

Abstract
Necropsy records and associated clinical histories from the rhesus macaque colony at the California National Primate Research Center were reviewed to identify mortality related to cardiac abnormalities involving left ventricular hypertrophy (LVH). Over a 21-y period, 162 cases (female, 90; male, 72) of idiopathic LVH were identified. Macaques presented to necropsy with prominent concentric hypertrophy of the left ventricle associated with striking reduction of the ventricular lumen. Among all LVH cases, 74 macaques (female, 39; male, 35), mostly young adults, presented for spontaneous (sudden) death; more than 50% of these 74 cases were associated with a recent history of sedation or intraspecific aggression. The risk of sudden death in the 6- to 9-y-old age group was significantly higher in male macaques. Subtle histologic cardiac lesions included karyomegaly and increased cardiac myocyte diameter. Pedigree analyses based on rhesus macaque LVH probands suggested a strong genetic predisposition for the condition. In humans, hypertrophic cardiomyopathy (HCM) is defined by the presence of unexplained left ventricular hypertrophy, associated with diverse clinical outcomes ranging from asymptomatic disease to sudden death. Although the overall risk of disease complications such as sudden death, end-stage heart failure, and stroke is low (1% to 2%) in patients with HCM, the absolute risk can vary dramatically. Prima facie comparison of HCM and LVH suggest that further study may allow the development of spontaneously occurring LVH in rhesus macaques as a useful model of HCM, to better understand the pathogenesis of this remarkably heterogeneous disease.
Abbreviations: HCM, hypertrophic cardiomyopathy; LVH, left ventricular hypertrophy
Naturally occurring hypertrophic cardiomyopathy (HCM) has been recognized in a variety of species including pigs,13 cats18,32 and humans. HCM emerged as an accepted clinical entity in humans during the late 1950s, with the publication of 2 key papers.5,46 Since then, a vast, complex, and sometimes contradictory body of research has developed around this clinically, phenotypically, and genotypically heterogeneous disease. HCM is defined as left ventricular hypertrophy without chamber dilation in the absence of either a systemic or other cardiac disease that would result in pressure overload and compensatory hypertrophy.22,39 The primary physiologic abnormality in HCM is reduced stroke volume due to impaired diastolic filling, which is secondary to reduced chamber size and impaired relaxation of the left ventricular myocardium during diastole; this impaired relaxation is due to the reduced compliance or increased stiffness of the hypertrophied left ventricle.49
Phenotypically, hearts affected by HCM exhibit a wide variety of changes summarized as left ventricular hypertrophy, which may be symmetric or asymmetric and which results in reduced left ventricular lumen volume, reduced stroke volume, and typically increased ejection fraction. These changes can be accompanied by valvular changes, some of which can be obstructive. Small intramural coronary arteries may have thickened walls and narrowed lumens due to proliferation of smooth muscle cells and collagen.30 Histologic changes in the left ventricle associated with—but not prerequisite for—HCM include myocyte hypertrophy and disarray, nuclear atypia, and expansion of the interstitial collagen compartment.14,22
In the 1980s, growing recognition that HCM was familial directed efforts toward identifying a genetic defect. Primary HCM in humans has been identified as an autosomal dominant disease with variable penetrance and a remarkable diversity in clinical presentation and disease course.27,40 The first mutation associated with HCM was a missense mutation in the gene encoding the β-myosin heavy chain.11 More than 2 decades of subsequent investigation has demonstrated the extensive heterogeneity of the HCM phenotype, with at least 11 causative genes and more than 1400 mutations identified. These genes primarily encode thick and thin myofilament proteins of the sarcomere or Z disc (sarcomere structural proteins). The majority of mutations occur in 3 genes—β-myosin heavy chain, myosin-binding protein C, and troponin T—whereas other genes, including troponin I, α-tropomyosin and α-actin, account for a smaller proportion of patients.22 Mutations in several additional sarcomere and calcium-handling genes have been proposed but with less evidence to support pathogenicity. At present, the precise mutation does not alter management. However, adverse outcomes (sudden death, stroke, progressive symptoms) are more prominent in patients with sarcomere mutations than in those without an identifiable mutation.37
In other animal species, naturally occurring familial HCM has been best characterized in cats. Maine coon cats18 and ragdoll cats32 develop HCM that is strongly similar to the human disease in clinical presentation and histopathology. Similar to humans, HCM in cats is inherited as an autosomal dominant trait, with the responsible gene, the cardiac myosin-binding protein C gene, recently identified.32,33 In addition, a genetically manipulated model has been developed in rabbits,21 as well as a vast array of mouse models.1,7,45
The purposes of this report were: 1) to identify cases of LVH diagnosed at necropsy in the rhesus macaque colony at the California National Primate Research Center since 1992; 2) to provide a preliminary pathologic characterization of these cases; and 3) to compare and contrast rhesus LVH and human HCM to assess the extent to which LVH might be developed as a model of HCM in a species closely related to humans.
Materials and Methods
The California National Primate Research Center is an AAALAC-accredited facility where animals were cared for in accordance with the USDA Animal Welfare Act and regulations and the Guide for the Care and Use of Laboratory Animals.2,3,15 All cases of LVH in rhesus macaques that were necropsied at the center since the entity was first recognized in 1983 were identified by conducting a search of the pathology database and by cross-referencing the cases with the necropsy log. Because cases prior to 1992 were sporadic and because some diagnoses could not be confirmed, these cases were excluded, as were diagnoses of right ventricular hypertrophy and generalized cardiac hypertrophy. The sex, cause of death, and age at death were recorded for each macaque, and the sires and dams of affected animals were identified when possible. For the macaques that had died spontaneously, any history of a possible precipitating cause, including trauma or sedation immediately prior to death, was noted from the animal records. Statistical analysis was performed in the R programming environment (version 3.1.3; https://www.r-project.org/). The function chisq.test was used to generate the χ2 contingency table tests. P values were computed by using the Pearson χ2 test with Yates continuity correction for data tables with sufficient sample size; otherwise, they were computed by using the Pearson χ2 test with simulated P value based on 2000 replicates. Statistical significance was defined as a P value less than 0.05.
A complete gross necropsy was conducted on each of the macaques included in this study. Prior to September 2007, LVH was defined by a markedly thickened left ventricular wall accompanied by near-complete obliteration of the left ventricular lumen. Although it is possible, even likely, that we missed less severe or early examples of LVH that we hope to identify in the future, we elected to be conservative and include only the most unequivocal cases in this report. Beginning in September 2007, the ratio of the left ventricular external diameter to internal luminal diameter (O:I ratio) has increasingly been used as a defining parameter in the diagnosis of LVH. To obtain this measure, each macaque heart was sectioned transversely midway between the apex and the base, and the diameters of the left ventricle and its lumen were measured (Figure 1). Similar measurements were taken from 132 (male, 50; female, 82) normal control macaques, in which the O:I ratio (mean ± 1 SD) was calculated to be 2.0 ± 0.3. Therefore an O:I ratio greater than 3 was considered to conservatively constitute LVH, given that this measure was more than 2 SD above the mean for the controls. In addition, in a preliminary effort to gather more data, a small cohort of 18 macaques—9 with LVH, including both animals that had died suddenly and those in which LVH was an incidental diagnosis at scheduled necropsy, and 9 controls, underwent more comprehensive evaluation. In this subset, the O:I ratio was measured and calculated as previously described (Figure 1), and then the thicknesses of the left and right ventricular free walls and the interventricular septum in the same plane of section were measured.
Figure 1.

Standardized assessment of ventricular thickness and lumen diameter involved a cross-section of the heart midway between the apex and the base and determined O (the external diameter of the left ventricle) and I (the internal diameter). An O:I ratio greater than 3 supported a diagnosis of left ventricular hypertrophy.
Routine histopathology performed on 85% of the cases of LVH included examination of the left ventricle, septum, and right ventricle, as well as atrioventricular, aortic, and pulmonary valves. However, standard histopathologic examination of our LVH cases revealed no overt lesions matching those associated with HCM. Therefore, in a preliminary effort to more fully characterize the histopathology of this condition, formalin-fixed hearts from 3 macaques with severe LVH who had died suddenly and 3 age-matched controls were serially sectioned transversely through both left and right ventricles, and alternating sections were stained with hematoxylin and eosin, Masson trichrome, and an immunohistochemical stain for Ki67, a marker of cell proliferation.
Results
During the period of January 1992 through May 2014, 162 cases (female, 90; male, 72) in rhesus macaques were identified as LVH at the time of necropsy. Diagnoses of right ventricular hypertrophy or generalized hypertrophy were rare (4 macaques in total) and were not included. Although sporadic cases were diagnosed as early as 1983, LVH has been diagnosed much more frequently since 1992. Of the total number of necropsies conducted from 1992 through 2014, LVH was diagnosed in 1.30% of the female macaques and 1.28% of the male animals. Figure 2 illustrates a pedigree assembled by using 8 generations of animal records; this is the largest of 4 family pedigrees. Some sires and dams have produced multiple offspring that were affected by LVH. In addition, 1 female and 12 male macaques (not shown) have produced 2 or more affected offspring, and 3 of the male macaques each has sired 3 or 4 affected offspring.
Figure 2.
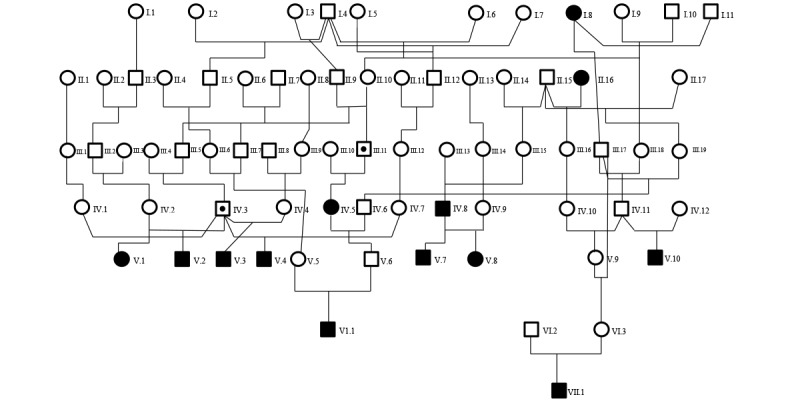
Pedigree of rhesus macaques with known left ventricular hypertrophy. For simplicity, only macaques directly involved in the transmission of the disease phenotype to the probands are displayed; many more unaffected macaques, including close and distant relatives, have been omitted from the pedigree. Open squares and circles represent unaffected male and female macaques, respectively; shaded squares and circles represent affected male and female macaques (or probands), respectively. Dots indicate carriers, but their absence does not necessarily indicate that the subject is not a carrier. Generations are labeled in Roman numerals, whereas subjects within each generation are indicated in Arabic numerals.
Sudden death.
Of the macaques diagnosed with LVH, 74 (46%) died spontaneously without evidence of any other life-threatening lesions (hereafter referred to as sudden death), whereas 88 (54%) were diagnosed after euthanasia for a scheduled necropsy. Of the 74 cases of sudden death, 39 were female and 35 male. When the ratio of males to females in the rhesus colony was considered, the estimated prevalence of sudden death was 0.06% in the female population and 0.09% in the male population (χ2 test, P = 0.13). Of the 74 macaques that died suddenly, 41 (55%) had a history of a precipitating event that might have increased their risk for sudden death in the face of cardiac insufficiency. Specifically, 14 macaques had received sedation just prior to death, and the remaining 27 macaques had indications (that is, characteristic musculocutaneous trauma) of aggressive behavior directed against them by other animals. Although macaques that died of sudden death ranged in age from a few weeks to older than 20 y, the largest number of cases occurred during early adulthood (mean age, 6.1 y). When sex was taken into account, the mean age at death was 7.2 y for females and 5.2 y for males; this sex-related difference was not statistically significant (unpaired t test, P = 0.13). However, the age distribution of the male and female animal populations within the colony differs quite markedly. Once this distribution was considered in context with the numbers of female and male sudden deaths by age group (0 to 3 y, 3 to 6 y, 6 to 9 y, 9 to 12 y, and older than 12 y), the number of sudden deaths in the 6- to 9 y-old group was greater for male macaques than female macaques (χ2 test: 6 to 9 y, P = 0.0014; 0 to 3 y, P = 0.77; 3 to 6 y, P = 0.21; 9 to 12 y, P = 0.43; older than 12 y, P = 0.32).
Gross pathology.
The position of the heart in the chests of macaques with LVH varied from a perpendicular diaphragmatic orientation to left deviation of the apex. Cardiac shape ranged from unaltered to elongated oval, and the hypertrophic left ventricle was markedly firm on palpation. On transverse section midway between apex and base, hearts with LVH showed circumferential thickening of the left ventricle with accompanying severe decrease in lumen size (Figure 3). Some cases showed hypertrophy of left ventricular papillary muscle, which has been suggested to represent either a subtype or early form of cardiac hypertrophy.4,19 No other structural abnormalities in the cardiac valves or great blood vessels were identified. In macaques from which measurements were obtained, a ratio of the ventricular diameter to luminal diameter (O:I ratio) that exceeded 3, together with observations at gross necropsy, was used to support the diagnosis of LVH. When a subgroup comprising 9 macaques with LVH and 9 controls was examined in detail, the left ventricular free wall and interventricular septum were found to be significantly thicker in the macaques with LVH, but the thickness of the right ventricular free wall did not differ between the 2 groups (unpaired t test: left ventricle, P = 0.0003; septum, P = 0.0002; right ventricle, P = 0.61). This finding correlated with a significant difference in the O:I ratio between LVH-diagnosed and control macaques (unpaired t test, P = 0.016; Figure 4).
Figure 3.

(A) A cross-section of the heart from a rhesus macaque diagnosed with left ventricular hypertrophy, where the left ventricle is concentrically thickened and the lumen markedly reduced such that it is difficult to identify, is compared with (B) that of an-age matched control, where the thickness of the left ventricular wall and the diameter of the lumen are considered to be within normal limits. Bar, 1 cm.
Figure 4.

Heart measurements from 9 rhesus macaques with LVH, including animals that died suddenly and animals with an incidental diagnosis of LVH at necropsy, and 9 control animals. All measurements were taken in the same transverse plane of section of the heart. (A) The left ventricular free wall and the interventricular septum were significantly (*, P < 0.05) thicker in the macaques with LVH compared with controls, whereas the thickness of the right ventricle did not differ between the groups. (B) The Tukey plot of these same populations shows that the O:I ratio was significantly increased in the macaques with LVH. The arrowhead denotes an outlier.
Histopathology.
Because routine histologic analysis of limited sections of the hearts of macaques with LVH revealed none of the changes typically reported in human cases of HCM, we examined in detail the hearts from 3 macaques with severe LVH that had died suddenly and 3 hearts from age-matched controls. In both LVH and control animals, serial sectioning of the heart demonstrated myocytes in close apposition, with myocyte profiles ranging from longitudinal to oblique and transverse. Although control animals had mild to moderate anisokaryosis, karyomegaly was prevalent and severe in the most severe cases of LVH. In addition, in extensive regions throughout the free wall of the left ventricle and the interventricular septum in LVH hearts, myocytes with an increased diameter were interspersed among smaller diameter fibers (Figure 5). Myocytes appeared to be viable, with cytoplasm striated by myofibrils and regularly spaced intercalated discs; there was no evidence of hypercontraction resulting in necrosis (contraction band necrosis). Neither control macaques nor those with LVH showed positive nuclear immunostaining for Ki67, a marker of cell proliferation, in cardiac myocytes. Myocardial interstitium was typically scant and inconspicuous in the hearts of both LVH and control macaques. Trichrome staining to highlight collagen did not demonstrate increased fibrosis or expansion of the interstitial matrix in the macaques with LVH, nor did it demonstrate areas of myocardial tissue loss and collapse. Examination of representative sections of the main organs of macaques that died revealed no lesions in other organ systems (for example, lungs) that indicated any degree of cardiac failure or hypertension prior to death.
Figure 5.

Histologic sections from the left ventricular free wall of (A) a macaque diagnosed with severe left ventricular hypertrophy after sudden death and (B) an age-matched control. The section from the macaque with severe left ventricular hypertrophy demonstrates karyomegaly and hypertrophied myocytes (arrows) interspersed among those within normal limits. Hematoxylin and eosin stain; bar, 50 µm.
Discussion
The current study is the first to report a large cohort of cases of LVH in rhesus macaques that presents as a pathologic entity resembling HCM. As in HCM, the primary lesion in LVH is left ventricular hypertrophy that often goes undetected until necropsy and that is associated with a substantial percentage of cases presenting as sudden death most likely related to cardiac insufficiency under stress.
In humans, HCM can be macroscopically symmetrical or asymmetric, with the symmetrical form accounting for approximately 42% of cases. Both forms occasionally (17% of cases) show right ventricular involvement.8 The classic lesions associated with HCM involve thickening of the basal anterior interventricular septum, which bulges beneath the aortic valve and causes narrowing of the left ventricular outflow tract.46 As a result, on cross section, the ventricular cavity loses its usual ovoid shape and may be compressed into a banana-like configuration. However, HCM is now understood as primarily a nonobstructive disease, and 75% of patients have no sizable resting out-flow tract gradient.22 In macaques with LVH, myocardial hypertrophy was symmetrical, with notable thickening of both left ventricular free wall and interventricular septum and with occasionally prominent papillary muscle hypertrophy. These changes were accompanied by a correspondingly marked decrease in the size of the ventricular lumen but no obstruction of the ventricular outflow tract. Although originally the gross description of thickened left ventricle was subjective, only cases with clear-cut hypertrophy were chosen for the current cohort (Figure 3). In addition, since 2007, we have gradually standardized the approach to examining the heart at gross necropsy, incorporating the O:I ratio of the left ventricle to support the diagnosis and prospectively, with more data, to detect cases of mild to moderate LVH in early stages of disease progression. Detailed data from a small cohort of 9 cases of LVH (Figure 4) demonstrated that the thicknesses of the left ventricular free wall and interventricular septum are increased significantly in these cases, supporting the use of the O:I ratio in the diagnosis of LVH (Figure 1).
The histologic lesion most often associated with HCM in humans is disorganization of the myocardial architecture in the left ventricle. Hypertrophied cardiac myocytes have bizarre shapes, vary in size and shape, and form multiple intercellular connections often arranged in chaotic alignment at oblique and perpendicular angles, producing a herringbone or pinwheel pattern.8,14,24,46 This disarray is accompanied by expansion of the interstitial collagen compartment,43 and collagen fibrils can be widely distributed, occupying a substantial portion of the left ventricular wall (average, 33%). Within myocytes, the myofibrillar architecture is disorganized, and nuclei may be bizarre; these changes are reported to be more extensive in young patients who die of their disease.24,47 Myocyte disarray, however, is not pathognomonic for HCM and can occur in the face of an array of other heart diseases, including syndromes where left ventricular hypertrophy is a feature (Noonan syndrome and Friedreich's ataxia), and in congenital heart disease. Another reported feature of HCM is small-vessel disease characterized by intramural coronary arteries narrowed by medial hypertrophy. The clinical significance of this lesion is uncertain, but small-vessel disease may contribute to myocardial ischemia in some patients.22,48
Our macaques demonstrated relatively subtle histologic lesions characterized predominantly by an increased prevalence of karyomegaly and of regions where myocytes with an enlarged diameter were interspersed between smaller myocytes. We did not note any myocyte disarray or interstitial fibrosis, nor did we see medial hypertrophy in intramural coronary arteries. We performed detailed examinations of 3 macaques with LVH, including evaluating numerous histologic sections and carefully comparing LVH macaques with 3 control animals, before we were confident in diagnosing subtle histologic lesions. The disorganized myocyte architecture and expanded collagenous matrix have been suggested to predispose subjects to the generation of arrhythmias, life-threatening electrical instability, and sudden death.28 However, inconsistencies between these reported histologic lesions and sudden death exist. Indeed, these lesions do not develop in our macaques, yet sudden death remains the primary clinical presentation. In humans, disorganized myocardial architecture is not confined to the regions of the left ventricle that exhibit maximal hypertrophy but can occur in regions with normal to mild thickening.31 This pattern challenges the theory that the degree of hypertrophy is related to the degree of architectural disorganization, which, in turn, is related to clinical outcome; these findings further suggest that rhesus macaques with LVH may be useful in unraveling these associations, where hypertrophy is seen in the absence of overt architectural disorganization or clear vascular changes. None of the animals in the current study population provided any evidence of pericardial, hypertensive, congenital, valvular or ischemic disease which might have resulted in secondary myocardial hypertrophy; we therefore were left to conclude that myocardial hypertrophy was indeed primary.
Most human patients with HCM have few (if any) symptoms, and the diagnosis is usually made during family screening or incidentally, as occurs in the majority of LVH cases identified in our colony. If symptoms do develop, the clinical course for patients with HCM can progress along several discrete pathways: sudden death, congestive heart failure, and atrial fibrillation (possibly with thrombosis and stroke). However, predicting which pathway disease might take in any patient or which subjects might remain symptom-free is not yet possible. In our colony, the only clinical presentation identified to date is sudden death, with the diagnosis of LVH made at necropsy and according to the appearance of the heart and the lack of evidence of other causes of morbidity or mortality. In humans, sudden death is the most devastating and unpredictable consequence of HCM, predominantly occurring in young adults, in whom sudden death can be the initial manifestation of HCM with no or mild prior symptoms.23 Most people who inherit a disease-causing mutation demonstrate HCM by early adulthood, with substantial remodeling of the left ventricle, with the development of left ventricular hypertrophy typically occurring with accelerated body growth during adolescence, and with increased risk of sudden death possibly coinciding with the pubertal growth phase.29,30 Patients with extreme hypertrophy appear to be at greatest risk of sudden death,44 and although not all cases are associated with vigorous physical exertion, HCM is the most common cause of sudden death in competitive athletes in the United States.25
As in humans with HCM,38 our macaques with LVH showed no significant difference in the overall incidence of sudden death between males and females. When age was taken into consideration, male macaques in the 6- to 9-y age group had a significantly increased risk of sudden death when compared with females of the same age. However, the male macaques in this age group might be particularly physically active and competing for position within the colony hierarchy. Consequently, the apparent male sex predilection for sudden death in this age group in our colony might be related to social and behavioral influences that make these macaques similar to competitive athletes. HCM has increasingly been identified in children prior to puberty,34 and although the causes appear to be more diverse than genetics alone, the gross lesions in these pediatric cases resemble the HCM of adult humans. Genetic mutations in genes routinely screened in adults with HCM account for approximately half of all cases of childhood-onset HCM.34,35 Syndromic and metabolic causes may account for most of the remaining cases. Similarly, LVH at the California National Primate Research Center has been diagnosed in several macaques prior to puberty and even in some that were younger than 1 y of age. Whether the pathogenesis of disease in this prepubertal, 0- to 3-y age group is similar to or different from that in the older animals remains to be investigated.
The pathogenesis of sudden death is thought to be ventricular arrhythmias (tachycardia and ventricular fibrillation), as suggested by evidence gathered from implantable defibrillators in patients with HCM,26 and the unstable electrophysiologic environment has been proposed to arise in response to the classic HCM histopathology (myocyte disarray and myocardial fibrosis) or to possible vascular lesions.9 However, this relationship is tenuous and inconsistent,6,47 rendering it controversial, because few rigorous studies have documented a positive correlation between myocyte disarray or coronary vascular disease and sudden death. In addition, data from a mouse model of HCM dissociate myocyte disarray and fibrosis from increased susceptibility to arrhythmia,50 instead suggesting that disruption of sarcoplasmic reticulum Ca2+ homeostasis is an important early event and common pathway in the pathogenesis of HCM. L-type Ca2+ channel inhibitors, which restore the endoplasmic reticular stores of calcium, significantly reduced the incidence of ventricular hypertrophy in a mouse model of HCM41 and have been used to treat HCM in humans. The fact that LVH occurs in the absence of myocyte disarray and fibrosis may make the naturally occurring LVH in rhesus macaques a useful model in which to study ventricular hypertrophy uncomplicated by these changes, particularly the correlation between cardiac myocyte hypertrophy and sudden death.
A genetic basis for HCM in humans was established after the first genetic linkage study in a French Canadian family in 1989.16 Although the majority of genes identified encode proteins in the cardiac sarcomere, the number of mutations implicated is confounding and has proved to be of little prognostic importance.20 Disease expression varies not only between unrelated persons but also between members of the same family, suggesting that HCM is a complex inherited trait whose phenotype is influenced by other modifier genes and environmental factors. A role for genetic modifiers in hypertrophic response to the Arg403Gln missense mutation in the cardiac myosin heavy chain has been demonstrated in a mouse model bred onto different genetic backgrounds.42 In our current cohort of macaques, several sires had multiple affected offspring, suggesting that LVH may be genetically transmitted within this colony. Further inspection of the pedigrees of 108 probands has suggested that the prevalence of LVH in this colony is dominated by founder effects, although currently there is no compelling support for a dominant mode of inheritance.17 Figure 2 illustrates a pedigree assembled based on 8 generations of animal records; this is the largest of 4 family pedigrees that have been assembled from these records. The lack of a sufficient number of siblings with a parent diagnosed with LVH has so far precluded further analysis to test the mode of inheritance.
One of the major challenges to developing macaque LVH as a model of HCM is the identification of animals with LVH prior to observations by a pathologist. In humans, unexplained thickening of the left ventricular wall in 2-dimensional echocardiography is generally sufficient to make the diagnosis; therefore a targeted ultrasound screening program to identify animals with LVH, which we have begun, will allow prospective monitoring of new cases. With the identification of more probands, pedigree analyses will facilitate studies in captive rhesus macaques especially with the availability of large numbers of half-sibs, detailed colony records, and genetic material from 8 or more generations for phenotypic scoring and genotypic sampling. In addition, this detailed pedigree information can be used to manipulate the colony mating structure to minimize inbreeding and maximize the genetic contribution from all founders to their descendants.10 Furthermore, biomarkers may prove informative for identifying macaques earlier in the disease process and may assist selective breeding programs for maintaining LVH pedigrees as a useful model for elucidating the genetic mechanism of the disease. In humans, norepinephrine, atrial natriuretic peptide, and β-type natriuretic peptide have been proposed as biomarkers for HCM,36 and in cats, β-type natriuretic peptide is highly sensitive in detecting severe HCM.12
Over the past 30 y, the study of HCM has shifted its perception from that of a rare disease of the young to an increasingly diverse disease with variable clinical expression and natural history. The genetic, pathologic, and clinical heterogeneity that is the hallmark of HCM makes its diagnosis and treatment a major challenge. As we have described here, the naturally occurring LVH in rhesus macaques (which includes sudden death as a clinical presentation), if developed as a model of HCM, could be important in advancing our understanding of HCM as a cause of sudden death in young adults. One of the apparent differences between HCM and LVH is the absence of histologic changes, including myocyte disarray and fibrosis, in macaque LVH. This difference, however, may permit evaluation of the condition without the complicating presence of—and associated debate regarding—these features, possibly allowing the uncoupling of histologic changes from other features of this condition, perhaps at a cellular biochemical level. Furthermore, developing the capacity to identify a population of LVH-affected macaques antemortem could provide the means for investigating both the pathophysiology and potential treatment of this multifaceted disease, both of which may have application to HCM.
Acknowledgments
We acknowledge the expertise of Michael Manser, who performed the immunohistochemistry for this study; Sarah Mills, who created the figures; and Ross Allen, who compiled the colony population data.
Funding acknowledgment: NIH/OD P51 OD011107 05/01/10-04/30/15
References
- 1.Alcalai R, Seidman JG, Seidman CE. 2008. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol 19:104–110. [DOI] [PubMed] [Google Scholar]
- 2.Animal Welfare Act as Amended. 2013. 7 USC §2131–2159.
- 3.Animal Welfare Regulations. 2013. 9 CFR § 3.129.
- 4.Anversa P, Olivetti G, Melissari M, Loud AV. 1980. Stereological measurement of cellular and subcellular hypertrophy and hyperplasia in the papillary muscle of adult rat. J Mol Cell Cardiol 12:781–795. [DOI] [PubMed] [Google Scholar]
- 5.Brock R. 1957. Functional obstruction of the left ventricle; acquired aortic subvalvar stenosis. Guys Hosp Rep 106:221–238. [PubMed] [Google Scholar]
- 6.Cecchi F, Olivotto I, Gistri R, Lorenzoni R, Chiriatti G, Camici PG. 2003. Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med 349:1027–1035. [DOI] [PubMed] [Google Scholar]
- 7.Chung MW, Tsoutsman T, Semsarian C. 2003. Hypertrophic cardiomyopathy: from gene defect to clinical disease. Cell Res 13:9–20. [DOI] [PubMed] [Google Scholar]
- 8.Davies MJ, McKenna WJ. 1995. Hypertrophic cardiomyopathy—pathology and pathogenesis. Histopathology 26:493–500. [DOI] [PubMed] [Google Scholar]
- 9.Dilsizian V, Bonow RO, Epstein SE, Fananapazir L. 1993. Myocardial ischemia detected by thallium scintigraphy is frequently related to cardiac arrest and syncope in young patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 22:796–804. [DOI] [PubMed] [Google Scholar]
- 10.Dyke B, Gage TB, VandeBerg JL, King RH, Mamelka PM, Cheng ML, Goodwin WJ. 1987. Decision making in genetic management of primate breeding colonies. Genetica 73:137–144. [DOI] [PubMed] [Google Scholar]
- 11.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. 1990. A molecular basis for familial hypertrophic cardiomyopathy: a β-cardiac myosin heavy-chain gene missense mutation. Cell 62:999–1006. [DOI] [PubMed] [Google Scholar]
- 12.Hsu A, Kittleson MD, Paling A. 2009. Investigation into the use of plasma NT–proBNP concentration to screen for feline hypertrophic cardiomyopathy. J Vet Cardiol 11 Suppl 1:S63–S70. [DOI] [PubMed] [Google Scholar]
- 13.Huang SY, Tsou HL, Chiu YT, Shyu JJ, Wu JJ, Lin JH, Liu SK. 1996. Heritability estimate of hypertrophic cardiomyopathy in pigs (Sus scrofa domestica). Lab Anim Sci 46:310–314. [PubMed] [Google Scholar]
- 14.Hughes SE. 2004. The pathology of hypertrophic cardiomyopathy. Histopathology 44:412–427. [DOI] [PubMed] [Google Scholar]
- 15.Institute for Laboratory Animal Research. 2011. Guide for the care and use of laboratory animals, 8th ed. Washington (DC): National Academies Press. [Google Scholar]
- 16.Jarcho JA, McKenna W, Pare JA, Solomon SD, Holcombe RF, Dickie S, Levi T, Donis-Keller H, Seidman JG, Seidman CE. 1989. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med 321:1372–1378. [DOI] [PubMed] [Google Scholar]
- 17.Kanthaswamy S, Reader R, Tarara R, Oslund K, Allen M, Ng J, Grinberg C, Hyde D, Glenn DG, Lerche N. 2014. Large-scale pedigree analysis leads to evidence for founder effects of hypertrophic cardiomyopathy in rhesus macaques. J Med Primatol 43:288–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kittleson MD, Meurs KM, Munro MJ, Kittleson JA, Liu SK, Pion PD, Towbin JA. 1999. Familial hypertrophic cardiomyopathy in Maine coon cats: an animal model of human disease. Circulation 99:3172–3180. [DOI] [PubMed] [Google Scholar]
- 19.Kobashi A, Suwa M, Ito T, Otake Y, Hirota Y, Kawamura K. 1998. Solitary papillary muscle hypertrophy as a possible form of hypertrophic cardiomyopathy. Jpn Circ J 62:811–816. [DOI] [PubMed] [Google Scholar]
- 20.Landstrom AP, Ackerman MJ. 2010. Mutation type is not clinically useful in predicting prognosis in hypertrophic cardiomyopathy. Circulation 122:2441–2449, discussion 2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marian AJ, Wu Y, Lim DS, McCluggage M, Youker K, Yu QT, Brugada R, DeMayo F, Quinones M, Roberts R. 1999. A transgenic rabbit model for human hypertrophic cardiomyopathy. J Clin Invest 104:1683–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maron BJ. 2002. Hypertrophic cardiomyopathy: a systematic review. JAMA 287:1308–1320. [DOI] [PubMed] [Google Scholar]
- 23.Maron BJ. 2010. Contemporary insights and strategies for risk stratification and prevention of sudden death in hypertrophic cardiomyopathy. Circulation 121:445–456. [DOI] [PubMed] [Google Scholar]
- 24.Maron BJ, Anan TJ, Roberts WC. 1981. Quantitative analysis of the distribution of cardiac muscle cell disorganization in the left ventricular wall of patients with hypertrophic cardiomyopathy. Circulation 63:882–894. [DOI] [PubMed] [Google Scholar]
- 25.Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller FO. 2009. Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980-2006. Circulation 119:1085–1092. [DOI] [PubMed] [Google Scholar]
- 26.Maron BJ, Maron MS. 2013. Hypertrophic cardiomyopathy. Lancet 381:242–255. [DOI] [PubMed] [Google Scholar]
- 27.Maron BJ, Maron MS, Semsarian C. 2012. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 60:705–715. [DOI] [PubMed] [Google Scholar]
- 28.Maron BJ, Shen WK, Link MS, Epstein AE, Almquist AK, Daubert JP, Bardy GH, Favale S, Rea RF, Boriani G, Estes NA, 3rd, Spirito P. 2000. Efficacy of implantable cardioverter–defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med 342:365–373. [DOI] [PubMed] [Google Scholar]
- 29.Maron BJ, Spirito P. 1998. Implications of left ventricular remodeling in hypertrophic cardiomyopathy. Am J Cardiol 81:1339–1344. [DOI] [PubMed] [Google Scholar]
- 30.Maron BJ, Spirito P, Wesley Y, Arce J. 1986. Development and progression of left ventricular hypertrophy in children with hypertrophic cardiomyopathy. N Engl J Med 315:610–614. [DOI] [PubMed] [Google Scholar]
- 31.Maron BJ, Wolfson JK, Roberts WC. 1992. Relation between extent of cardiac muscle cell disorganization and left ventricular wall thickness in hypertrophic cardiomyopathy. Am J Cardiol 70:785–790. [DOI] [PubMed] [Google Scholar]
- 32.Meurs KM, Norgard MM, Ederer MM, Hendrix KP, Kittleson MD. 2007. A substitution mutation in the myosin binding protein C gene in ragdoll hypertrophic cardiomyopathy. Genomics 90:261–264. [DOI] [PubMed] [Google Scholar]
- 33.Meurs KM, Sanchez X, David RM, Bowles NE, Towbin JA, Reiser PJ, Kittleson JA, Munro MJ, Dryburgh K, Macdonald KA, Kittleson MD. 2005. A cardiac myosin binding protein C mutation in the Maine coon cat with familial hypertrophic cardiomyopathy. Hum Mol Genet 14:3587–3593. [DOI] [PubMed] [Google Scholar]
- 34.Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, Towbin JA, Seidman JG, Seidman CE. 2008. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med 358:1899–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nugent AW, Daubeney PE, Chondros P, Carlin JB, Colan SD, Cheung M, Davis AM, Chow CW, Weintraub RG, National Australian Childhood Cardiomyopathy Study. 2005. Clinical features and outcomes of childhood hypertrophic cardiomyopathy: results from a national population-based study. Circulation 112:1332–1338. [DOI] [PubMed] [Google Scholar]
- 36.Ogino K, Ogura K, Kinugawa T, Osaki S, Kato M, Furuse Y, Kinugasa Y, Tomikura Y, Igawa O, Hisatome I, Shigemasa C. 2004. Neurohumoral profiles in patients with hypertrophic cardiomyopathy: differences to hypertensive left ventricular hypertrophy. Circ J 68:444–450. [DOI] [PubMed] [Google Scholar]
- 37.Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, Ommen SR, Theis JL, Vaubel RA, Re F, Armentano C, Poggesi C, Torricelli F, Cecchi F. 2008. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc 83:630–638. [DOI] [PubMed] [Google Scholar]
- 38.Olivotto I, Maron MS, Adabag AS, Casey SA, Vargiu D, Link MS, Udelson JE, Cecchi F, Maron BJ. 2005. Gender-related differences in the clinical presentation and outcome of hypertrophic cardiomyopathy. J Am Coll Cardiol 46:480–487. [DOI] [PubMed] [Google Scholar]
- 39.Robbins SL, Kumar V, Cotran RS. 2010. Robbins and Cotran pathologic basis of disease, 8th ed. Philadelphia (PA): Saunders–Elsevier. [Google Scholar]
- 40.Seidman CE, Seidman JG. 2011. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ Res 108:743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Semsarian C, Ahmad I, Giewat M, Georgakopoulos D, Schmitt JP, McConnell BK, Reiken S, Mende U, Marks AR, Kass DA, Seidman CE, Seidman JG. 2002. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest 109:1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Semsarian C, Healey MJ, Fatkin D, Giewat M, Duffy C, Seidman CE, Seidman JG. 2001. A polymorphic modifier gene alters the hypertrophic response in a murine model of familial hypertrophic cardiomyopathy. J Mol Cell Cardiol 33:2055–2060. [DOI] [PubMed] [Google Scholar]
- 43.Shirani J, Pick R, Roberts WC, Maron BJ. 2000. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 35:36–44. [DOI] [PubMed] [Google Scholar]
- 44.Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ. 2000. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med 342:1778–1785. [DOI] [PubMed] [Google Scholar]
- 45.Tardiff JC. 2005. Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail Rev 10:237–248. [DOI] [PubMed] [Google Scholar]
- 46.Teare D. 1958. Asymmetrical hypertrophy of the heart in young adults. Br Heart J 20:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Varnava AM, Elliott PM, Mahon N, Davies MJ, McKenna WJ. 2001. Relation between myocyte disarray and outcome in hypertrophic cardiomyopathy. Am J Cardiol 88:275–279. [DOI] [PubMed] [Google Scholar]
- 48.Varnava AM, Elliott PM, Sharma S, McKenna WJ, Davies MJ. 2000. Hypertrophic cardiomyopathy: the interrelation of disarray, fibrosis, and small-vessel disease. Heart 84:476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wigle ED, Rakowski H, Kimball BP, Williams WG. 1995. Hypertrophic cardiomyopathy. Clinical spectrum and treatment. Circulation 92:1680–1692. [DOI] [PubMed] [Google Scholar]
- 50.Wolf CM, Moskowitz IP, Arno S, Branco DM, Semsarian C, Bernstein SA, Peterson M, Maida M, Morley GE, Fishman G, Berul CI, Seidman CE, Seidman JG. 2005. Somatic events modify hypertrophic cardiomyopathy pathology and link hypertrophy to arrhythmia. Proc Natl Acad Sci USA 102:18123–18128. [DOI] [PMC free article] [PubMed] [Google Scholar]