

Abstract
Many observational epidemiologic studies suggest an association between high-fat-diet (HFD) and colon cancer risk. However, the lack of controlled experimental studies that examine this relationship and the mechanisms involved weaken the basis for inferring a causal relationship. Inflammation plays a role in colon cancer progression and HFDs have been reported to increase inflammation; however, the inflammatory effects of HFD in colon cancer have yet to be firmly established. We examined the effects of a novel HFD that closely mimics the standard American diet (12% and 40% of total caloric intake from saturated fat and total fat, respectively) on macrophage markers and inflammatory mediators in a mouse model of intestinal tumorigenesis and relate this to polyp characteristics as well as measures of adiposity. Male ApcMin/+ mice (7–8/group) were fed a Control Diet (Con) or novel high-fat-diet (HFD) from 4 to 12 weeks of age. Body weight and body composition were measured weekly and monthly, respectively. Intestinal tissue was analyzed for polyp burden (number and size). Gene expression of macrophage markers and inflammatory mediators were examined in the adipose tissue and polyps. The HFD increased the expression of macrophage markers and inflammatory mediators in the adipose tissue (F4/80, CD11c, TLR-4 and MCP-1) and tumor microenvironment (IL-12, MCP-1, IL-6 and TNF-α). As expected, the HFD increased body weight, body fat percent, fat mass and blood glucose (P < 0.05), and was associated with an increase in the number of large polyps (P < 0.05) but not total polyps. In summary, consumption of a HFD, similar in macronutrient composition to the standard American diet, altered the expression of macrophage phenotypic markers and inflammatory mediators in adipose tissue and intestinal polyps and this was associated with increased tumorigenesis.
Keywords: High-fat-diet, Obesity, Intestinal tumorigenesis, Inflammation, Macrophages
1. Introduction
The etiology of colon cancer is a complex phenomenon that involves the interaction of genetic and environmental factors. However, the vast majority of cases can be ascribed to environmental causes as they account for more than 80% of all incidences [1]. High-fat-diet (HFD) induced obesity has emerged as one of the leading environmental risk factors for the development of colon cancer [2–4] as supported by epidemiological studies as well as controlled experimental studies in mice [5–10].
Animal studies provide evidence for a link between HFD consumption and colon cancer risk [8–11]. For example, Baltgalvis et al. reported a 75% increase in polyp number in the Apcmin/+ mouse model of intestinal tumorigenesis following treatment with a Western Style diet (40% calories from fat) [8]. Similarly, HFD-induced obesity (60% of total calories from fat) has been shown to increase development of AOM-induced aberrant crypt foci in the colon [10]. In addition, an increase in proliferation and a decrease in apoptosis have been reported in the colonic epithelium following HFD-induced obesity (60% of total calories from fat) in mice [12]. While there is reasonable evidence for an association between HFD-induced obesity and colon cancer risk, there is a fundamental gap in the understanding of underlying mechanisms for such a relationship. Further, many of the HFDs that have been used to test this relationship contain unreasonably high levels of both total fat and saturated fat and typically contain fat from only one food source making it difficult to assess the clinical relevance.
While the pathophysiological mechanisms that link HFD-induced obesity to colon cancer risk are not well understood, it is believed that inflammation plays a key role. Consumption of a HFD can lead to accumulation of excess body fat that is associated with adipose tissue dysfunction and a chronic state of low-grade inflammation [13], which is known to promote tumor development and growth [13]. Inflammation associated with HFD-induced obesity is thought to be mediated, at least in part, through numerical and functional alterations in adipose tissue macrophages [13–15]. It has been reported that in HFD-induced obese mice, approximately 45–60% of adipose tissue cells express the macrophage marker EMR-1 (F4/80), whereas only 10–15% of cells from lean mice express this marker [14]. In addition, adipose tissue macrophages in HFD-induced obese mice exhibit a pro-inflammatory classical phenotype (M1) while those from lean mice have an alternatively activated anti-inflammatory phenotype (M2) [15]. While there is an abundance of evidence that implicates inflammation as a link between HFD-induced obesity and cancer, there are few studies that have examined the inflammatory response in a mouse model of colon cancer in the settings of HFD-induced obesity.
The purpose of this study was to examine the effects of a novel HFD closely mimicking the standard American diet [16] (12% and 40% of total caloric intake from saturated and total fat, respectively, a 2:1 monounsaturated: polyunsaturated fatty-acid ratio, and a 20:1 omega-6:omega-3 fatty-acid ratio) on the expression of macrophage markers and inflammatory mediators in adipose tissue and the tumor microenvironment in a mouse model of intestinal tumorigenesis and to relate this to polyp characteristics as well as measures of adiposity. We used the Apcmin/+ mouse, the most widely used genetic mouse model for cancer studies that involve the gastro-intestinal tract [17]. Since the Apc gene is mutated in a large percentage of human colon cancer cases, this is a common model for studying factors that may influence progression of colon cancer. A novel HFD, designed by our laboratory, was used to mimic a typical American diet. We hypothesized that this novel HFD would alter the expression of macrophage phenotypic markers and increase the expression of inflammatory meditators in the adipose tissue and tumor microenvironment in association with increased adiposity and enhanced tumorigenesis.
2. Materials and methods
2.1. Animals
ApcMin/+ on a C57BL/6 background and C57BL/6 wild-type mice were originally purchased from Jackson Laboratories (Bar Harbor, ME). ApcMin/+ male mice were bred with female C57BL/6 mice in the University of South Carolina’s Center for Colon Cancer Research (CCCR) mouse core facility to generate ApcMin/+ mice. Offspring were genotyped as heterozygotes by RT-PCR for the Apc gene by taking tail snips at weaning. The primer sequences were sense: 5′-TGAGAAAGACAGAAGTTA-3′; and antisense: 5′-TTCCACTTTGGCATAAGGC-3′. Mice were maintained on a 12:12 h light–dark cycle in a low-stress environment (22 °C, 50% humidity and low noise) and provided food and water ad libitum. All animal experimentation was approved by the University of South Carolina’s Institutional Animal Care and Use Committee.
2.2. High-fat-diet treatment
ApcMin/+ mice (7–8/group) were assigned to one of two treatment groups: (1) an AIN-76A Control Diet (Con) or (2) a HFD (12% and 40% of total caloric intake from saturated and total fat, respectively) resembling the standard American Diet (HFD) (Bio-Serv, Frenchtown, NJ) (Table 1). Treatments began at 4 weeks of age and continued until 12 weeks of age. The HFD retained the same vitamin and mineral content as the control diet. Food and water was available ad libitum and measured on a weekly basis.
Table 1.
Diet composition.
| AIN-76A | HFD | |
|---|---|---|
| Ingredient (g/kg) | ||
| Casein | 200 | 165 |
| DL Methionine | 3 | 3 |
| Lard | 0 | 35.4 |
| Coconut Oil | 0 | 30 |
| Corn Oil | 50 | 49.9 |
| Soybean Oil | 0 | 9.3 |
| Olive Oil | 0 | 78.4 |
| Corn Starch | 150 | 50 |
| Maltodextrin | 0 | 100 |
| Sucrose | 500 | 381.5 |
| Cellulose | 50 | 50 |
| Vitamin Mix (AIN-76A) | 10 | 10 |
| Mineral Mix (AIN-76A) | 35 | 35 |
| Choline Bitartrate | 2 | 2.5 |
| Energy (kcal/g) | 3.79 | 4.57 |
| Energy (% kcal) | ||
| Carbohydrate | 68.8 | 47 |
| Fat | 12.2 | 40 |
| Protein | 19.0 | 13 |
| Fatty Acid Profile (g/kg) | ||
| Caprylic Acid (C8:0) | 0 | 2.3 |
| Capric Acid (C10:0) | 0 | 1.8 |
| Lauric Acid (C12:0) | 0 | 13.5 |
| Myristic Acid (C14:0) | 0 | 5.5 |
| Palmitic Acid (C16:0) | 5.3 | 26 |
| Palmitoleic Acid (C16:1) | 0 | 2 |
| Stearic Acid (C18:0) | 0 | 8.5 |
| Oleic Acid (C18:1) | 13.7 | 88 |
| Linoleic Acid (C18:2) | 26.8 | 43.2 |
| α-Linolenic Acid (C18:3) | 0.6 | 2.2 |
| % of Total Calories from SFAs | 1.4% | 12% |
| % of Total Calories from MCFAs (C6:0-C12:0) | - | 3.6% |
| % of Total Calories from LCSFAs (C14:0-C20:0) | 1.4% | 8.4% |
| % of Total Calories from USFAs | 10.8% | 28% |
| % of Total Calories from MUFAs | 3.6% | 18.6% |
| % of Total Calories from PUFAs | 7.2% | 9.4% |
| % of Total Calories from n-3 FAs | .15% | .45% |
| % of Total Calories from n-6 FAs | 7.0% | 8.9% |
| Cholesterol (mg/kg) | 0 | 34 |
| Ratio: MUFA:PUFA | 1:2 | 2:1 |
| Ratio: n-6:n-3 FA | 45:1 | 20:1 |
2.3. Body composition
Body weight was measured weekly and body composition was measured at baseline (4 weeks), midpoint (8 weeks), and at the 12-week endpoint via a Dual Energy X-ray Absorptiometry (DEXA) (Lunar PIXImus, Madison, WI) scan under isofluorane anesthesia.
2.4. Blood collection
At 8 weeks and 12 weeks of age, blood samples were collected from the tip of the tail after a 5-h fast. Blood glucose concentrations were determined in whole blood using a glucometer (Bayer Contour, Michawaka, IN). At sacrifice, blood was collected from the inferior vena cava using a heparinized syringe and was spun in a microcentrifuge at 10,000 relative centrifugal force for 10 min. Plasma was then stored at −80 °C until analysis.
2.5. Tissue collection
Mice were sacrificed for tissue collection by isoflurane overdose. Epididymal, mesenteric, and retroperitoneal fat pads were removed and weighed. The small intestine was carefully dissected distally to the stomach and proximal to the cecum. Sections 1 and 4 of the small intestine and the large intestine (Section 5) were fixed in 10% buffered formalin (Fisher Scientific, Pittsburg, PA) for 24 h as previously described [18]. For intestinal Section 3, polyps were excised, immediately frozen in liquid nitrogen and then stored at −80 °C until analysis for mRNA expression of macrophage phenotype markers and inflammatory mediators. Previously reported findings from our laboratory has shown that intestinal Section 3, a region of high polyp incidence, has a larger inflammatory response than Section 2, an area of low polyp occurrence [18], and therefore we included only Section 3 for these analyses.
2.6. Circulating adipokines and cytokines
Circulating leptin, adiponectin and markers of inflammation (MCP-1 and IL-6) were examined using an enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN). All samples were run in duplicate.
2.7. Polyp counts
Formalin-fixed intestinal sections from all animals were rinsed in deionized water, briefly stained in 0.1% methylene blue, and counted by the same investigator who was blinded to the treatments. Polyps were counted under a dissecting microscope, using tweezers to pick through the intestinal villi and identify polyps. Polyps were categorized by size (>2 mm, 1–2 mm, and <1 mm).
2.8. Adipocyte size
To identify adipocyte enlargement, formalin-fixed, paraffin-embedded adipose tissue was cut on a microtome in 4-μm sections. Paraffin sections were stained with hematoxylin and eosin (H&E). At 40× magnification, two images per sample were obtained and surface area (manual trace) was determined for all intact adipocytes [19], which were then averaged to represent mean adipocyte size (Infinity Analyze software, Lumenera, Ottawa, ON).
2.9. mRNA gene expression
Quantification of gene expression for select macrophage markers (adipose tissue: F4/80, CD11c & CD206; polyps: IL-12, IL-23, IL-13, CD206 & CCL17) and inflammatory mediators (adipose tissue: TLR-4, MCP-1 & TNF-α; polyps: MCP-1, IL-6 & TNF-α) were performed as previously described [18]. Briefly, polyps were homogenized under liquid nitrogen with a polytron, and total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA). For isolation of RNA from adipose tissue, we used an RNeasy Lipid Tissue Mini Kit (Qiagen, Valencia, CA). The extracted RNA (2.5 μL of sample) was dissolved in diethyl pyrocarbonate (DEPC)-treated water and quantified spectrophotometrically at 260 nm wavelength. RNA was reverse transcribed into cDNA and quantitative RT-PCR analysis was done per the manufacturer’s instructions (Applied Biosystems, Foster City, CA) using TaqMan® Gene Expression Assays. Quantification of gene expression for was calculated using the ΔΔCT method.
2.10. Statistical analysis
All data were analyzed using commercial software (SigmaStat, SPSS, Chicago, IL). Body weight, body composition outcomes and fasting blood glucose were analyzed using a repeated measures two-way ANOVA. All other data were analyzed using a Student’s t-test. Student–Newman–Keuls test was used for all post hoc analyses. Statistical significance was set with an alpha value of P ≤ 0.05. Data are presented as mean (±SEM).
3. Results
3.1. Food and water intake
Food and water intake were measured weekly. There were no group differences in total food (grams) ingested, or in water consumed (data not shown).
3.2. Body weight
Body weights were recorded weekly from 4 to 12 weeks of age (Fig. 1A). Mice fed the HFD had increased body weights from 10 weeks of age until sacrifice at 12 weeks of age (P < 0.05).
Fig. 1.

Effects of HFD (12% and 40% of total caloric intake from saturated and total fat, respectively) on (A) weekly mean body weight, (B) epididymal, (C) mesentery, (D) retroperitoneal, and (E) total visceral fat pad weights in the ApcMin/+ mouse model of intestinal tumorigenesis (n = 7–8). *Significantly different from Con (control mice) (P < 0.05).
3.3. Visceral fat weights
At sacrifice, epididymal, mesenteric, and retroperitoneal fat pads were collected and weighed. The HFD group had elevated epididymal fat mass weight compared to Con (990 ± 153 mg vs. 487 ± 37 mg) (P < 0.05) (Fig. 1B). Although mesentery fat was greater with the HFD, there were no significant differences detected (505 ± 54 mg vs. 432 ± 19 mg) (Fig. 1C). Similar to epididymal fat, retroperitoneal fat weight was significantly greater with the HFD (292 ± 47 mg vs. 138 ± 18 mg) (P < 0.05) (Fig. 1D). Lastly, when all three fat weights were combined, the HFD had an increase in total visceral fat (1787 ± 251 mg vs. 1058 ± 69 mg) (P < 0.05) (Fig. 1E).
3.4. Body composition
At 4 (baseline), 8, and 12 weeks of age, mice underwent a DEXA scan to determine body composition (body fat percentage, fat mass and lean mass) (Fig. 2). As expected there were no differences between the groups at baseline (4 weeks). However, by 8 weeks of age the HFD had increased body fat percentage (22.1 ± 1.2% vs. 19.1 ± 1.1%) and this continued through the 12 week time-point (25.1 ± 1.7% vs. 19.3 ± 1.1%) (P < 0.05) (Fig. 2A). Similarly, fat mass in the HFD group was greater than the Con group at 8 weeks (4.8 ± 0.3 g vs. 3.8 ± 0.2 g) and 12 weeks (6.23 ± 0.6 g vs. 4.2 ± 0.3 g) (P < 0.05) (Fig. 2B). There were no significant group differences detected for lean mass at any of the time points (Fig. 2C).
Fig. 2.

HFD (12% and 40% of total caloric intake from saturated and total fat, respectively) increases (A) body fat percent, (B) fat mass, and (D) adipocyte size in the ApcMin/+ mouse model of intestinal tumorigenesis but does not alter lean mass (C) (n = 7–8). *Significantly different from Con (control mice) (P < 0.05).
3.5. Adipocyte size
Epididymal fat pads were formalin-fixed and paraffin-embedded for H&E staining to determine potential differences in adipocyte enlargement between the groups. As expected, the HFD had increased adipocyte size compared to Con (2927 ± 248 μm2 vs. 1904 ± 200 μm2) (P < 0.05) (Fig. 2D).
3.6. Circulating adipokines and cytokines
Circulating levels of leptin and adiponectin were determined using ELISAs. The HFD increased plasma leptin (19.3 ± 5.0 ng/ml vs. 12.4 ± 0.7 ng/ml); however, this did not reach statistical significance (P = 0.1) (Fig. 3A). Interestingly, the HFD had a greater concentration of plasma adiponectin compared to Con (5659 ± 640 ng/ml vs. 3854 ± 453 ng/ml) (P < 0.05) (Fig. 3B). Protein concentration of inflammatory mediators (MCP-1 & IL-6) was also examined in the plasma at 12 weeks of age. There were no significant increases in plasma concentration of MCP-1 or IL-6 with the HFD feedings (data not shown).
Fig. 3.

Effects of HFD (12% and 40% of total caloric intake from saturated and total fat, respectively) on (A) plasma leptin, (B) plasma adiponectin, and (C) blood glucose in the ApcMin/+ mouse model of intestinal tumorigenesis (n = 7–8). *Significantly different from Con (control mice) (P < 0.05).
3.7. Fasting blood glucose
Fasting blood glucose was assessed at 8 and 12 weeks of age following a 5-h fast (Fig. 3C). The HFD had higher blood glucose levels at 8 weeks (177 ± 11 mg/dl vs. 136 ± 9 mg/dl) (P < 0.05) and this continued through the 12 week time-point (218 ± 19 mg/dl vs. 167 ± 19 mg/dl) (P < 0.05).
3.8. Polyp incidence
At 12 weeks of age, ApcMin/+ mice were sacrificed, intestinal tissue was collected and polyps were counted on formalin-fixed, methylene blue-stained sections. While there was no difference in overall polyp number between the groups (HFD: 14.0 ± 1.9 polyps vs. Con: 15.8 ± 2.4 polyps) (Fig. 4A), the HFD did have a greater number of large polyps (2.2 ± 0.6 vs. 0.6 ± 0.6) (P < 0.05) but fewer small polyps (6.3 ± 1.6 vs. 11.0 ± 1.6) (P < 0.05) (Fig. 4B) and there was no change in medium polyps (5.5 ± 1.5 vs. 4.2 ± 1.0) (Fig. 4B).
Fig. 4.
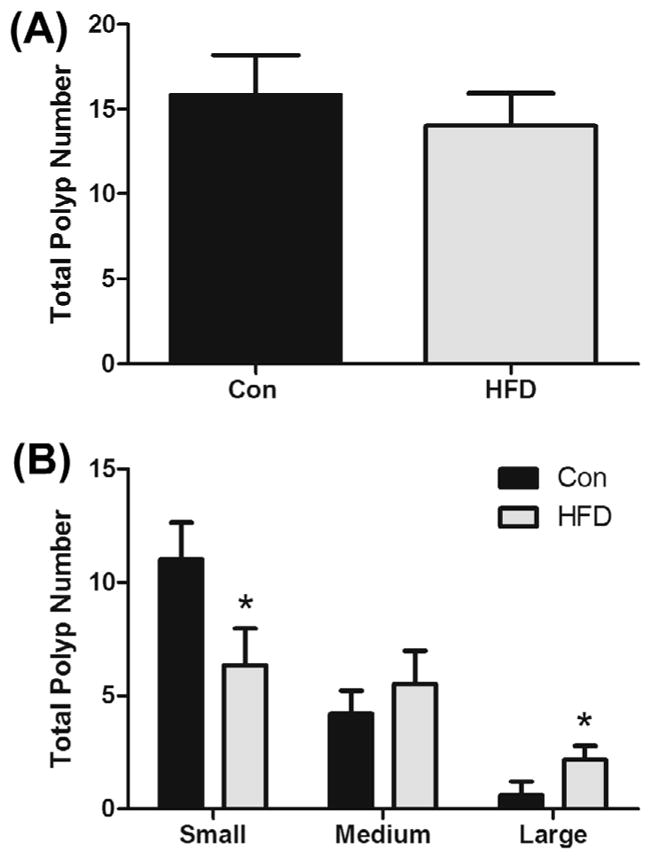
HFD (12% and 40% of total caloric intake from saturated and total fat, respectively) does not affect overall polyp number (A), but increases large polyp number and decreases small polyp number (B) in the ApcMin/+ mouse model of intestinal tumorigenesis (n = 7–8). *Significantly different from Con (control mice) (P < 0.05).
3.9. Adipose tissue gene expression
Gene expression of macrophage markers (EMR-1 (overall), CD11c (M1) and CD206 (M2)) and inflammatory mediators (TLR-4, MCP-1, and TNF-α) were examined in the epididymal fat (Fig. 5). All data was normalized to fold-change from Con mice. The HFD increased expression of EMR-1 (2.2-fold) (P < 0.05) (Fig. 5A). Similarly, when examining the M1 sub-population of macrophages, we found that CD11c expression levels were significantly increased with the HFD (1.9-fold) (P < 0.05) (Fig. 5B). As expected, there were no significant differences found for the M2 subpopulation of macrophages (CD206) (Fig. 5C). TLR-4, a trans-membrane receptor capable of binding saturated fatty acids, and that is responsible for signal transduction events and the downstream release of inflammatory mediators, was also examined (Fig. 5D). As hypothesized, the HFD increased TLR-4 expression levels (1.5-fold) compared to Con (P < 0.05). Similarly, the HFD elevated expression of MCP-1 (3.2-fold) in epididymal tissue (P < 0.05). However, there were no significant group differences detected for TNF-α (Fig. 5F).
Fig. 5.

Effects of HFD (12% and 40% of total caloric intake from saturated and total fat, respectively) on mRNA expression of (A) EMR-1, (B) CD11c, (C) CD206, (D) TLR-4, (E) MCP-1 and (F) TNF-α in the adipose tissue of the ApcMin/+ mouse model of intestinal tumorigenesis (n = 7–8). *Significantly different from Con (control mice) (P < 0.05).
3.10. Intestinal polyp gene expression
Gene expression of macrophage markers including M1 (IL-12 & IL-23) and M2 (IL-13, CD206 & CCL17) markers as well as inflammatory mediators (MCP-1, IL-6 & TNF-α) were examined in the polyps (Fig. 6). Intestinal polyp data was normalized to fold-change from Con mice. The HFD increased mRNA expression of IL-12 (7.8-fold) (P < 0.05) (Fig. 6A) but there was no effect on IL-23 (Fig. 6B). There were no significant changes in any of the M2 markers measured including IL-13 (Fig. 6C), CD206 (Fig. 6D) or CCL17 (Fig. 6E). All inflammatory mediators (MCP-1, IL-6 & TNF-α) were increased with the HFD (Fig. 6F, G & H, respectively); there was a 1.8-fold increase in MCP-1 (P = 0.06), a 1.5-fold increase in IL-6 (P = 0.1) and a 1.7-fold increase in TNF-α (P < 0.05).
Fig. 6.

HFD (12% and 40% of total caloric intake from saturated and total fat, respectively) alters mRNA expression of select macrophages markers and inflammatory mediators in the polyps of ApcMin/+ mice (n = 7–8). (A) IL-12, (B) IL-23, (C) IL-13, (D) CD206, (E) CCL17, (F) MCP-1, (G) IL-6 and (H) TNF-α. *Significantly different from Con (control mice) (P < 0.05) **trend to be different from Con (P < 0.1).
4. Discussion
Although epidemiological evidence supports a link between HFD-induced obesity and colon cancer risk, there have been limited controlled experimental studies in rodents that have examined this relationship. Further, the HFDs that have been used to test this association contain unreasonably high levels of both total fat and saturated fat making it difficult to assess the clinical relevance. Therefore, the purpose of this study was to examine the effects of a novel HFD, closely mimicking the standard American diet, on the expression of macrophage markers and inflammatory mediators in adipose tissue as well as the tumor microenvironment in a mouse model of intestinal tumorigenesis and to relate this to polyp characteristics as well as measures of adiposity. As hypothesized, the HFD increased the expression of select macrophage markers and inflammatory mediators in the adipose tissue (EMR1, CD11c, TLR4 and MCP-1) and tumor microenvironment (IL-12, MCP-1 and TNF-α), which was associated with an increase in the number of large polyps. These effects are likely mediated by the HFD-induced increase in body weight, body fat percent, fat mass and blood glucose.
HFDs are strongly linked to the accumulation of excess body fat, chronic inflammation, and metabolic perturbations, ultimately leading to poorer health outcomes including cancer. We show that a novel HFD, similar to the standard American diet, increases body weight, percent body fat, fat mass, visceral fat weight, adipocyte size, and fasting blood glucose in a mouse model of intestinal tumorigenesis. While leptin levels were increased with HFD, this did not quite reach statistical significance, which is likely due to the small sample size and/or the limited treatment duration of the HFD (8 weeks). Similar findings of a non-significant increase in leptin following HFD feedings in the same mouse model were reported in a recent study [8]. Interestingly, we observed an increase in adiponectin with the HFD. This is contrary to what was hypothesized as it is well established that adiponectin decreases with obesity and is inversely associated with insulin resistance and hyperinsulinemia [20,21]. Further, it has been proposed to have beneficial effects on tumorigenesis [11]. Because ApcMin/+ mice develop hyperglycemia that is exacerbated with HFD feedings, the observed increase in adiponectin with the HFD may be explained as an attempt to regulate glucose levels in these mice [22]. A potential limitation of this study is the shorter duration of exposure to the HFD, as we have found much larger effects on adiposity when this same diet was administered to cancer-free C57BL/6 mice [23]. However, because ApcMin/+ mice can become cachectic between 3 and 6 months of age, an early sacrifice time point (12 weeks of age) was used in an attempt to minimize the influence of cachexia-related losses in fat mass on our findings [24]. Therefore, it is possible that our findings may actually underestimate the effects of this HFD on adiposity in this mouse model.
As macrophages are thought to play a role in the link between obesity and colon cancer, we sought to examine the influence of this novel HFD on macrophage behavior in the adipose tissue and tumor microenvironment. The HFD increased overall expression of macrophages (EMR-1) in the adipose tissue. Further examination of macrophage populations suggests that this increase is primarily due to an elevation in mRNA expression of M1 proinflammatory subset (CD11c) as the M2 population (CD206) was unchanged. This is in agreement with previously reported literature as adipose tissue macrophages are known to exhibit a proinflammatory classical phenotype (M1) in response to obesity development [14]. In the tumor microenvironment, macrophages can represent up to 50% of the tumor mass and are known to produce a wide array of inflammatory mediators with pro-tumoral functions [25,26]. Thus, high levels of macrophages are associated with poor prognosis in colon cancer [27–29]. It is generally thought that M1 macrophages are cytotoxic against neoplastic cells, whereas M2 macrophages exert pro-tumoral functions [26]. We report the novel finding that the HFD increased mRNA expression of the M1 marker IL-12 in the polyp tissue but there was no change in IL-13, IL-23, CD206 or CCL17. While our data suggest that the HFD can alter macrophage behavior in the adipose tissue and the tumor microenvironment, a greater understanding of the roles of each macrophage subset in colon cancer in the setting of HFD-induced obesity is necessary.
Saturated fatty acids can act as ligands for TLR-4 on macrophages, preadipocytes, and adipocytes and thereby induce gene transcription of pro-inflammatory mediators via NF-κB [30]. Consistent activation of this pathway can lead to insulin resistance and a chronic state of inflammation [31]. Further, TLR-4 has been associated with poorer prognosis in colon cancer, presumably due to its ability to increase inflammatory processes [32]. We show an increase in expression of TLR-4 in the adipose tissue following consumption of a diet similar in macronutrient composition to the standard American diet. Similarly, MCP-1 expression was increased with the HFD in both the adipose tissue and the tumor microenvironment. This chemokine was measured as it has been strongly associated with obesity and tumorigenesis [18,33–35]. In fact, our laboratory has recently reported that knocking out MCP-1 can lead to a reduction in overall polyp number as well as large polyp number in ApcMin/+ mice [33]. IL-6 expression was measured in the tumor microenvironment as it has previously been shown that its overexpression can increase polyp burden in the ApcMin/+ mouse [24]. We show here that IL-6 is increased in the polyps with HFD feedings, although this did not quite reach statistical significance. Lastly, mRNA expression of TNF-α was measured in the adipose tissue and tumor microenvironment as it has been reported to increase with obesity and has been linked with pro-tumoral processes [36]. While, we did not detect any changes in adipose tissue TNF-α expression with this diet, it was increased in the polyp tissue.
The increase in mRNA expression of macrophage markers and inflammatory mediators was associated with an increase in the number of large polyps but it did not change total polyp number. And in fact, there was a decrease in the number of small polyps. We interpret this to mean that the HFD-induced alterations in inflammatory processes may play a larger role in the progression of polyp growth as opposed to the initiation of polyp development. Alternatively, it is also possible that other HFD-induced factors may have caused the elevated number of large polyps resulting in increased expression of inflammatory cytokines due to ulceration or traumatization that is characteristic of larger polyps. These findings are partially supported by recent reports on the effects of HF feeding on colon cancer. For example, Moon et al. reported an increase in the number of large tumors (230%) following 5 months of feedings with a Western Diet (60% calories from fat) in an implantation model [11]. Similarly, a study by Baltgalvis et al. reported an increase in the number of large polyps following 6 weeks of feedings with a Western Diet (41% calories from fat) in the ApcMin/+ mouse [8]. However, an increase in the total number of polyps was also reported in that same study. The most likely explanation for these convergent findings is the composition of the diets; our diet had similar levels of total fat but differed greatly in the composition and source of the fat.
It is important to point out that in general, the magnitude of increase for the measured macrophage markers and inflammatory mediators is lower than previously shown in the literature [37]. For example, an 8-fold increase in expression of F4/80 has been reported in adipose tissue of cancer-free mice following HFD feedings [37] whereas here we report a ~2-fold increase in ApcMin/+ mice. Similarly, we found an ~8-fold increase with this same diet (HFD-12%) when administered to a C57BL/6 wild-type mouse [23]. As previously mentioned, this is likely explained by the short duration of feedings as well as the fact that ApcMin/+ mice are pre-disposed to cachexia. In order to more accurately assess the effects of HFD-induced obesity on colon cancer progression, a longer duration of HFD feedings may be necessary as well as utilization of an alternate model in which obesity can be induced prior to initiation of the cancer. Of additional importance in understanding the link between HFD-induced obesity and colon cancer is the type of HFD used to induce obesity. A recent study in our laboratory reported divergent findings in obesity, inflammatory signaling, macrophage behavior and metabolism across three HFDs when the content, but not total percentage, of fat was altered [23]. Given that various fatty acids can differ in their propensity to activate inflammatory signaling, it is certainly likely that altering the fat content of the diet can impact inflammatory processes and subsequent tumorigenesis.
In summary, we report that a diet mimicking the macronutrient content of the standard American diet can increase the expression of select macrophage markers and inflammatory mediators within the adipose tissue and tumor microenvironment, which was associated with an increase in the number of large polyps. These effects are likely mediated by the HFD-induced increase in body weight, body fat percent, fat mass and blood glucose. Overall these findings provide evidence for an influence of HFD feedings on the expression of macrophage markers and inflammatory mediators in the progression of colon cancer.
Acknowledgments
This work was supported by grants from the National Institutes of Health (National Cancer Institute R21CA167058 to E.A.M.; National Institute of General Medical Sciences P20GM103641) and the University of South Carolina (Advanced Support Programs for Innovative Research Excellence (ASPIRE) to E.A.M.).
Contributor Information
Stani D. Day, Email: sgilmer@erskine.edu.
Reilly T. Enos, Email: enosr@email.sc.edu.
Jamie L. McClellan, Email: mcclelll@mailbox.sc.edu.
J.L. Steiner, Email: jls2tc@virginia.edu.
Kandy T. Velázquez, Email: kvelazquez@uscupstate.edu.
E.A. Murphy, Email: angela.murphy@uscmed.sc.edu.
References
- 1.Rustgi AK. The genetics of hereditary colon cancer. Genes Dev. 2007;21(20):2525–38. doi: 10.1101/gad.1593107. [DOI] [PubMed] [Google Scholar]
- 2.Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4(8):579–91. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- 3.Yehuda-Shnaidman E, Schwartz B. Mechanisms linking obesity, inflammation and altered metabolism to colon carcinogenesis. Obes Rev. 2012;13(12):1083–95. doi: 10.1111/j.1467-789X.2012.01024.x. [DOI] [PubMed] [Google Scholar]
- 4.Sung MK, et al. Obesity-induced metabolic stresses in breast and colon cancer. Ann N Y Acad Sci. 2011;1229:61–8. doi: 10.1111/j.1749-6632.2011.06094.x. [DOI] [PubMed] [Google Scholar]
- 5.Giovannucci E, Michaud D. The role of obesity and related metabolic disturbances in cancers of the colon, prostate, and pancreas. Gastroenterology. 2007;132(6):2208–25. doi: 10.1053/j.gastro.2007.03.050. [DOI] [PubMed] [Google Scholar]
- 6.Gunter MJ, Leitzmann MF. Obesity and colorectal cancer: epidemiology, mechanisms and candidate genes. J Nutr Biochem. 2006;17(3):145–56. doi: 10.1016/j.jnutbio.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 7.Thomson CA, et al. Nutrition and diet in the development of gastrointestinal cancer. Curr Oncol Rep. 2003;5(3):192–202. doi: 10.1007/s11912-003-0110-y. [DOI] [PubMed] [Google Scholar]
- 8.Baltgalvis KA, et al. The interaction of a high-fat diet and regular moderate intensity exercise on intestinal polyp development in ApcMin/+ mice. Cancer Prev Res (Phila) 2009;2(7):641–9. doi: 10.1158/1940-6207.CAPR-09-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newmark HL, Lipkin M, Maheshwari N. Colonic hyperplasia and hyperproliferation induced by a nutritional stress diet with four components of Western-style diet. J Natl Cancer Inst. 1990;82(6):491–6. doi: 10.1093/jnci/82.6.491. [DOI] [PubMed] [Google Scholar]
- 10.Padidar S, et al. High-Fat Diet Alters Gene Expression in the Liver and Colon: Links to Increased Development of Aberrant Crypt Foci. Dig Dis Sci. 2012;57(7):1866–74. doi: 10.1007/s10620-012-2092-9. [DOI] [PubMed] [Google Scholar]
- 11.Moon HS, et al. Salutary effects of adiponectin on colon cancer: in vivo and in vitro studies in mice. Gut. 2013;62(4):561–70. doi: 10.1136/gutjnl-2012-302092. [DOI] [PubMed] [Google Scholar]
- 12.Liu Z, et al. Diet-induced obesity elevates colonic TNF-alpha in mice and is accompanied by an activation of Wnt signaling: a mechanism for obesity-associated colorectal cancer. J Nutr Biochem. 2012;23(10):1207–13. doi: 10.1016/j.jnutbio.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Kruijsdijk RC, van der Wall E, Visseren FL. Obesity and cancer: the role of dysfunctional adipose tissue. Cancer Epidemiol Biomarkers Prev. 2009;18(10):2569–78. doi: 10.1158/1055-9965.EPI-09-0372. [DOI] [PubMed] [Google Scholar]
- 14.Weisberg SP, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–84. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grotto D, Zied E. The standard American diet and its relationship to the health status of Americans. Nutr Clin Pract. 2010;25(6):603–12. doi: 10.1177/0884533610386234. [DOI] [PubMed] [Google Scholar]
- 17.Tammariello AE, Milner JA. Mouse models for unraveling the importance of diet in colon cancer prevention. J Nutr Biochem. 2010;21(2):77–88. doi: 10.1016/j.jnutbio.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McClellan JL, et al. Intestinal inflammatory cytokine response in relation to tumorigenesis in the Apc(Min/+) mouse. Cytokine. 2012;57(1):113–9. doi: 10.1016/j.cyto.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen HC, Farese RV., Jr Determination of adipocyte size by computer image analysis. J Lipid Res. 2002;43(6):986–9. [PubMed] [Google Scholar]
- 20.Hecker PA, et al. Role of adiponectin in the development of high fat diet-induced metabolic abnormalities in mice. Horm Metab Res. 2011;43(2):100–5. doi: 10.1055/s-0030-1269898. [DOI] [PubMed] [Google Scholar]
- 21.Ziemke F, Mantzoros CS. Adiponectin in insulin resistance: lessons from translational research. Am J Clin Nutr. 2010;91(1):258S–61S. doi: 10.3945/ajcn.2009.28449C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Q, et al. Adiponectin regulates expression of hepatic genes critical for glucose and lipid metabolism. Proc Natl Acad Sci U S A. 2012;109(36):14568–73. doi: 10.1073/pnas.1211611109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Enos RT, et al. Influence of Dietary Saturated Fat Content on Adiposity, Macrophage Behavior, Inflammation, and Metabolism: Composition Matters. J Lipid Res. 2013;54(1):152–63. doi: 10.1194/jlr.M030700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baltgalvis KA, et al. Interleukin-6 and cachexia in ApcMin/+ mice. Am J Physiol Regul Integr Comp Physiol. 2008;294(2):R393–401. doi: 10.1152/ajpregu.00716.2007. [DOI] [PubMed] [Google Scholar]
- 25.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22(2):231–7. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 26.Solinas G, et al. Tumor-associated macrophages (TAMs) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86(5):1065–73. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- 27.Jedinak A, Dudhgaonkar S, Sliva D. Activated macrophages induce metastatic behavior of colon cancer cells. Immunobiology. 2010;215(3):242–9. doi: 10.1016/j.imbio.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 28.Kaler P, Augenlicht L, Klampfer L. Macrophage-derived IL-1β stimulates Wnt signaling and growth of colon cancer cells: a crosstalk interrupted by vitamin D3. Oncogene. 2009;28(44):3892–902. doi: 10.1038/onc.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang JC, et al. Intratumoral macrophage counts correlate with tumor progression in colorectal cancer. J Surg Oncol. 102(3):242–8. doi: 10.1002/jso.21617. [DOI] [PubMed] [Google Scholar]
- 30.Lee JY, et al. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276(20):16683–9. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- 31.Holland WL, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest. 2011;121(5):1858–70. doi: 10.1172/JCI43378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fukata M, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. 2007;133(6):1869–81. doi: 10.1053/j.gastro.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McClellan JL, et al. Linking tumor associated macrophages, inflammation, and intestinal tumorigenesis: Role of MCP-1. Am J Physiol Gastrointest Liver Physiol. 2012;303(10):G1087–95. doi: 10.1152/ajpgi.00252.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4(7):540–50. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 35.Bailey C, et al. Chemokine expression is associated with the accumulation of tumour associated macrophages (TAMs) and progression in human colorectal cancer. Clin Exp Metast. 2007;24(2):121–30. doi: 10.1007/s10585-007-9060-3. [DOI] [PubMed] [Google Scholar]
- 36.Flores MB, et al. Obesity-Induced Increase in Tumor Necrosis Factor-alpha Leads to Development of Colon Cancer in Mice. Gastroenterology. 2012;143(3):741–53. doi: 10.1053/j.gastro.2012.05.045. [DOI] [PubMed] [Google Scholar]
- 37.Fujisaka S, et al. Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes. 2009;58(11):2574–82. doi: 10.2337/db08-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]