

Abstract
For years the human microbiota has been implicated in the etiology of colorectal cancer (CRC). However, identifying the molecular mechanisms for how aneuploidy and chromosomal instability (CIN) arise in sporadic and colitis-associated CRC has been difficult. In this Addendum we review recent work from our laboratory that explore mechanisms by which intestinal commensals polarize colon macrophages to an M1 phenotype to generate a bystander effect (BSE) that leads to mutations, spindle malfunction, cell cycle arrest, tetraploidy, and aneuploidy in epithelial cells. BSE represents the application of a phenomenon initially described in the radiation biology field. The result of commensal-driven BSE on colon epithelial cells is aneuploidy, chromosomal instability (CIN), expression of stem cell and tumor stem cell markers and, ultimately, malignant transformation. Our findings provide a conceptual framework for integrating the microbiota with aging, cyclooxygenase (COX)-2, and inflammation as risk factors for CRC.
Keywords: Bystander effects, carcinogenesis, colorectal cancer, commensal bacteria
This decade has increasingly witnessed the importance of commensal microbiota in the etiology of CRC.1,2 However, despite a better understanding of associations between the human microbiota and CRC, much remains to be learned of how commensals initiate cancer. Although several lines of investigation, including our own,3-8 suggest mechanisms by which wayward commensals, or so-called pathobioants, can damage epithelial cell DNA, large gaps remain in our knowledge of how bacteria induce aneuploidy and CIN in sporadic and colitis-associated CRCs. One example of an intestinal commensal that can lead to DNA damage is Escherichia coli. Strains that express hybrid peptide-polyketide or cytolethal distending toxins generate double-strand DNA breaks, induce G2/M cell cycle arrest, promote invasive carcinoma in mice, and occur at increased frequency in patients with CRC and inflammatory bowel disease.9 These findings directly implicate one member of the complex and diverse human commensal microbiota in CRC initiation. The genome landscape for CRC includes cancer cells and premalignant precursors that often express highly variable and abnormally structured genomes that consist of non-diploid chromosome numbers (i.e., aneuploidy) that constantly evolve on a background of complex recombinations that include gene duplications, deletions, inversions, and translocations (i.e., CIN).10,11 Despite recent advances, the role of the microbiota in CRC and origin of initiating events for aneuploidy and CIN remain obscure.
We have reported in several papers a novel mechanism by which the intestinal microbiota induces aneuploidy and CIN in primary colon epithelial cells.3-5 We termed this mechanism as macrophage-induced BSE (Fig. 1). Using a human intestinal commensal, Enterococcus faecalis, to test this theory, we linked the polarization of macrophages to the induction of aneuploidy and CIN in chromosomally stable epithelial cells. In addition, we most recently showed that BSE induced stem cell and tumor stem cell markers during malignant transformation.5
Figure 1.
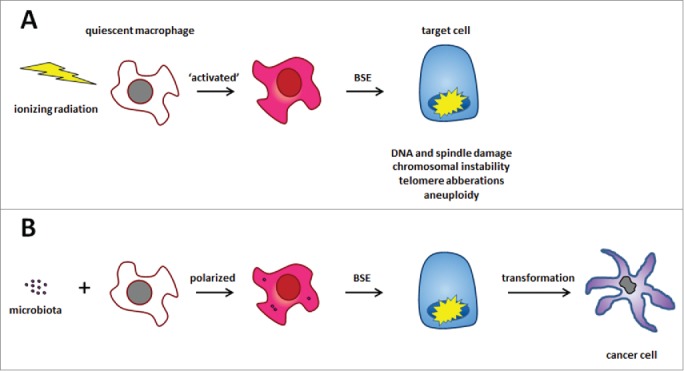
Radiation- and macrophage-induced bystander effects (BSE). (A) Ionizing radiation activates cells to generate diffusible clastogens that damage genomic DNA in non-irradiated target cells leading to CIN. (B) Similarly, polarization of quiescent macrophages by commensals generates BSE in a fashion similar to irradiation. In our recent paper,5 we show this causes malignant transformation of primary colonic epithelial cells.
Plausible theories for CRC should explain the clinical and epidemiological evidence that is strongly associated with these cancers. Any theory should be able to describe the role of cyclooxygenase (COX)-2, inflammation, and aging as risk factors for CRC.12,13 Merging these diverse observations into a coherent theory for microbiota-driven CRC carcinogenesis has been challenging, in part, due to the complex ecology of the colon. However, work by our group now provides a theoretical foundation for linking these data. In a series of publications using both in vitro and in vivo models of colitis-associated CRC,3,4,6-8 we demonstrated that normally quiescent colon macrophages can be polarized by E. faecalis. As a consequence, COX-2 is induced to produce BSE that results in mutations, aneuploidy, and CIN in target cells during malignant transformation.5 We use colonization of IL-10 knockout mice with E. faecalis to polarize colon macrophages in vivo and generate molecular signatures for BSE.7 In this Addendum, we briefly review BSE as a concept and describe how this particular phenomenon, when triggered by commensals, initiates cellular transformation. We also speculate on BSE as a mechanism for endogenous mutagenesis leading to CRC.
Bystander Effects and DNA Damage
BSE was first observed as chromosomal damage in non-irradiated cells that was caused by factors detected in the plasma of irradiated human subjects. The two initial reports described subjects who were accidentally or therapeutically exposed to whole-body radiation.14,15 Investigators found “substances” in plasma that damaged chromosomes of normal non-irradiated leukocytes. These non-targeted effects of radiation were noted to occur years after exposure. Since the initial descriptions of these findings, a large body of evidence has accumulated that confirm the ability of irradiated cells (typically myeloid cells or fibroblasts) to generate diffusible clastogens (i.e., chromosome-breaking factors) that can damage DNA in non-irradiated cells leading to CIN, telomere aberrations, and micronucleation.16 Radiation-induced BSE has been observed in plasma from survivors of the atomic bombing of Hiroshima, Japan, and the catastrophic nuclear disaster at Chernobyl.17,18 It has also been noted in vitro using grids to shield target cells,16 and confirmed in vivo using mice in sophisticated congenic sex-mismatch bone marrow transplant studies.19 Another form of radiation-induced BSE involves gap junction signaling but is not further considered here.
Work by our laboratory and others has shown that ionizing radiation is not the only trigger for BSE. In the 1980s Emerit et al. reported on extracellular superoxide, an anionic radical with a short half-life, as an initiator of cellular events leading to clastogenesis.20 Investigation by this group suggested that clastogens were <10 kDal in weight. One particular mediator was identified as trans-4-hydroxy-2-nonenal (4-HNE), a breakdown product of ω-6 polyunsaturated fatty acids. These findings were important to our later work.
Bystander Effects and Commensal Bacteria
We used these various observations to explore the role of E. faecalis in generating genomic DNA damage leading to CRC. Enterococci are Gram-positive cocci that colonize the intestinal tract of animals and often cause opportunistic infections in hospitalized patients.21 A unique characteristic of this bacterium is the ability to generate extracellular superoxide. Genetic studies by us showed that this radical was generated through the rapid non-enzymatic reduction of dioxygen by membrane-associated demethylmenaquinones.22 The rate of extracellular superoxide production, when rates were normalized to surface cell membrane area, was comparable to neutrophils undergoing a respiratory burst.23 This unique physiology is shared by few other intestinal bacteria.24 However, among humans enterococcal colonization is common if not ubiquitous.21 In a prospective study of fecal carriage, we found 64 (40%) of 159 samples tested positive for superoxide-producing enterococci.25 Although we were unable to find any association between enterococcal colonization and the occurrence of large adenomas or CRC, we were surprised to find that colonization with enterococcal strains was not stable over time in this elderly cohort of subjects. This likely confounded our ability to detect an association.
In another study using real-time PCR to quantify fecal bacteria that are potentially associated with CRC, increased colonization by E. faecalis was reported in patients with CRC compared to healthy volunteers.26 Butyrate-producing bacteria and Desulfovibrio spp. were, in contrast, reduced in number or no different than controls. Overall, we were strongly led to further investigate the potential role of E. faecalis in CRC given its unique pro-oxidant phenotype that could potentially generate DNA damage in epithelial cells.4 We believed any such evidence would provide a nexus for microbiota-triggered CRC.
Initially, we used an aromatic hydroxylation assay to measure superoxide production in vivo in rats colonized with E. faecalis.27 We detected radical generation but did not observe colon inflammation. This suggested that normal host defenses readily control the oxidant stress generated by colonizing enterococci. However, our study design did not address the question of whether E. faecalis could damage the genomic DNA in colon epithelial cells. To initially approach this issue we utilized an ultra-sensitive alkaline single cell gel electrophoresis (comet) assay to measure DNA damage. We again colonized rats with superoxide-producing E. faecalis and detected significantly increased levels of DNA damage in colon epithelial cells compared to controls.28
These results led us to study host responses to redox stress and DNA damage produced by E. faecalis. We acutely exposed the colons of wild-type mice to superoxide-producing E. faecalis. As with the rat experiments, we found no histological abnormalities in colon biopsies at 6 hours.29 However, immunohistochemical staining showed that NF-κB was activated and induced COX-2 in stromal macrophages. Gene array analyses identified one highly significant interaction network that included genes for NF-κB signaling, regulation of apoptosis, and cell cycle control. These results indicated that E. faecalis was able to strongly induce pro-inflammatory responses in colon macrophages. This led to a series of experiments using macrophages that were polarized by E. faecalis.
It was at this point that we used DNA damage at a distance due to radiation-induced BSE to guide our thinking. We sought evidence for whether E. faecalis could induce CIN through a macrophage-induced BSE. We measured CIN in target cells using a validated ALN-CD59 complement-lysis assay developed by radiation biologists.30 This assay uses CD59 on human chromosome 11 in transformed Chinese hamster ovary cells as targets for mutagenesis. In the presence of anti-CD59 antibody that fixes complement, cells with normal CD59 expression are lysed allowing for positive selection of cells with CD59 mutations.
In a series of experiments, we exposed ALN cells to E. faecalis and found mutations at rates comparable to 2 Gray of ionizing radiation.4 Importantly, E. faecalis-treated ALN cells were protected from DNA damage by superoxide dismutase, γ-tocopherol, and COX-2 inhibitors, indicating that superoxide and COX-2 contributed to this effect. We developed a dual-chamber tissue culture system using macrophages polarized by E. faecalis to mimic BSE with ALN cells as targets. We found increased mutant fractions in ALN cells. These fractions decreased when COX-2 was silenced using short interfering RNA. For comparison, we tested Escherichia coli, a Gram-negative intestinal commensal that produces negligible extracellular superoxide, and detected only modest increases in mutant fractions. These in vitro observations linked the pro-oxidant phenotype of E. faecalis to macrophage-induced BSE. Of great significance was the role COX-2 appeared to play in generating DNA damage.
We then more closely examined the genomic damage generated by BSE using the near-diploid HCT116 cell line, derived from a human CRC with deficient mismatch repair, as a cellular target in the dual-chamber system.3 The average percent of aneuploid cells increased when the multiplicity of infection for macrophages was 1,000, but this effect was not observed at lower levels of infection. Using flow cytometry, double-strand DNA breaks were detected in target cells using antibody to γH2AX. In these experiments we saw anaphase bridging, rapid phosphorylation of ATM—a DNA damage sensor protein31—and G2/M cell cycle arrest. Superoxide dismutase, catalase, and tocopherols attenuated, while caffeine and glutathione synthase inhibitors exacerbated, these aneugenic and cell cycle changes.
To characterize potential mediators for macrophage-induced BSE we focused on 4-HNE, a highly reactive aldehyde arising from lipid peroxidation.32,33 We purified 4-HNE from macrophages polarized by E. faecalis and found that it damaged genomic DNA and promoted CIN.6 We also noted that colonic epithelial cells exposed to 4-HNE developed cell cycle arrest and a failure of cytokinesis due to mitotic spindle damage. The result of these changes was tetraploidy, a genomic state considered to be an initial step toward aneuploidy and CIN.34 Studies by other investigators show that 4-HNE can form bulky DNA adducts that preferentially generate CIN, instead of microsatellite instability,35 and lead to mutations in genes associated with CRC (e.g., TP53, APC, and KRAS).36 Finally, 4-HNE-protein adducts are a sensitive tissue marker for the production of this aldehyde and were found in colon macrophages from biopsies of interleukin-10 knockout (Il10−/−) mice colonized with E. faecalis.6 Il10−/− mice are a useful model for CRC research involving the microbiota because colonization with pathobioants like E. coli or E. faecalis specifically cause colitis, dysplasia, and cancer.6,9 When these knockout mice are housed in germ-free or specific-pathogen free environments they fail to develop colon pathology. This demonstrates the key role selected commensals can play in CRC initiation. At this point, our findings indicated that 4-HNE was generated by polarized macrophages and acted as a diffusible endogenous mutagen and spindle poison for epithelial cells.
Next, using specific inhibitors of COX-2 and siRNA, we found that 4-HNE was generated by macrophages that were polarized by E. faecalis in a COX-2 dependent manner.37 Colonization of Il10−/− mice with E. faecalis produced 4-HNE-protein adducts in colon macrophages expressing COX-2. These findings reinforced COX-2 as a procarcinogenic enzyme that generated clastogens and damaged DNA in target cells.37 Immunohistochemical staining of colon biopsies confirmed an M1 polarization of macrophages by E. faecalis.7 M1 cells arise from quiescent macrophages that have been exposed to bacteria, interferon-γ, or tumor necrosis factor (TNF)-α.38 The phenotype is usually considered microbicidal and/or tumoricidal but an emerging role in cancer initiation has also been described by us and others.7,39 In contrast, M2 macrophages are generated by anti-inflammatory factors such as IL-10, IL-13, IL-4, IL-1ra, or transforming growth factor-β. These cells release anti-inflammatory cytokines that induce immune tolerance, angiogenesis, and tissue remodeling and, for existing cancers, promote tumor growth. Depletion of tissue macrophages using encapsulated liposomal clodronate blocked BSE and suppressed COX-2 and TNF-α. 4-HNE-protein adduct formation, colitis, and CRC were no longer detected. These findings provided strong evidence that colon macrophages were key effector cells for commensal-triggered BSE, inflammation, and cancer initiation.
We believe the overall evidence from these studies shows 4-HNE as a potent endogenous mutagen generated by M1 polarized macrophages. 4-HNE is a diffusible mediator for BSE that contributes to carcinogenesis by damaging DNA, inhibiting DNA repair, activating NF-κB, and helping generate its own production by inducing COX-2.6,40-43 Finally, we take note that E. faecalis-polarized macrophages also produce a multitude of other inflammatory cytokines that potentially act as mediators for BSE.8 For example, TNF-α is classically produced by M1 polarized macrophages and can promote carcinogenesis by activating NF-κB and damaging DNA.44 Our own work suggests that TNF-α contributes to macrophage-induced BSE by inducing netrin-1 in colon epithelial cells. This anti-apoptotic protein promotes intestinal epithelial cell proliferation and is considered an oncogene for CRC.8,45 In sum, these observations show the importance of BSE as a mechanism for driving microbiota-triggered colorectal carcinogenesis.
Bystander Effects and Malignant Transformation
We most recently investigated whether repetitive exposure of non-transformed colonic epithelial cells to macrophages that had been polarized by E. faecalis, or purified 4-HNE, could lead to heritable CIN and cellular transformation. Using tsA58 as a target gene for mutagenesis in YAMC cells—a primary murine colon epithelial cell line—we found that mutant fractions increased significantly after only a few treatments.5 As noted previously,3 aneuploidy increased following these treatments with clones becoming malignant after only 10 weeks. In NOD/scid mice, 8 of 25 treated clones formed poorly differentiated carcinomas with 3 tumors invading skin and/or muscle. Gene expression profiling of these clones showed strong upregulation of Ly6A/E stem cell markers. Products of these same genes were found by immunohistochemical staining in colon epithelial cells from areas of inflamed or dysplastic tissue in Il10−/− mice colonized with E. faecalis. These findings provide evidence for macrophage-induced BSE as an endogenous mechanism for commensal-driven malignant transformation in CRC.
Resident colon macrophages are important innate immune effector cells that help maintain immunological tolerance to commensals and resistance to invading pathogens. Because these professional phagocytes are not typically polarized by commensals, any loss of regulatory control over their activation could drive malignant transformation. This might occur if aging leads to significant cellular senescence, loss of regulatory controls over tolerance, or deficiencies in host defense (e.g., as represented by the knockout of IL-10 in our murine model).46 We have shown that colonization with E. faecalis polarizes quiescent macrophages to an M1 phenotype in IL-10 deficient mice. The M1 state is characterized by expression of inducible nitric oxide synthase 2 and TNF-α and the absence of arginase 1 activity.5,7 Notably, Helicobacter pylori, a group 1 carcinogen, polarizes gastric macrophages to an M1 phenotype.39 In a recent study, we depleted colon macrophages in IL-10 knockout mice colonized with E. faecalis and found that this prevented the development of colitis and cancer.7 These findings strongly suggested that M1-polarized macrophages were primary effector cells that drove inflammation and malignant transformation in our model.
In our Gut study5 we found malignant transformation of a primary colon epithelial cell line associated with changes in “driver” genes that promote carcinogenesis.10 Treated clones that formed malignant tumors in NOD/scid mice each had differential expression of 3 to 7 CRC driver genes. Although we did not investigate events leading to altered expression of these genes in these clones, we speculate that changes likely arose from direct mutations, chromosome copy number variations, epigenetic modifications, and/or altered transcriptional regulation. Several mechanisms for altered gene expression have been noted in our model of macrophage-induced BSE.3-5
A longstanding question exists about whether CIN is a driving force for malignant transformation versus a consequence of that transformation. The results from our current study do not settle this issue. Instead, we note that a majority of aneuploid clones, especially clones with higher percentages of aneuploidy, did not form tumors in NOD/scid mice. This may reflect a tumor-suppressive effect of aneuploidy that has been described by others.47 Interestingly, although multiple mutations in APC, SMAD4, TP53, KRAS, and PI3CA were reported in one study using transformed organoids from normal human colonic epithelial cells, CIN was essential for macrometastases suggesting this phenotype is necessary for cancer invasion.48
An unexpected finding from our Gut publication concerned the induction of stem cell markers by BSE.5 We found that Ly6A and Ly6E showed increased expression in transformed clones. In addition, these same proteins were seen by immunohistochemical staining in colon biopsies from E. faecalis-colonized Il10−/− mice. Ly6A and Ly6E are stem/progenitor cell markers that belong to a multigene family of glycosyl phosphatidylinositol-anchored cell surface proteins.49 Unlike Ly6E, Ly6A has no human homolog. Overexpression of LY6E has been reported in many human malignancies including breast, lung, gastric, ovarian, pancreatic, and colon.50,51 The potential importance of LY6E as a target for therapy was noted in one model that used antibody-drug conjugates to induce tumor regression.50
Surprisingly, doublecortin-like kinase 1 (Dclk1), a newly identified colon cancer stem cell marker,52,53 was also strongly expressed in malignant allografts and colon epithelial and stromal cells from E. faecalis-colonized Il10−/− mice. Analysis of transformed clones failed to identify increased gene expression. We believe this may reflect altered regulation of post-translational processing for this gene product. Under normal physiological circumstances, colon epithelial cells that express Dclk1 are quiescent and show long in vivo life spans.53 This may be important to colon epithelial cells accumulating multiple mutations over time during malignant transformation. Our findings suggest that Dclk-positive cells may be important targets for BSE (Fig. 2). Taken together, the increase in expression of stem cell markers by macrophage-induced BSE conforms to a cancer stem cell theory for CRC.54
Figure 2.
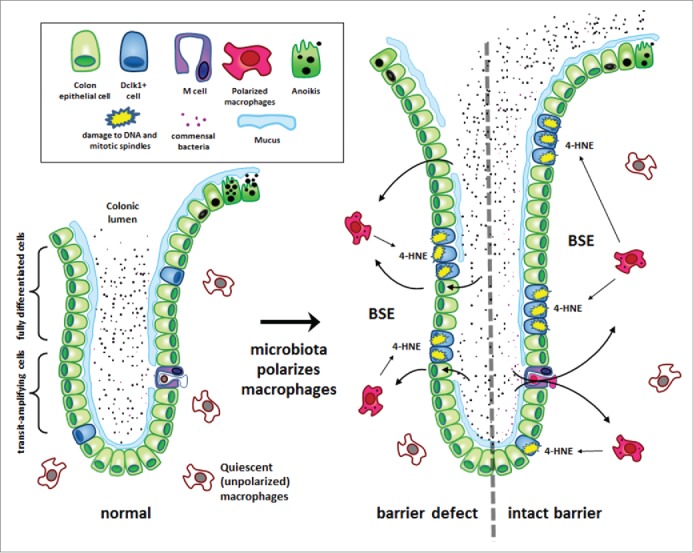
Macrophage-induced bystander effects (BSE) induce colorectal cancer. This is a commensal-triggered model for CRC involving endogenous mutagenesis caused by macrophage-induced BSE that drives cellular proliferation, mutagenesis, transformation, and colorectal cancer. (Left) Under normal physiological conditions, mucus layers, intraepithelial tight junctions, and quiescent colon macrophages protect the colonic epithelium from adverse effects of the microbiota. (Right) The loss of mucus, disruption of tight junctions, or dysregulation of macrophages can result in chronic macrophage polarization. Commensals initiate polarization by direct invasion when barrier defects are present (left) or through M cell portals when mucus layers are intact (right). Macrophage polarization generates bystander effects (BSE). One mediator is trans-4-hydroxy-2-nonenol (4-HNE), a clastogen and spindle poison (see text for details). Four-HNE diffuses into colon epithelial cells and causes mutations, double-strand breaks, and spindle dysfunction. A consequence of BSE is colon crypt hypertrophy, reduced anoikis, chromosomal instability, and malignant transformation. One potential target for BSE are epithelial cells that express doublecortin-like kinase 1 (Dclk1). These long-lived cells are scattered throughout the crypt, dramatically increase in number during inflammation, and appear to be good candidates for tumor stem cell progenitors. It remains to be determined whether this cell type, and/or others, are proximate target for BSE-mediated DNA damage, aneuploidy, and chromosomal instability leading to CRC.52,53
To these findings we would like to add that the transformed clones generated by BSE included differentially expressed genes for stromal cell-derived factor 1 (SDF-1/Cxcl12), insulin-like growth factor 2 (Igf2), pleiotrophin (Ptn), and secreted frizzled-related protein 2 (Sfrp2) (Fig. 3). A recent study showed that a combination of SDF-1/Cxcl12, IGF2, PTN, and ephrin B1 led to the differentiation of human embryonic stem cells to neurons.55 Although ephrin B1 was not altered in our transformed clones, perhaps decreased expression of these growth factors leads to de-differentiation or stem cell enriched states that give rise to tumor stem cells. We found that Sfrp2, a soluble Wnt antagonist, was significantly decreased in all transformed clones. Activation of Wnt signaling by NF-κB induces de-differentiation of intestinal epithelial cells and can help initiate tumorigenesis.56 Hypermethylation of the Sfrp2 promoter and/or decreases in Sfrp2 expression have been seen in many human cancers including CRC.57 Sfrp2 is also involved in stem cell fate and cell migration.58 Speculatively, down-regulation of these or similar genes by macrophage-induced BSE, in a mutagenic environment promoted by macrophage-induced BSE, could lead to tumor stem cell formation. Further work is needed to investigate mechanisms by which BSE contributes to the de-differentiation of colonic epithelial cells.
Figure 3.
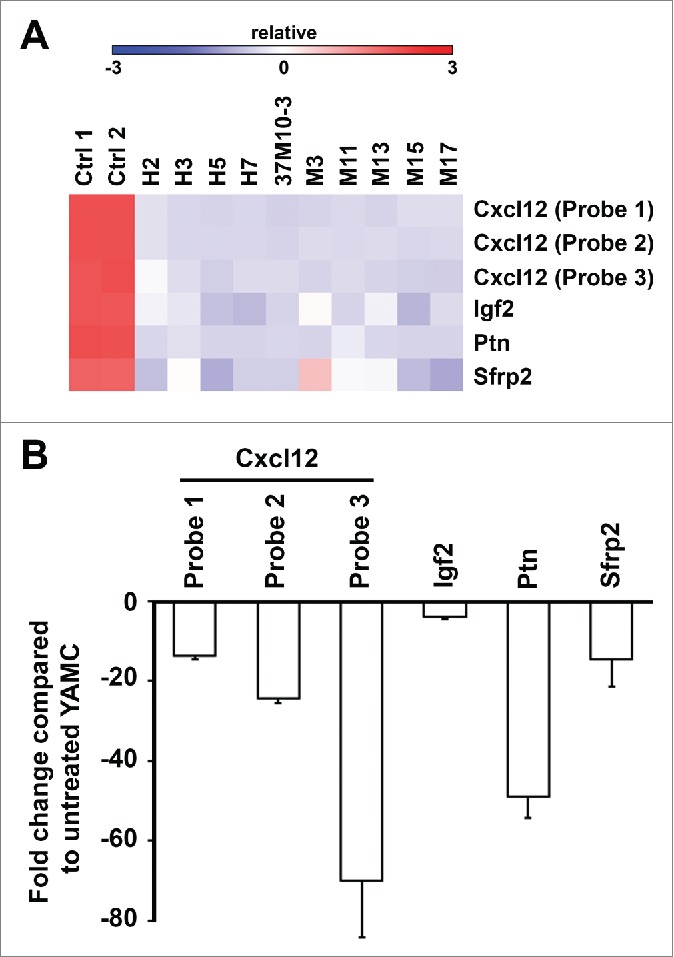
Gene expression of Cxcl12, Igf2, Ptn, and Sfrp2 in transformed clones. (A). Heat map of gene expression. (B). Fold changes for Cxcl12, Igf2, Ptn, and Sfrp2 normalized to untreated YAMC cells. Expression of these genes is significantly decreased in transformed clones compared to untreated YAMC controls.
Finally, we must pause and recognize that the vast majority of intestinal commensals are beneficial symbiotes that co-exist with their hosts to promote health and exclude potentially pathogenic exogenous bacteria.59 E. faecalis, however, appears to be an unusual member of the colonic microbiome with the potential to initiate DNA damage leading to aneuploidy, CIN, and inflammation-associated CRC. Much work remains to be done before this commensal should be considered causally linked to human CRC. Other wayward commensals undoubtedly exist that can also damage epithelial cell DNA and potentially contribute to CRC. E. faecalis is just one example and extracellular superoxide one mechanism.
Summary
Radiation- and macrophage-induced BSE appear similar if not identical in consequence on affected genomes, differing only in triggering events and leading to pathways involving clastogenesis, DNA damage, aneuploidy, CIN, and malignant transformation. Commensal bacteria represent a trigger for polarizing colonic macrophages in a promising theory for CRC that links the intestinal microbiota to COX-2, inflammation, and, potentially, aging. This represents an integrated etiology for these cancers. We selected E. faecalis as a model microorganism for our work because of its unusual redox properties. We still do not know whether E. faecalis, or any of the many other members of the microbiota, act singly or in concert to chronically polarize colon macrophages to generate BSE. Despite such gaps in our knowledge, BSE represents an important mechanism for cellular transformation and provides a rich field for hypothesis testing. The results of future investigation in this area should identify new targets for prevention.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Supported in part by the National Cancer Institute, Department of Veterans Affairs, Oklahoma Center for the Advancement of Science and Technology and Francis Duffy Endowment.
References
- 1.Garrett WS. Cancer and the microbiota. Science 2015; 348:80-6; PMID:25838377; http://dx.doi.org/ 10.1126/science.aaa4972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe 2014; 15:317-28; PMID:24629338; http://dx.doi.org/ 10.1016/j.chom.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X, Allen TD, May RJ, Lightfoot S, Houchen CW, Huycke MM. Enterococcus faecalis induces aneuploidy and tetraploidy in colonic epithelial cells through a bystander effect. Cancer Res 2008; 68:9909-17; PMID:19047172; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Huycke MM. Extracellular superoxide production by Enterococcus faecalis promotes chromosomal instability in mammalian cells. Gastroenterology 2007; 132:551-61; PMID:17258726; http://dx.doi.org/ 10.1053/j.gastro.2006.11.040 [DOI] [PubMed] [Google Scholar]
- 5.Wang X, Yang Y, Huycke MM. Commensal bacteria drive endogenous transformation and tumour stem cell marker expression through a bystander effect. Gut 2015; 64:459-68; PMID:24906974; http://dx.doi.org/ 10.1136/gutjnl-2014-307213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Yang Y, Moore DR, Nimmo SL, Lightfoot SA, Huycke MM. 4-Hydroxy-2-nonenal mediates genotoxicity and bystander effects caused by Enterococcus faecalis-infected macrophages. Gastroenterology 2012; 142:543-51; PMID:22108198; http://dx.doi.org/ 10.1053/j.gastro.2011.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Y, Wang X, Huycke T, Moore DR, Lightfoot SA, Huycke MM. Colon macrophages polarized by commensal bacteria cause colitis and cancer through the bystander effect. Transl Oncol 2013; 6:596-606; PMID:24151540; http://dx.doi.org/ 10.1593/tlo.13412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Y, Wang X, Moore DR, Lightfoot SA, Huycke MM. TNF-a mediates macrophage-induced bystander effects through netrin-1. Cancer Res 2012; 72:5219-29; PMID:22915753; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012; 338:120-3; PMID:22903521; http://dx.doi.org/ 10.1126/science.1224820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW. Cancer genome landscapes. Science 2013; 339:1546-58; PMID:23539594; http://dx.doi.org/ 10.1126/science.1235122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet 2012; 13:189-203; PMID:22269907 [DOI] [PubMed] [Google Scholar]
- 12.Wu WK, Sung JJ, Lee CW, Yu J, Cho CH. Cyclooxygenase-2 in tumorigenesis of gastrointestinal cancers: an update on the molecular mechanisms. Cancer Lett 2010; 295:7-16; PMID:20381235; http://dx.doi.org/ 10.1016/j.canlet.2010.03.015 [DOI] [PubMed] [Google Scholar]
- 13.Terzič J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology 2010; 138:2101-14; PMID:20420949; http://dx.doi.org/ 10.1053/j.gastro.2010.01.058 [DOI] [PubMed] [Google Scholar]
- 14.Hollowell JG Jr., Littlefield LG. Chromosome damage induced by plasma of x-rayed patients: an indirect effect of x-ray. Proc Soci Exp Biol Med 1968; 129:240-4; http://dx.doi.org/ 10.3181/00379727-129-33295 [DOI] [PubMed] [Google Scholar]
- 15.Goh K, Sumner H. Breaks in normal human chromosomes: are they induced by a transferable substance in the plasma of persons exposed to total-body irradiation? Radiat Res 1968; 35:171-81; PMID:5659668; http://dx.doi.org/ 10.2307/3572443 [DOI] [PubMed] [Google Scholar]
- 16.Lorimore SA, Wright EG. Radiation-induced genomic instability and bystander effects: related inflammatory-type responses to radiation-induced stress and injury? A review. Int J Radiat Biol 2003; 79:15-25; PMID:12556327; http://dx.doi.org/ 10.1080/0955300021000045664 [DOI] [PubMed] [Google Scholar]
- 17.Pant GS, Kamada N. Chromosome aberrations in normal leukocytes induced by the plasma of exposed individuals. Hiroshima J Med 1977; 26:149-54 [PubMed] [Google Scholar]
- 18.Marozik P, Mothersill C, Seymour CB, Mosse I, Melnov S. Bystander effects induced by serum from survivors of the Chernobyl accident. Exp Hematol 2007; 35:55-63; PMID:17379088; http://dx.doi.org/ 10.1016/j.exphem.2007.01.029 [DOI] [PubMed] [Google Scholar]
- 19.Watson GE, Lorimore SA, Macdonald DA, Wright EG. Chromosomal instability in unirradiated cells induced in vivo by a bystander effect of ionizing radiation. Cancer Res 2000; 60:5608-11; PMID:11059747 [PubMed] [Google Scholar]
- 20.Emerit I. Clastogenic factors as potential biomarkers of increased superoxide production. Biomark Insights 2007; 2:429-38; PMID:19662223 [PMC free article] [PubMed] [Google Scholar]
- 21.Gilmore MS, Clewell DB, Ike Y, Shankar N, eds. Enterococci: From Commensals to Leading Causes of Drug Resistant Infection. Boston, 2014; PMID: 24649513.11722738 [PubMed] [Google Scholar]
- 22.Huycke MM, Moore D, Shepard L, Joyce W, Wise P, Kotake Y, Gilmore MS. Extracellular superoxide production by Enterococcus faecalis requires demethylmenaquinone and is attenuated by functional terminal quinol oxidases. Mol Microbiol 2001; 42:729-40; PMID:11722738; http://dx.doi.org/ 10.1046/j.1365-2958.2001.02638.x [DOI] [PubMed] [Google Scholar]
- 23.Moore DR, Kotake Y, Huycke MM. Effects of iron and phytic acid on production of extracellular radicals by Enterococcus faecalis. Exp Biol Med (Maywood) 2004; 229:1186-95; PMID:15564446 [DOI] [PubMed] [Google Scholar]
- 24.Huycke MM, Joyce W, Wack MF. Augmented production of extracellular superoxide production by blood isolates of Enterococcus faecalis. J Infect Dis 1996; 173:743-6; PMID:8627044; http://dx.doi.org/ 10.1093/infdis/173.3.743 [DOI] [PubMed] [Google Scholar]
- 25.Winters MD, Schlinke TL, Joyce WA, Glore SR, Huycke MM. Prospective case-cohort study of intestinal colonization with enterococci that produce extracellular superoxide and the risk for colorectal adenomas or cancer. Am J Gastroenterol 1998; 93:2491-500; PMID:9860414; http://dx.doi.org/ 10.1111/j.1572-0241.1998.00710.x [DOI] [PubMed] [Google Scholar]
- 26.Balamurugan R, Rajendiran E, George S, Samuel GV, Ramakrishna BS. Real-time polymerase chain reaction quantification of specific butyrate-producing bacteria, Desulfovibrio and Enterococcus faecalis in the feces of patients with colorectal cancer. J Gastroenterol Hepatol 2008; 23:1298-303; PMID:18624900; http://dx.doi.org/ 10.1111/j.1440-1746.2008.05490.x [DOI] [PubMed] [Google Scholar]
- 27.Huycke MM, Moore DR. In vivo production of hydroxyl radical by Enterococcus faecalis colonizing the intestinal tract using aromatic hydroxylation. Free Radic Biol Med 2002; 33:818-26; PMID:12208369; http://dx.doi.org/ 10.1016/S0891-5849(02)00977-2 [DOI] [PubMed] [Google Scholar]
- 28.Huycke MM, Abrams V, Moore DR. Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis 2002; 23:529-36; PMID:11895869; http://dx.doi.org/ 10.1093/carcin/23.3.529 [DOI] [PubMed] [Google Scholar]
- 29.Allen TD, Moore DR, Wang X, Casu V, May R, Lerner MR, Houchen C, Brackett DJ, Huycke MM. Dichotomous metabolism of Enterococcus faecalis induced by haematin starvation modulates colonic gene expression. J Med Microbiol 2008; 57:1193-204; PMID:18809545; http://dx.doi.org/ 10.1099/jmm.0.47798-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou H, Randers-Pehrson G, Waldren CA, Vannais D, Hall EJ, Hei TK. Induction of a bystander mutagenic effect of alpha particles in mammalian cells. Proc Natl Acad Sci USA 2000; 97:2099-104; PMID:10681418; http://dx.doi.org/ 10.1073/pnas.030420797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su TT. Cellular responses to DNA damage: one signal, multiple choices. Annu Rev Genet 2006; 40:187-208; PMID:16805666; http://dx.doi.org/ 10.1146/annurev.genet.40.110405.090428 [DOI] [PubMed] [Google Scholar]
- 32.Emerit I, Khan SH, Esterbauer H. Hydroxynonenal, a component of clastogenic factors? Free Radic Biol Med 1991; 10:371-7; PMID:1909988; http://dx.doi.org/ 10.1016/0891-5849(91)90045-5 [DOI] [PubMed] [Google Scholar]
- 33.Catalá A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chemistry and physics of lipids 2009; 157:1-11; PMID:Can't; http://dx.doi.org/ 10.1016/j.chemphyslip.2008.09.004 [DOI] [PubMed] [Google Scholar]
- 34.Storchova Z, Kuffer C. The consequences of tetraploidy and aneuploidy. J Cell Sci 2008; 121:3859-66; PMID:19020304; http://dx.doi.org/ 10.1242/jcs.039537 [DOI] [PubMed] [Google Scholar]
- 35.Breivik J, Gaudernack G. Genomic instability, DNA methylation, and natural selection in colorectal carcinogenesis. Cancer Biol 1999; 9:245-54; http://dx.doi.org/ 10.1006/scbi.1999.0123 [DOI] [PubMed] [Google Scholar]
- 36.Glei M, Schaeferhenrich A, Claussen U, Kuechler A, Liehr T, Weise A, Marian B, Sendt W, Pool-Zobel BL. Comet fluorescence in situ hybridization analysis for oxidative stress-induced DNA damage in colon cancer relevant genes. Toxicol Sci 2007; 96:279-84; PMID:17192441; http://dx.doi.org/ 10.1093/toxsci/kfl197 [DOI] [PubMed] [Google Scholar]
- 37.Wang X, Allen TD, Yang Y, Moore DR, Huycke MM. Cyclooxygenase-2 generates the endogenous mutagen trans-4-hydroxy-2-nonenal in Enterococcus faecalis-infected macrophages. Cancer Prev Res (Phila) 2013; 6:206-16; PMID:23321929; http://dx.doi.org/ 10.1158/1940-6207.CAPR-12-0350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2015; 122:787-95; http://dx.doi.org/ 10.1172/JCI59643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quiding-Jarbrink M, Raghavan S, Sundquist M. Enhanced M1 macrophage polarization in human helicobacter pylori-associated atrophic gastritis and in vaccinated mice. PLoS One 2010; 5:e15018; PMID:21124899; http://dx.doi.org/ 10.1371/journal.pone.0015018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng Z, Hu W, Tang MS. Trans-4-hydroxy-2-nonenal inhibits nucleotide excision repair in human cells: a possible mechanism for lipid peroxidation-induced carcinogenesis. Proc Natl Acad Sci USA 2004; 101:8598-602; PMID:15187227; http://dx.doi.org/25592152 10.1073/pnas.0402794101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bastide NM, Chenni F, Audebert M, Santarelli RL, Tache S, Naud N, Baradat M, Jouanin I, Surya R, Hobbs DA, et al. A central role for heme iron in colon carcinogenesis associated with red meat intake. Cancer Res 2015; 75:870-9; PMID:25592152; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-2554 [DOI] [PubMed] [Google Scholar]
- 42.Liu W, Kato M, Itoigawa M, Murakami H, Yajima M, Wu J, Ishikawa N, Nakashima I. Distinct involvement of NF-kappaB and p38 mitogen-activated protein kinase pathways in serum deprivation-mediated stimulation of inducible nitric oxide synthase and its inhibition by 4-hydroxynonenal. J Cell Biochem 2001; 83:271-80; PMID:11573244; http://dx.doi.org/ 10.1002/jcb.1234 [DOI] [PubMed] [Google Scholar]
- 43.Zarrouki B, Soares AF, Guichardant M, Lagarde M, Geloen A. The lipid peroxidation end-product 4-HNE induces COX-2 expression through p38MAPK activation in 3T3-L1 adipose cell. FEBS Lett 2007; 581:2394-400; PMID:17481611; http://dx.doi.org/ 10.1016/j.febslet.2007.04.048 [DOI] [PubMed] [Google Scholar]
- 44.Onizawa M, Nagaishi T, Kanai T, Nagano K, Oshima S, Nemoto Y, Yoshioka A, Totsuka T, Okamoto R, Nakamura T, Sakamoto N, Tsuchiya K, Aoki K, Ohya K, Yagita H, Watanabe M. Signaling pathway via TNF-a/NF-kB in intestinal epithelial cells may be directly involved in colitis-associated carcinogenesis. Am J Physiol Gastrointest Liver Physiol 2009; 296:G850-9; PMID:19179628; http://dx.doi.org/ 10.1152/ajpgi.00071.2008 [DOI] [PubMed] [Google Scholar]
- 45.Mehlen P, Delloye-Bourgeois C, Chédotal A. Novel roles for slits and netrins: axon guidance cues as anticancer targets? Nat Rev Cancer 2011; 11:188-97; PMID:21326323; http://dx.doi.org/ 10.1038/nrc3005 [DOI] [PubMed] [Google Scholar]
- 46.Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, Khosla S, Jensen MD, Kirkland JL. Fat tissue, aging, and cellular senescence. Aging Cell 2010; 9:667-84; PMID:20701600; http://dx.doi.org/17189716 10.1111/j.1474-9726.2010.00608.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 2007; 11:25-36; PMID:17189716; http://dx.doi.org/ 10.1016/j.ccr.2006.12.003 [DOI] [PubMed] [Google Scholar]
- 48.Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, Watanabe T, Kanai T, Sato T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat Med 2015; 21:256-62; PMID:25706875 [DOI] [PubMed] [Google Scholar]
- 49.Holmes C, Stanford WL. Concise review: stem cell antigen-1: expression, function, and enigma. Stem Cells 2007; 25:1339-47; PMID:17379763; http://dx.doi.org/ 10.1634/stemcells.2006-0644 [DOI] [PubMed] [Google Scholar]
- 50.Asundi J, Crocker L, Tremayne J, Chang P, Sakanaka C, Tanguay J, Spencer S, Chalasani S, Luis E, Gascoigne K, et al. An antibody-drug conjugate directed against lymphocyte antigen 6 complex, locus E (LY6E) provides robust tumor killing in a wide range of solid tumor malignancies. Clin Cancer Res 2015; 21(14):3252-62; PMID:25862760 [DOI] [PubMed] [Google Scholar]
- 51.Bresson-Mazet C, Gandrillon O, Gonin-Giraud S. Stem cell antigen 2: a new gene involved in the self-renewal of erythroid progenitors. Cell Prolif 2008; 41:726-38; PMID:18823497; http://dx.doi.org/ 10.1111/j.1365-2184.2008.00554.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakanishi Y, Seno H, Fukuoka A, Ueo T, Yamaga Y, Maruno T, Nakanishi N, Kanda K, Komekado H, Kawada M, et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat Genet 2012; 45:98-103; PMID:23202126; http://dx.doi.org/24487592 10.1038/ng.2481 [DOI] [PubMed] [Google Scholar]
- 53.Westphalen CB, Asfaha S, Hayakawa Y, Takemoto Y, Lukin DJ, Nuber AH, Brandtner A, Setlik W, Remotti H, Muley A, et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J Clin Invest 2014; 124:1283-95; PMID:24487592; http://dx.doi.org/ 10.1172/JCI73434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zeki SS, Graham TA, Wright NA. Stem cells and their implications for colorectal cancer. Nat Rev Gastroenterol Hepatol 2011; 8:90-100; PMID:21293509; http://dx.doi.org/ 10.1038/nrgastro.2010.211 [DOI] [PubMed] [Google Scholar]
- 55.Vazin T, Becker KG, Chen J, Spivak CE, Lupica CR, Zhang Y, Worden L, Freed WJ. A novel combination of factors, termed SPIE, which promotes dopaminergic neuron differentiation from human embryonic stem cells. PLoS One 2009; 4:e6606; PMID:19672298; http://dx.doi.org/ 10.1371/journal.pone.0006606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013; 152:25-38; PMID:23273993; http://dx.doi.org/ 10.1016/j.cell.2012.12.012 [DOI] [PubMed] [Google Scholar]
- 57.Sui C, Wang G, Chen Q, Ma J. Variation risks of SFRP2 hypermethylation between precancerous disease and colorectal cancer. Tumour Biol 2014; 35:10457-65; PMID:25053594; http://dx.doi.org/ 10.1007/s13277-014-2313-2 [DOI] [PubMed] [Google Scholar]
- 58.Skah S, Nadjar J, Sirakov M, Plateroti M. The secreted Frizzled-Related Protein 2 modulates cell fate and the Wnt pathway in the murine intestinal epithelium. Exp Cell Res 2015; 330:56-65; PMID:25447442; http://dx.doi.org/ 10.1016/j.yexcr.2014.10.014 [DOI] [PubMed] [Google Scholar]
- 59.Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host-gut microbiota metabolic interactions. Science 2012; 336:1262-7; PMID:22674330; http://dx.doi.org/ 10.1126/science.1223813 [DOI] [PubMed] [Google Scholar]