, Waguri S. Measurement of autophagy in cells and tissues. Methods Mol Biol. 2010;648:193–214. doi: 10.1007/978-1-60761-756-3_13. [DOI] [PubMed] [Google Scholar]
- 22.Lazova R, Klump V, Pawelek J. Autophagy in cutaneous malignant melanoma. J Cutan Pathol. 2010;37:256–68. doi: 10.1111/j.1600-0560.2009.01359.x. [DOI] [PubMed] [Google Scholar]
- 22.Dolled-Filhart M, McCabe A, Giltnane J, Cregger M, Camp RL, Rimm DL. Quantitative in situ analysis of beta-catenin expression in breast cancer shows decreased expression is associated with poor outcome. Cancer Res. 2006;66:5487–94. doi: 10.1158/0008-5472.CAN-06-0100. [DOI] [PubMed] [Google Scholar]
- 23.Ma X, Piao S, Wang DW, et al. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res. 2011 doi: 10.1158/1078-0432.CCR-10-2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 25.Isaka Y, Kimura T, Takabatake Y. The protective role of autophagy against aging and acute ischemic injury in kidney proximal tubular cells. Autophagy. 2011;7 doi: 10.4161/auto.7.9.16465. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 26.Czaja MJ. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology. 2011;140:1895–908. doi: 10.1053/j.gastro.2011.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–29. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato K, Tsuchihara K, Fujii S, et al. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res. 2007;67:9677–84. doi: 10.1158/0008-5472.CAN-07-1462. [DOI] [PubMed] [Google Scholar]
- 30.Yoshioka A, Miyata H, Doki Y, et al. LC3, an autophagosome marker, is highly expressed in gastrointestinal cancers. Int J Oncol. 2008;33:461–8. [PubMed] [Google Scholar]
- 31.Fujii S, Mitsunaga S, Yamazaki M, et al. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci. 2008;99:1813–9. doi: 10.1111/j.1349-7006.2008.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.