

ABSTRACT
Tetraploidy has been proposed as an intermediate state in neoplastic transformation due to the intrinsic chromosome instability of tetraploid cells. Despite the identification of p53 as a major factor in growth arrest of tetraploid cells, it is still unclear whether the p53-dependent mechanism for proliferation restriction is intrinsic to the tetraploid status or dependent on the origin of tetraploidy. Substrate adherence is fundamental for cytokinesis completion in adherent untransformed cells. Here we show that untransformed fibroblast cells undergoing mitosis in suspension produce binucleated tetraploid cells due to defective cleavage furrow constriction that leads to incomplete cell abscission. Binucleated cells obtained after loss of substrate adhesion maintain an inactive p53 status and are able to progress into G1 and S phase. However, binucleated cells arrest in G2, accumulate p53 and are not able to enter mitosis as no tetraploid metaphases were recorded after one cell cycle time. In contrast, tetraploid metaphases were found following pharmacological inhibition of Chk1 kinase, suggesting the involvement of the ATR/Chk1 pathway in the G2 arrest of binucleated cells. Interestingly, after persistence in the G2 phase of the cell cycle, a large fraction of binucleated cells become senescent. These findings identify a new pathway of proliferation restriction for tetraploid untransformed cells that seems to be specific for loss of adhesion-dependent cytokinesis failure. This involves Chk1 and p53 activation during G2. Inhibition of growth and entrance into senescence after cytokinesis in suspension may represent an important mechanism to control tumor growth. In fact, anchorage independent growth is a hallmark of cancer and it has been demonstrated that binucleated transformed cells can enter a cycle of anchorage independent growth.
KEYWORDS: Binucleated cells, cytokinesis failure, loss of adherence, p53, senescence, tetraploidy
Introduction
Eukaryotic organisms usually contain a diploid complement of chromosomes. However, developmentally regulated formation of polyploid cells occurs in some mammalian tissues, such as hepatic tissues or megakaryocytes in blood and usually coincides with terminal differentiation.1 Unscheduled polyploidy, although tolerated in plants, is detrimental to mammals, so that triploid and tetraploid embryos are not vital in humans and represent approximately 10% of total miscarriages.2 In addition, several lines of evidence converge to indicate that aberrant polyploidy can promote cell transformation. Cytogenetic analyses of tumor samples have shown that 26% of solid tumors are near-polyploid or near triploid.3 The great variability of chromosome number in polyploid tumors, already observed in the early times of cytology, has led to a model that envisions tetraploidy, resulting from a whole genome doubling, as an intermediate stage in the development of cancer.1,4 In this model, unstable polyploid cells have greater survival chances, as compared to chromosomally unstable diploid cells, since the presence of several copies of the same chromosome may counteract the negative effects of chromosome loss. This idea is supported by a recent study that demonstrated a higher tolerance of polyploid cells to chromosome instability.5
Tetraploid cells are generated by 3 main mechanisms: cell fusion, mitotic exit without chromosome segregation or cytokinesis failure induced by different stimuli.1 In this last case, tetraploid cells possess 2 nuclei and 2 centrosomes within a single cytoplasm and are, therefore, called binucleated. Due to the intrinsic instability of tetraploid cells and their tumorigenic capacity, 6-9 several groups have investigated whether control mechanisms exist that limit the proliferation of tetraploid cells.4 Early works showed that cells arrested by spindle poisons proceed to interphase without chromosome segregation after a variable time period in a process called "mitotic slippage" and that these tetraploid cells are arrested in the following G1 by a p53-mediated process.10,11,12 Similarly, other work showed that binucleated tetraploid cells obtained by a treatment with the actin inhibitor dihydrocytochalasin B arrested in the first G1 following treatment in a p53-dependent manner.13 However, it was successively demonstrated that arrest of binucleated cells was dependent on drug concentration, indicating that drug-induced cellular damage was possibly responsible for the G1 arrest.14,15 Nevertheless, tetraploids arising in untransformed cultures from mitotic slippage, cell fusion or cytokinesis failure induced by chemical treatment or depletion of proteins required for cytokinesis are usually limited in their proliferation by a p53-mediated pathway.6,16-18 Recent studies have linked p53 activation in tetraploids to the induction of oxidative stress leading to ATM activation at the first tetraploid mitosis19 or to the activation of the tumor suppressor Hippo pathway.20 However, it is still unclear whether the p53-dependent pathway restricting binucleated cell proliferation is inherent to the binucleation condition or other pathways may intervene, depending on the origin of cell binucleation.
Cell anchorage is required for proliferation of untransformed adherent cells and, upon loss of substrate adherence, cells arrest in the G1 phase of the cell cycle.21 Several studies have identified another anchorage-dependent restriction point acting during cytokinesis so that growth in suspension (i.e. in soft-agar) causes cytokinesis failure and produces binucleated tetraploid cells in primary human fibroblasts.22-24 To shed new light on the mechanism(s) limiting proliferation of tetraploid cells obtained by loss of anchorage, we produced binucleated cells by maintaining mitotic cells in suspension during cytokinesis and followed cell cycle progression of these binucleated cells.
Substrate-independent growth is a hallmark of cancer cells and a fundamental feature of metastatic cancer cells.25 Notably, metastatic tumors, which release in the blood stream circulating tumor cells or CTC are very frequently treated with microtubule inhibitors. However, it is still unknown whether mitotic spindle damage may promote binucleation in released cancer cells experiencing no spatial cues in the blood stream. Moreover, the proliferation ability of tetraploid cells originated under these circumstances is still unexplored. We, therefore, combined loss of substrate adherence during cytokinesis with spindle poison treatment and followed the fate of the induced tetraploid cells.
Results and discussion
Cytokinesis in suspension produces binucleated cells through defective cleavage furrow constriction
Previous studies have shown that growth in suspension of untransformed adherent cells produces binucleated cells.22-24,26 To investigate the fate of binucleated cells induced by loss of substrate adhesion, human fibroblast MRC-5 mitotic cells were isolated by mitotic shake off, maintained in suspension for 30 min to allow mitotic progression, seeded in growth medium and analyzed for several parameters at different time points. Parallel cultures were treated with the microtubule inhibitor nocodazole (NOC) for 3 hrs, then released in drug-free medium for 30 min to allow reformation of the mitotic spindle and subjected to the same procedure (Figure 1A). Approximately 20% of MRC-5 cells undergoing mitosis in loss of adhesion were found to be binucleated at early times after re-seeding from shake off (Figure 1B). Interestingly, NOC pre-treatment induced the formation of binucleated cells at a much higher rate, i.e., about 40% of the cells were binucleated. Increased binucleation was characteristic of the combined procedure of NOC treatment and mitotic shake off, since NOC treatment of MRC-5 adherent cells did not induce binucleation over control values (Figure 1B). Similar results were obtained in adherent epithelial cells of marsupial origin (data not shown) suggesting that the process of binucleation following substrate loss during cytokinesis occurs independently of species and tissue of origin and is likely to be common to all untransformed cells growing in adherence. Microscopic observation of the mitotic population after shake off showed higher frequencies of late stages of mitosis (ana-telophase and cytokinesis) in NOC treated and released cells as compared to cells exposed to the NOC solvent DMSO prior to shake off (Figure 1C). This result suggests that increased binucleation is associated to a synchronization effect in the prometaphase stage of the NOC treatment and supports the results obtained in previous studies indicating that ana-telophase and cytokinesis are the critical stages for the formation of binucleated cells in suspension conditions.24 However, the magnitude of the NOC effect on the frequency of binucleated cells (42.3 ± 8.5 % in NOC pre-treated vs. 17.8 ± 2.4 % in shake off cells) strongly suggests that other mechanisms may be at work when cytokinesis intervenes in suspension in cells undergoing defective chromosome segregation, as after spindle poison treatment. On the whole, this finding suggests that induction of a genetically unstable tetraploid state by exposure to spindle poisons of non-adherent cancer cells may represent an unwanted side effect of the use of these drugs as chemotherapeutic agents.
Figure 1.

Cytokinesis in suspension produces binucleated cells. (A) Experimental schedule. (B) Frequency of binucleated cells in MRC-5 cultures grown in adherence with or without a 3hr NOC treatment (Adherence) or after mitotic shake off and cytokinesis in suspension (Shake off) with or without a 3hr NOC pre-treatment (see experimental schedule). Binucleated cells were identified by actin and tubulin staining. DNA was counterstained by DAPI. Results shown are the mean ± SEM of 4 independent experiments. **P <0.01 (Student's t-test, vs. untreated). (C) Frequencies of the different mitotic stages after detachment from the substrate with or without a 3hr NOC pre-treatment. The different mitotic stages are depicted in the images. PM: prometaphase, M: metaphase, A: anaphase, T: telophase. Results shown are the mean ± SEM of 4 independent experiments. Scale bar = 5 μm.
To investigate the mechanisms responsible for binucleated cell production during loss of substrate adherence, MRC-5 cells detached from the substrate by mitotic shake off were cytospun on coverslips at the end of the suspension time and cells in cytokinesis (identified by a constricting cleavage furrow) were analyzed for their morphology and microtubule organization by phase-contrast and fluorescence microscopy. Remarkably, cells that underwent cytokinesis in suspension had regularly shaped microtubule-based midbody or midbody remnants, as shown by tubulin immunostaining, but exhibited a defective ingression of the actomyosin contractile ring, the compact structure that appears under phase contrast illumination as a dense bar at the constriction site (Figure 2A). Quantitative measurements demonstrated that the actomyosin ring was significantly wider in cells undergoing cytokinesis in suspension with or without a NOC pretreatment in comparison with cells that underwent mitosis in adherence (Figure 2B). These results suggest that defective cleavage furrow constriction leads to the formation of binucleated cells in the absence of substrate adhesion. Concordantly, inhibition of intercellular bridge abscission and fusion of connected daughter cells have been demonstrated by live cell imaging of dermal fibroblasts performing cytokinesis on experimentally generated soft substrates.26
Figure 2.
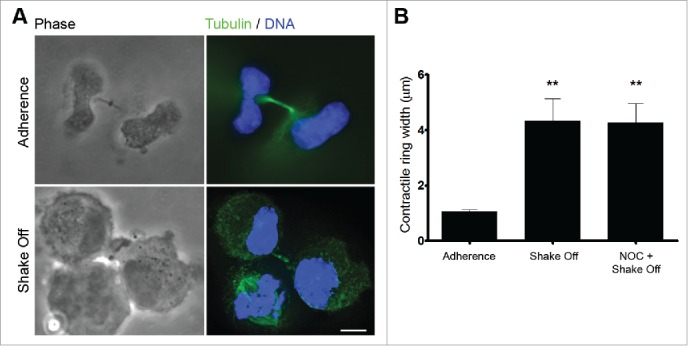
Cytokinesis in suspension produces defective cleavage furrow constriction. (A) Representative images of late telophase cells grown in adherence or cytospun on slides after mitotic shake off and cytokinesis in suspension. Scale bar = 5 μm. (B) Contractile ring width in late telophase cells grown on adherence (Adherence), after cytokinesis in suspension (Shake off) or after cytokinesis in suspension with a 3hr NOC pre-treatment (NOC + Shake off). Late telophase cells were identified by the presence of decondensed chromatin and of a midbody following α tubulin and DNA staining; contractile ring width was measured on phase contrast images. Ten to twenty cells were measured for each condition. **P <0.01 (Student's t-test, vs. adherence).
Binucleated cells produced by cytokinesis in suspension preferentially arrest in the G2 phase
We then followed progression through the cell cycle of mononucleated and binucleated cells from the same preparation after cytokinesis in suspension. Analysis of BrdU incorporation into DNA by antibody staining in mononucleated and binucleated cells from the same culture revealed that both cell types were regularly progressing through S-phase 20 hrs after re-seeding from cytokinesis in suspension (Figure 3A). Indeed, the frequency of BrdU positive cells was similar in binucleated and mononucleated cells and did not differ from the frequency observed in MRC-5 cells grown in adherence (Figure 3B). On the contrary, NOC pre-treatment resulted in a significantly lower rate of BrdU incorporation in binucleated cells, so that only 40% of NOC pre-treated binucleates incorporated BrdU in the preceding 20 hrs as compared to 80% BrdU-positive mononucleated cells from the same culture (Figure 3B). These findings clearly suggest that cells becoming binucleated due to loss of substrate adherence during cytokinesis are not inhibited in entering S phase whereas the combined procedure of NOC treatment and shake off strongly impairs cell cycle progression during G1. Notably, MRC-5 cells grown in adherence did not progress into a tetraploid S phase in response to cytochalasin B treatment (Figure S1). This confirms that MRC-5 binucleated cells obtained in adherence conditions are subjected to the G1 checkpoint activation described in literature4 while loss-of-adhesion allows the same cells to enter DNA replication. A single cell analysis of p53 accumulation was then performed through the evaluation of p53 nuclear stain 20 hrs after cytokinesis in suspension (Figure 3C), since activation and stabilization of this tumor suppressor protein has been proposed as the major mechanism to limit the proliferation of tetraploid cells.4 Consistent with the lack of G1 arrest (see above), no significant increase in the number of binucleated cells showing p53 accumulation in the nucleus was observed over mononucleated cells from the same culture, while a significant increase in p53 positive nuclei was recorded in NOC pre-treated binucleated cells, as compared to mononucleated cells (Figure 3D). Interestingly, more than 90% of the cells were positive to the p53 staining after the DNA damaging agent camptothecin (CPT) (Figure 3C and D). These results are consistent with the idea that activation of p53 is much weaker following spindle damage as compared to agents producing DNA breaks. In the case of spindle inhibitors, damage to DNA or cellular stress intervening during the prolonged prometaphase time may be responsible for p53 activation.27 Altogether, these data suggest that cytokinesis in suspension does not activate a p53-dependent G1 arrest whereas spindle damage-dependent mitotic arrest does.10,13,28
Figure 3.
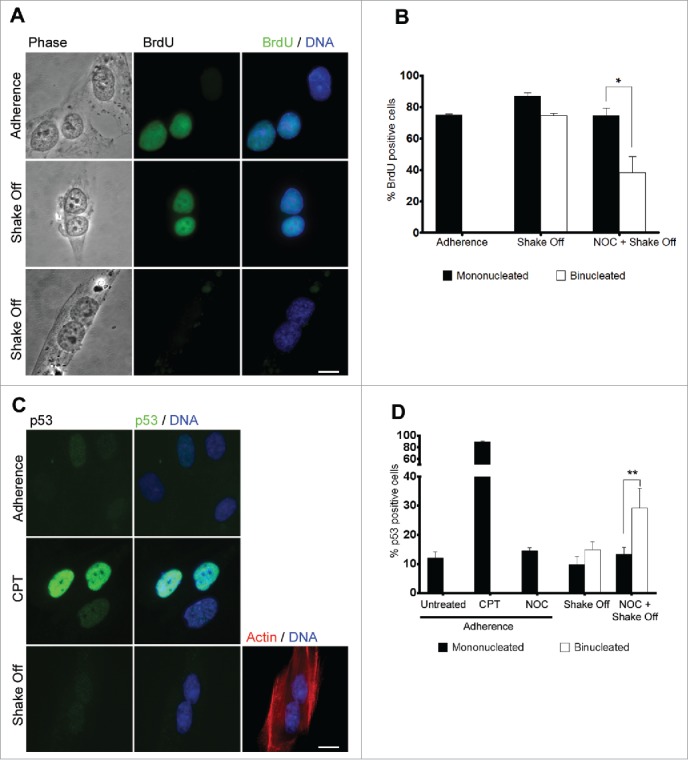
Binucleated cells induced by cytokinesis in suspension progress through S phase. (A) Representative images of BrdU incorporation in mononucleated cells grown in adherence or in binucleated cells obtained 20 hrs after cytokinesis in suspension. (B) Frequency of BrdU-positive cells among mononucleated cells grown in adherence and mononucleated or binucleated cells obtained 20 hrs after cytokinesis in suspension (Shake off) or after cytokinesis in suspension with a 3 hr NOC pre-treatment (NOC + Shake off). Results shown are the mean ± SEM of 3 independent experiments. For each experiment at least 200 mononucleated cells were analyzed in adherence condition, 200 mononucleated and 50 binucleated cells after cytokinesis in suspension. (C) Representative images of p53 immunostaining in cells grown in adherence (Adherence), after camptothecin treatment (CPT), or in binucleated cells obtained 20 hrs after cytokinesis in suspension (Shake off). Binucleation was confirmed by actin immunostaining. (D) Frequency of p53-positive cells in cells grown in adherence (untreated), after camptothecin treatment (CPT) or nocodazole exposure (NOC) in adherence, and in mononucleated or binucleated cells obtained 20 hrs after cytokinesis in suspension (Shake off) or after cytokinesis in suspension with a 3hr NOC pre-treatment (NOC + Shake off). Results shown are the mean ± SEM of 3 independent experiments. For each experiment at least 500 mononucleated cells were analyzed in adherence conditions, 200 mononucleated and 50 binucleated cells after cytokinesis in suspension. *P<0.05; **P<0.01 (Student's t-test). Scale bar = 5 μm.
In order to follow the progression into cell cycle of binucleated cells after DNA replication, we analyzed cyclin B expression 40 hrs after cytokinesis in suspension to identify G2 phase cells (Figure 4A). Consistent with the activation of a G2/M checkpoint preventing mitotic entry of binucleated cells, 50% of binucleated cells accumulated cyclin B in their nuclei. In contrast, only 20% of mononucleated cells from the same culture stained positive to cyclin B, demonstrating that binucleated cells significantly accumulated in the G2 phase. Interestingly, a moderate but statistically significant increase in the nuclear p53 fluorescence intensity was recorded using quantitative image analysis in cyclin B-positive mononucleated and binucleated cells in comparison with untreated cultures when cultures were co-stained for cyclin B and p53 at 40 hrs from cytokinesis in suspension (Figure 4B and C). Interestingly, binucleated cells accumulated significantly more p53 than mononucleates (Figure 4C). Thus, these data suggest that cytokinesis in suspension can eventually lead to the accumulation of cyclin B-expressing, G2 phase binucleated interphases accompanied with a moderate p53 stabilization after DNA replication.
Figure 4.
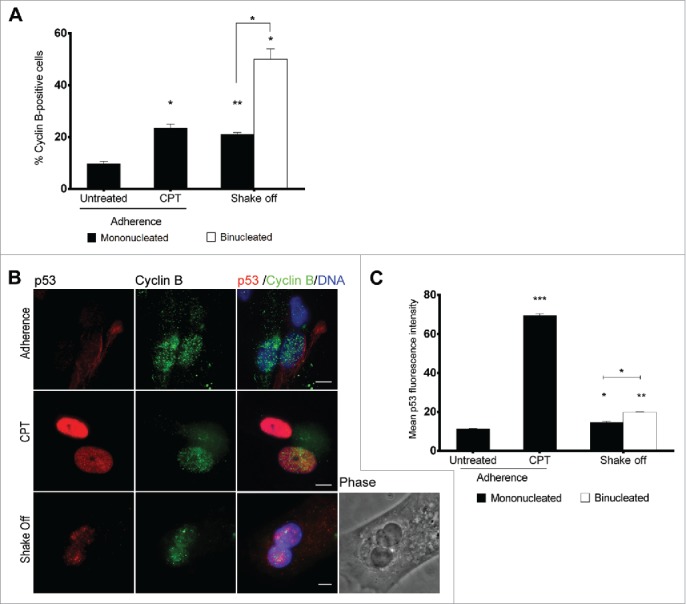
Binucleated cells induced by cytokinesis in suspension arrest in G2 and accumulate p53. (A) Frequency of cyclin B-positive cells among mononucleated cells grown in adherence (Untreated), after camptothecin treatment (CPT) and in mononucleated or binucleated cells obtained 40 hrs after cytokinesis in suspension (Shake off). For each of the 2 experiments at least 500 mononucleated cells were analyzed in adherence conditions, 200 mononucleated and 50 binucleated cells after cytokinesis in suspension. (B) Representative images of p53 and cyclin B immunostaining in cells grown in adherence (Adherence), after camptothecin treatment (CPT), or in binucleated cells obtained 40 hrs after cytokinesis in suspension (Shake off). (C) The graph shows the mean ± SEM of p53 fluorescence intensity (arbitrary units) in cyclin B positive cells grown in adherence (untreated), after camptothecin treatment (CPT), and in mononucleated or binucleated cells obtained 40 hrs after cytokinesis in suspension (Shake off). For each condition, 2 replicate cultures (at least 50 cells/culture) were analyzed. *P<0.05; **P<0.01 ***P<0.001 (Student's t-test). Scale bar = 5 μm.
To test whether DNA damage was involved in p53 activation of G2 arrested binucleated cells, we immunostained for nuclear γ-H2AX foci, as an indicator of DNA damage (Figure S2A).29 A minor increase in the number of cells showing more than 5 γ-H2AX foci was observed in both mononucleated and binucleated cells at 40 hrs from cytokinesis in suspension (Figure S2B), suggesting that a subtle induction of DNA damage is present following cytokinesis in suspension but excluding increased levels of DNA damage as cause of G2 accumulation of the binucleated cells. Furthermore, since p53 phosphorylation at Ser15 is a sensitive marker of p53-mediated response to DNA damage30 or chromosome mis-segregation in diploid cells,31 Phospho-Ser15 p53 was determined by immunostaining. No nuclear accumulation of Phospho-Ser15 p53 was observed in binucleated cells (Figure S2C), confirming that DNA damage has very little, if any, effect on the G2 arrest of binucleated cells.
Binucleated cells are arrested in G2 by a ATR-Chk1-dependent pathway and undergo senescence
On the whole, the above data indicate that binucleated cells are arrested at the G2 phase of the cell cycle and do not enter mitosis. To further investigate this point, we assessed chromosome numbers in metaphase cells harvested 2 or 4 days after re-seeding from cytokinesis in suspension. Chromosome counts revealed that binucleated cells were not able to enter mitosis, since a similar number of 4N metaphases was recorded 48 or 96 hrs from re-seeding after shake off and in an asynchronously growing population of MRC-5 cells (Figure 5A and B). Therefore, it can be inferred that activation of a G2/M checkpoint prevents binucleated cells from progressing into mitosis.
Figure 5.

Binucleated cells induced by cytokinesis in suspension are arrested in G2 through a ATR/Chk1 dependent pathway and enter senescence. (A) Representative images of near 2N and near 4N metaphase spreads. (B) Frequency of near 4N metaphases in chromosome spreads obtained from exponentially growing cultures (Adherence), 48 or 96 hrs after cytokinesis in suspension in the absence (Shake Off ) or in the presence of 5 mM caffeine (Shake Off + CAF) for 6 hr prior to harvesting. Results shown are the mean ± SEM of 2 independent experiments (200-400 metaphases were analyzed per each sample). (C) Frequency of near 4N metaphases in chromosome spreads obtained 48 hrs after cytokinesis in suspension in the absence (Shake Off) or in the presence of 5 μM LY2603618 (Shake Off + LY2603618) for 6 hrs prior to harvesting. Results shown are the mean ± SEM of 2 independent experiments (200-400 metaphases were analyzed per each sample). (D) Representative images of mononucleated and binucleated cells identified by phase contrast and stained for the senescence marker β-Galactosidase (SA-β Gal). SA-β Gal positive and negative cells are indicated by black and white arrows, respectively. (E) Frequency of SA-β Gal positive cells in exponentially growing cultures (untreated), 48 or 96 hrs after H2O2 treatment of adherent cells, and 48 or 96 hrs after cytokinesis in suspension (Shake off). Results shown are the mean ± SEM of 2 independent experiments. (500-1000 mononucleated and 300-400 binucleated cells were counted per sample). *P <0.05 (Student's t-test). Scale bar = 5 μm.
Major activators of the G2/M checkpoint are the 2 protein-kinases ATM and ATR.32 To verify whether the ATM/ATR pathways were involved in the G2 arrest of binucleated cells, cells were treated with caffeine, a well-known inhibitor of these kinases. A 6 hr exposure to caffeine prior to cell harvesting produced significant increases in tetraploid metaphases at both 2 and 4 days from re-seeding (Figure 5B), suggesting that binucleated cells induced by loss of substrate contact may be limited in their proliferation by an ATM/ATR dependent mechanism. However, since ATM directly phosphorylates p53 at Ser1530 and no such phosphorylation was recorded in our experimental conditions (Figure S2C), we decided to address the contribution of the ATR/Chk1 pathway to the G2 arrest of binucleated cells by exposing cells to LY2603618, a selective Chk1 inhibitor that has been shown to induce a premature entry into mitosis by abrogating the Chk1-dependent G2/M checkpoint.33 Inhibition of Chk1 kinase by LY2603618 6 hrs prior to cell harvesting was able to overcome the G2/M arrest of binucleated cells as demonstrated by the increase in tetraploid metaphases observed in chromosome counts (Figure 5C). The frequency of tetraploid metaphases recorded after Chk1 inhibition was similar to the one observed after caffeine treatment, strongly suggesting that the G2/M checkpoint that blocks mitotic entry of binucleated cells is under the control of the ATR/Chk1 pathway.
Cellular senescence represents a final state of cell withdrawal from the cell cycle, as cells lose their capability to proliferate in response to growth factors or mitogens. Moreover, acquisition of a senescent phenotype is an important physiological anti-tumor response that is activated to counteract oncogenic insults and DNA damage.34 In light of the connection between senescence and cell cycle arrest, we decided to assess whether G2 accumulated binucleated cells were prone to undergo senescence. To this aim, we monitored the activity of senescence-associated β-galactosidase (SA-β-gal), a widely accepted general marker of the senescent phenotype, in binucleated and mononucleated cells from the same preparation (Figure 5D). Analysis of SA-β-gal staining showed that shake off induced binucleated cells expressed the senescence marker at significantly higher frequencies compared with mononucleated cells from the same culture, when assayed at 48 or 96 hrs from cyctokinesis in suspension (Figure 5E). Interestingly, the frequencies of senescence positive binucleated cells were similar to the frequencies obtained after exposure of adherent cells to H2O2, a model inducer of senescence, indicating that activation of a senescence pathway is a main route to eliminate tetraploid cells from the proliferating cell population in untransformed cells.
In conclusion, the present work confirms that cytokinesis in suspension leads to cleavage furrow regression and cytokinesis failure. Our data suggest that ana-telophase and cytokinesis are critical stages for binucleation during mitotic progression in suspension conditions. Binucleated human fibroblasts originated by failure of cytokinesis during suspension do not activate p53 in the G1 phase, undergo DNA replication but fail to enter mitosis, as shown by the absence of tetraploid metaphases at the first post-shake off mitosis. This suggests that tetraploid cell proliferation after substrate detachment is limited by a mechanism that intervenes during the G2 phase, as shown by the accumulation of cyclin B-positive binucleated cells 40 hrs after cytokinesis in suspension. Exposure to caffeine or the Chk1 inhibitor LY2603618 during the G2 phase produces a significant increase in tetraploid metaphases suggesting that binucleated cells are limited in their proliferation by an ATR/Chk1 dependent mechanism acting during G2. In this scenario, p53 accumulation in G2 cells could be attributed to a Chk1-dependent phosphorylation on p53, as the protein is known to be phosphorylated by Chk1 on Ser20.35 Thus, this work shows that the G2 arrest of binucleated cells after cytokinesis in suspension is likely to be sustained by overlapping p53-independent and p53-dependent mechanisms, similarly to what has been described for the DNA damage-dependent G2/M checkpoint.36
Overall, this study indicates that binucleated cells induced by substrate detachment of non-transformed human cells can survive in cell culture. However, after persistence for 1 or 2 cell cycle times in the G2 phase of the cell cycle, binucleated cells become senescent. This pathway of proliferation restriction seems to be specific for loss of adhesion-dependent cytokinesis failure that may involve perturbation of integrin signaling.23,37,38 Conversely, binucleated cells induced by the actin inhibitors cytochalasins or the myosin inhibitor blebbistatin have been found to arrest at the first or second mitosis from drug treatment through accumulation of p53/p21,16,19,20 demonstrating that the pathways activated to arrest cell cycle progression after loss of substrate adhesion or inhibition of cytokinesis in adherence conditions are fundamentally distinct. Interestingly, recent work on primary rat embryonic and human fibroblasts demonstrates that non-transformed cells respond to drug-induced tetraploidy by entering senescence from a tetraploid G1 stage.18 These findings together with the present results suggest that induction of senescence is a common mechanism for limiting tetraploidy in non-transformed cells. In contrast, the mechanism which triggers tetraploidy dictates the cell cycle phase at which tetraploid cells arrest. In the vast majority of tetraploidy-inducing conditions, senescence intervenes during a p53-dependent G1 arrest. Conversely, when binucleation is induced by loss of adhesion during cytokinesis, a condition permissive for tetraploid DNA replication, senescence ensues from the G2 phase. More generally, induction of a G2 arrest triggering senescence may represent a very potent back-up control mechanism to avoid proliferation of loss of adhesion-induced binucleated cells in non-transformed human cells. The findings presented in this paper give new clues to identify potential mechanisms controlling tumor growth, since binucleated cells induced by cytokinesis failure in the absence of substrate adhesion were previously shown to enter a cycle of anchorage independent growth when expressing an oncogenic H-Ras mutation24 and have been found to induce oncogenic transformation in vivo.39
Materials and methods
Cell culture and treatments
Normal human fibroblast (MRC-5) cells (ATCC CCL-171) were grown in MEM medium supplemented with 10% fetal bovine serum (Cambrex), 1% L-Glutamine, 1% Hepes, 1% non-essential amino acids and antibiotics. Mitotic cells from an asynchronously growing population treated with DMSO for 3 hrs were isolated by gentle shake off and held in a centrifuge tube for 30 min to allow completion of mitosis in suspension. Parallel cultures were incubated in 0.1μM NOC for 3hrs, washed twice in saline and maintained in complete medium for 30 min to allow mitotic progression. Cultures were then subjected to mitotic shake off as above. Thereafter, cells were centrifuged at 1100 rpm for 7 min and seeded on 22×22 mm coverslips for further analyses. For midbody analysis cells were cytospun on Poly-L-Lysine coated coverslips at the end of the suspension time. Re-seeded cells were harvested 4 hrs later to count binucleated cell frequencies. At that time 30 μM BrdU was supplied to cultures that were harvested 16 hrs later for BrdU immunostaining. At the same time, parallel cultures were harvested for p53 immunostaining. Cyclin B and p53 co-staining was performed on cultures grown for 40 hrs after cytokinesis in suspension Adherent cells treated with 5 μM camptothecin for 24 hrs served as positive control for p53 induction.
Immunostaining and microscopy analyses
Cells were rinsed in PHEM (60 mM PIPES, 25 mM Hepes, 10 mM EGTA, 2 mM MgCl2), fixed for 10 min with 3.7% formaldehyde in PHEM and permeabilized 5 min with 0.1% Triton-X100. For BrdU detection, fixed cells were treated with 1N HCl solution for 30 min at room temperature. Thereafter, coverslips were blocked in PBS containing 20% goat serum for 30 min at 37°C, before being processed for immunofluorescence. Antibodies were used at the following dilutions: anti-α-Tubulin FITC-conjugate (F2168, Sigma-Aldrich, 1:100), anti-p53 (M7001, Dako 1:100), anti-BrdU (M0744, Dako 1:50), anti cyclin B (sc-245, Santa Cruz, 1:50), anti-γH2AX (1:400). Secondary antibodies conjugated to Alexa-488 (Molecular Probes) or X-Red (Jackson Laboratories) were used as recommended by the supplier. Detection of actin fiber was obtained by rhodamine-conjugate phalloidin staining (R415, Molecular Probes). DNA was counterstained with 0.1 μg/ml 4'-6'-Diamidino-2-phenylindole (DAPI, Sigma-Aldrich). All preparations were examined under an Olympus Vanox microscope equipped with a 100X (1.35 NA) oil immersion objective and a SPOT CCD camera (Diagnostic Instruments). Color-encoded images were acquired using ISO 2000 software (Deltasistemi). Mononucleated cells in adherence cultures and mononucleated and binucleated cells obtained 20 hrs after cytokinesis in suspension were analyzed for BrdU incorporation and p53 nuclear accumulation by visual inspection. For p53 analysis on cyclin B positive cells at 40 hrs, all images were acquired at identical exposure settings and fluorescence intensity of the nuclear area was measured using NIH ImageJ 1.3 software. Cleavage furrow width was measured on phase contrast images using the same software.
Metaphase preparation and chromosome analysis
Metaphase preparations of MRC-5 cells were obtained 48 and 96 hrs after re-seeding from cytokinesis in suspension, since previous work on the same cell line showed the presence of the first mitotic peak at 48 hrs after seeding on coverslips.40 Cells were harvested after 3 hr incubation in 30 μM colchicine, washed with PBS buffer and treated with an hypotonic KCl solution (5.6 g/l) for 10 min at 37°C. Thereafter, cells were fixed by several incubations in a 3:1 MeOH/acetic acid mixture followed by centrifugation at 1100 rpm for 7 min. Metaphase spreads were obtained by dropping cell suspension on glass slides that were successively air dried at room temperature. Slides were stained in a 5% Giemsa solution for 20 min and rinsed with water. At least 100 metaphases were observed for each experimental point and chromosome numbers were classified as ≈2n (41-50) or ≈4n (80-96).
Supplementary Material
Abbreviations
- NOC
nocodazole
- SA-β-gal
senescence-associated β-galactosidase
- CPT
camptothecin
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Mario Fiore for his excellent technical assistance. We also thank I. Vitale and R. Cozzi for providing reagents.
Funding
Work in our laboratory is supported by grants from Consiglio Nazionale delle Ricerche (CNR Interomics Flagship Project, grant IBISA). LG received a postdoctoral fellowship from the Fund for Scientific Research–Flanders (FWO-Vlaanderen).
References
- 1.Davoli T, de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol 2011; 27:585-610; PMID:21801013; http://dx.doi.org/ 10.1146/annurev-cellbio-092910-154234 [DOI] [PubMed] [Google Scholar]
- 2.Storchova Z, Kuffer C. The consequences of tetraploidy and aneuploidy. J Cell Sci 2008; 121:3859-66; PMID:19020304; http://dx.doi.org/ 10.1242/jcs.039537 [DOI] [PubMed] [Google Scholar]
- 3.Zasadil LM, Britigan EM, Weaver BA. 2n or not 2n: Aneuploidy, polyploidy and chromosomal instability in primary and tumor cells. Sem Cell Dev Biol 2013; 24:370-9; http://dx.doi.org/ 10.1016/j.semcdb.2013.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganem NJ, Pellman D. Limiting the proliferation of polyploid cells. Cell 2007; 131:437-40; PMID:17981108; http://dx.doi.org/ 10.1016/j.cell.2007.10.024 [DOI] [PubMed] [Google Scholar]
- 5.Dewhurst SM, McGranahan N, Burrell RA, Rowan AJ, Gronroos E, Endesfelder D, Joshi T, Mouradov D, Gibbs P, Ward RL, et al.. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov 2014; 4:175-85; PMID:24436049; http://dx.doi.org/ 10.1158/2159-8290.CD-13-0285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005; 437:1043-7; PMID:16222300; http://dx.doi.org/ 10.1038/nature04217 [DOI] [PubMed] [Google Scholar]
- 7.Duelli DM, Padilla-Nash HM, Berman D, Murphy KM, Ried T, Lazebnik Y. A virus causes cancer by inducing massive chromosomal instability through cell fusion. Curr Biol 2007; 17:431-7; PMID:17320392; http://dx.doi.org/ 10.1016/j.cub.2007.01.049 [DOI] [PubMed] [Google Scholar]
- 8.Davoli T, de Lange T. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell 2012; 21:765-76; PMID:22698402; http://dx.doi.org/ 10.1016/j.ccr.2012.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valente D, Bossi G, Moncada A, Tornincasa M, Indelicato S, Piscuoglio S, Karamitopoulou ED, Bartolazzi A, Pierantoni GM, Fusco A, et al.. HIPK2 deficiency causes chromosomal instability by cytokinesis failure and increases tumorigenicity. Oncotarget 2015; 6:10320-34; PMID:25868975; http://dx.doi.org/ 10.18632/oncotarget.3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lanni JS, Jacks T. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol Cell Biol 1998; 18:1055-64; PMID:9448003; http://dx.doi.org/ 10.1128/MCB.18.2.1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casenghi M, Mangiacasale R, Tuynder M, Caillet-Fauquet P, Elhajouji A, Lavia P, Mousset S, Kirsch-Volders M, Cundari E. p53-independent apoptosis and p53-dependent block of DNA rereplication following mitotic spindle inhibition in human cells. Exp Cell Res 1999; 250:339-50; PMID:10413588; http://dx.doi.org/ 10.1006/excr.1999.4554 [DOI] [PubMed] [Google Scholar]
- 12.Demidenko ZN, Kalurupalle S, Hanko C, Lim CU, Broude E, Blagosklonny MV. Mechanism of G1-like arrest by low concentrations of paclitaxel: next cell cycle p53-dependent arrest with sub G1 DNA content mediated by prolonged mitosis.Oncogene. 2008; 27:4402-10; PMID:18469851; http://dx.doi.org/ 10.1038/onc.2008.82 [DOI] [PubMed] [Google Scholar]
- 13.Andreassen PR, Lohez OD, Lacroix FB, Margolis RL. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell 2001; 12:1315-28; PMID:11359924; http://dx.doi.org/ 10.1091/mbc.12.5.1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uetake Y, Sluder G. Cell cycle progression after cleavage failure: mammalian somatic cells do not possess a "tetraploidy checkpoint." J Cell Biol 2004; 165:609-15; PMID:15184397; http://dx.doi.org/ 10.1083/jcb.200403014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong C, Stearns T. Mammalian cells lack checkpoints for tetraploidy, aberrant centrosome number, and cytokinesis failure. BMC Cell Biol 2005; 6:6; PMID:15713235; http://dx.doi.org/ 10.1186/1471-2121-6-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krzywicka-Racka A, Sluder G. Repeated cleavage failure does not establish centrosome amplification in untransformed human cells. J Cell Biol 2011; 194:199-207; PMID:21788368; http://dx.doi.org/ 10.1083/jcb.201101073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho CC, Hau PM, Marxer M, Poon RY. The requirement of p53 for maintaining chromosomal stability during tetraploidization. Oncotarget 2010; 1:583-95; PMID:21317454; http://dx.doi.org/ 10.18632/oncotarget.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panopoulos A, Pacios-Bras C, Choi J, Yenjerla M, Sussman MA, Fotedar R, Margolis RL. Failure of cell cleavage induces senescence in tetraploid primary cells. Mol Biol Cell 2014; 25:3105-18; PMID:25143403; http://dx.doi.org/ 10.1091/mbc.E14-03-0844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuffer C, Kuznetsova AY, Storchova Z. Abnormal mitosis triggers p53-dependent cell cycle arrest in human tetraploid cells. Chromosoma 2013; 122:305-18; PMID:23624524; http://dx.doi.org/ 10.1007/s00412-013-0414-0 [DOI] [PubMed] [Google Scholar]
- 20.Ganem NJ, Cornils H, Chiu SY, O'Rourke KP, Arnaud J, Yimlamai D, Thery M, Camargo FD, Pellman D. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 2014; 158:833-48; PMID:25126788; http://dx.doi.org/ 10.1016/j.cell.2014.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thullberg M, Stromblad S. Anchorage-independent cytokinesis as part of oncogenic transformation? Cell Cycle 2008; 7:984-8; PMID:18414025; http://dx.doi.org/ 10.4161/cc.7.8.5674 [DOI] [PubMed] [Google Scholar]
- 22.Ben-Ze'ev A, Raz A. Multinucleation and inhibition of cytokinesis in suspended cells: reversal upon reattachment to a substrate. Cell 1981; 26:107-15; PMID:6799205; http://dx.doi.org/ 10.1016/0092-8674(81)90038-6 [DOI] [PubMed] [Google Scholar]
- 23.Reverte CG, Benware A, Jones CW, LaFlamme SE. Perturbing integrin function inhibits microtubule growth from centrosomes, spindle assembly, and cytokinesis. J Cell Biol 2006; 174:491-7; PMID:16908668; http://dx.doi.org/ 10.1083/jcb.200603069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thullberg M, Gad A, Le Guyader S, Stromblad S. Oncogenic H-Ras V12 promotes anchorage-independent cytokinesis in human fibroblasts. Proc Natl Acad Sci USA 2007; 104:20338-43; PMID:18077377; http://dx.doi.org/ 10.1073/pnas.0706609105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 26.Sambandamoorthy S, Mathew-Steiner S, Varney S, Zuidema JM, Gilbert RJ, Van De Water L, LaFlamme SE. Matrix compliance and the regulation of cytokinesis. Biol Open 2015; 4:885-92; PMID:26002930; http://dx.doi.org/ 10.1242/bio.011825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uetake Y, Sluder G. Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr Biol 2010; 20:1666-71; PMID:20832310; http://dx.doi.org/ 10.1016/j.cub.2010.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ciciarello M, Mangiacasale R, Casenghi M, Zaira Limongi M, D'Angelo M, Soddu S, Lavia P, Cundari E. p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J Biol Chem 2001; 276:19205-13; PMID:11376010; http://dx.doi.org/ 10.1074/jbc.M009528200 [DOI] [PubMed] [Google Scholar]
- 29.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273:5858-68; PMID:9488723; http://dx.doi.org/ 10.1074/jbc.273.10.5858 [DOI] [PubMed] [Google Scholar]
- 30.Giaccia AJ, Kastan MB. The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev 1998; 12:2973-83; PMID:9765199; http://dx.doi.org/ 10.1101/gad.12.19.2973 [DOI] [PubMed] [Google Scholar]
- 31.Li M, Fang X, Baker DJ, Guo L, Gao X, Wei Z, Han S, van Deursen JM, Zhang P. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc Natl Acad Sci USA 2010; 107:14188-93; PMID:20663956; http://dx.doi.org/ 10.1073/pnas.1005960107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O'Connell MJ, Cimprich KA. G2 damage checkpoints: what is the turn-on? J Cell Sci 2005; 118:1-6; PMID:15615778; http://dx.doi.org/ 10.1242/jcs.01626 [DOI] [PubMed] [Google Scholar]
- 33.King C, Diaz H, Barnard D, Barda D, Clawson D, Blosser W, Cox K, Guo S, Marshall M. Characterization and preclinical development of LY2603618: a selective and potent Chk1 inhibitor. Invest New Drugs 2014; 32:213-26; PMID:24114124; http://dx.doi.org/ 10.1007/s10637-013-0036-7 [DOI] [PubMed] [Google Scholar]
- 34.Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell 2007; 130:223-33; PMID:17662938; http://dx.doi.org/ 10.1016/j.cell.2007.07.003 [DOI] [PubMed] [Google Scholar]
- 35.Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev 2000; 14:289-300; PMID:10673501 [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene 2001; 20:1803-15; PMID:11313928; http://dx.doi.org/ 10.1038/sj.onc.1204252 [DOI] [PubMed] [Google Scholar]
- 37.Pellinen T, Tuomi S, Arjonen A, Wolf M, Edgren H, Meyer H, Grosse R, Kitzing T, Rantala JK, Kallioniemi O, et al.. Integrin trafficking regulated by Rab21 is necessary for cytokinesis. Dev Cell 2008; 15:371-85; PMID:18804435; http://dx.doi.org/ 10.1016/j.devcel.2008.08.001 [DOI] [PubMed] [Google Scholar]
- 38.Wood S, Sivaramakrishnan G, Engel J, Shafikhani SH. Cell migration regulates the kinetics of cytokinesis. Cell Cycle 2011; 10:648-54; PMID:21293189; http://dx.doi.org/ 10.4161/cc.10.4.14813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hognas G, Tuomi S, Veltel S, Mattila E, Murumagi A, Edgren H, Kallioniemi O, Ivaska J. Cytokinesis failure due to derailed integrin traffic induces aneuploidy and oncogenic transformation in vitro and in vivo. Oncogene 2012; 31:3597-606; PMID:22120710; http://dx.doi.org/ 10.1038/onc.2011.527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cimini D, Antoccia A, Tanzarella C, Degrassi F. Topoisomerase II inhibition in mitosis produces numerical and structural chromosomal aberrations in human fibroblasts. Cytogenet Cell Genet 1997; 76:61-7; PMID:9154130; http://dx.doi.org/ 10.1159/000134517 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.