

Atrial myocytes show a wider diversity of physiological calcium (Ca2+) signals than their ventricular counterparts. A study by Hohendanner and colleagues1 examines the fine tuning of Ca2+ signals within atrial myocytes.
Beating mammalian hearts require the repetitive firing of action potentials from the pace-making sino-atrial node, and the coordinated contraction and relaxation of many millions of cardiac myocytes as action potentials sweep through the atrial and ventricular chambers. When an action potential reaches a myocyte within the atrial or ventricular chambers it causes their cell membrane (the sarcolemma) to depolarise, and thereby triggers a rapid, transient, Ca2+ signal. Subsequently, the Ca2+ signal causes the actin and myosin fibres to engage, and thus elicits myocyte contraction.2
The depolarisation-evoked Ca2+ signals are caused by the interplay of different types of Ca2+ channels. Specifically, sarcolemmal depolarisation triggers voltage-activated Ca2+ channels, allowing Ca2+ to enter from the external medium. This Ca2+ entry triggers the opening of other Ca2+ channels known as ryanodine receptors (RyRs), which sit on the sarcoplasmic reticulum, by a process known as Ca2+-induced Ca2+ release (CICR).
Action potentials cause both atrial and ventricular myocytes to contract by evoking sarcolemmal depolarisation and Ca2+ signals, but with differences. Mammalian ventricular myocytes respond to an action potential with a Ca2+ signal that arises simultaneously across the whole cell. With atrial myocytes from many mammalian species action potential-evoked Ca2+ signals are not simultaneous, whole-cell events. Rather, they occur as Ca2+ waves that arise at the edge of a myocyte and propagate in a centripetal manner inside the cell3. A critical difference between ventricular and atrial myocytes is that the former cells have distinctive thin (100 – 200 nm) sarcolemma invaginations called transverse tubules (T-tubules) that convey action potentials deep into the cells.4
In terms of T-tubule expression, mammalian atrial myocyes are much more diverse than their ventricular counterparts. Some species express extensive T-tubules in their atrial cells. Whereas, others have no T-tubules, a few dispersed T-tubules, or a less organised series of sarcolemma invaginations that run transversely and axially.5 In those atrial myocytes that do not have sarcolemma invaginations, action potential-evoked Ca2+ signals will solely arise at the edge of the cells and propagate inwards. So, to understand the regulation of atrial contraction, it is important to determine what controls the propagation of centripetal Ca2+ waves.
Hohendanner et al. used rabbit atrial myocytes, which do not express T-tubules and therefore rely entirely on centripetal Ca2+ waves. They observed that centripetal Ca2+ wave propagation was faster at the cell periphery than in the cell centre. This two-speed Ca2+ wave propagation was due to mitochondria (Fig. 1). By sequestering Ca2+, mitochondria dampen CICR and inhibit Ca2+ wave propagation. Their observations provoke a number of intriguing questions; what purpose would mitochondrial Ca2+ uptake serve? Why dampen the propagation of a Ca2+ wave when it is needed to trigger contraction?
Firstly, mitochondrial respiration is regulated by the concentration of Ca2+ within the mitochondrial matrix. So, by sequestering Ca2+ mitochondria can match cellular activity and energy production. Secondly, the ventricular chambers, which are larger and stronger muscles, are responsible for the bulk of blood pumping. The atrial chambers enhance the filling of ventricular chambers with blood, and are particularly important during periods of exercise. It has been demonstrated that the extent of the inward centripetal Ca2+ wave propagation modulates the contraction of atrial myocytes. So, by dampening Ca2+ wave propagation, mitochondria serve to limit contraction, and conserve energy, when forceful contraction is not needed, but their dampening effect must be overcome when stronger contraction is required.
One way in which the dampening effect of mitochondrial Ca2+ uptake can be overcome is to activate additional Ca2+ channels during Ca2+ wave propagation. Hohendanner et al demonstrated that inositol 1,4,5-trisphosphate receptors (InsP3Rs) augment Ca2+ signalling within atrial myocytes. To trigger the specific opening of InsP3Rs, Hohendanner et al used photolytic uncaging of InsP3. They found that depolarisation-evoked Ca2+ transients were larger following uncaging of InsP3 inside an atrial myocyte. In essence, InsP3Rs act to boost CICR as a Ca2+ wave passes by.
Although InsP3 enhanced centripetal Ca2+ wave propagation in atrial myocytes, uncaging InsP3 did not trigger Ca2+ waves by itself unless mitochondrial Ca2+ uptake was blocked. This observation alludes to another function of mitochondrial Ca2+ uptake; preventing arrhythmic Ca2+ signals. As described earlier, a beating heart requires the coordinated activation of Ca2+ release following myocyte depolarisation. However, InsP3Rs can activate solely upon the binding of InsP3, and unlike RyRs don't have to wait for cellular depolarisation to trigger CICR. InsP3Rs can therefore release Ca2+ during periods when a myocyte should be silent. Promiscuous Ca2+ release in the heart is dangerous, as it disrupts myocyte beating6. Atrial myocytes use InsP3Rs as a means to boost Ca2+ signals and thereby enhance contraction, but need mitochondrial Ca2+ uptake to prevent InsP3Rs from triggering arrhythmic Ca2+ waves.
The responses to uncaging of InsP3 observed by Hohendanner et al were not the same throughout an atrial myocyte. The nucleus was more responsive to InsP3 than the remainder of the cell. Uncaging InsP3 evoked mini Ca2+ waves that were restricted to the nucleus. This nuclear restriction was also due to the Ca2+ uptake by mitochondria. InsP3 is highly diffusible inside cells and can pass through nuclear pore complexes, thereby triggering nucleoplasmic Ca2+ signals. Any Ca2+ escaping through nuclear pores would be sequestered by perinuclear mitochondria, thus making the nucleus an autonomous Ca2+ signalling domain. It is likely that nuclear Ca2+ signals caused by InsP3Rs will modulate myocyte gene expression, and that the particular genes will be different to those affected by the centripetal Ca2+ waves that also invade the nucleoplasm.
Atrial myocytes are amazing cells in which to study Ca2+ signalling because of their ability to show diverse responses. Their regular structure and geometry belies a Ca2+ signalling toolkit with multiple interacting components to enable discrete subcellular and whole-cell Ca2+ signals that are highly plastic and suit particular physiological needs.
Figure 1.
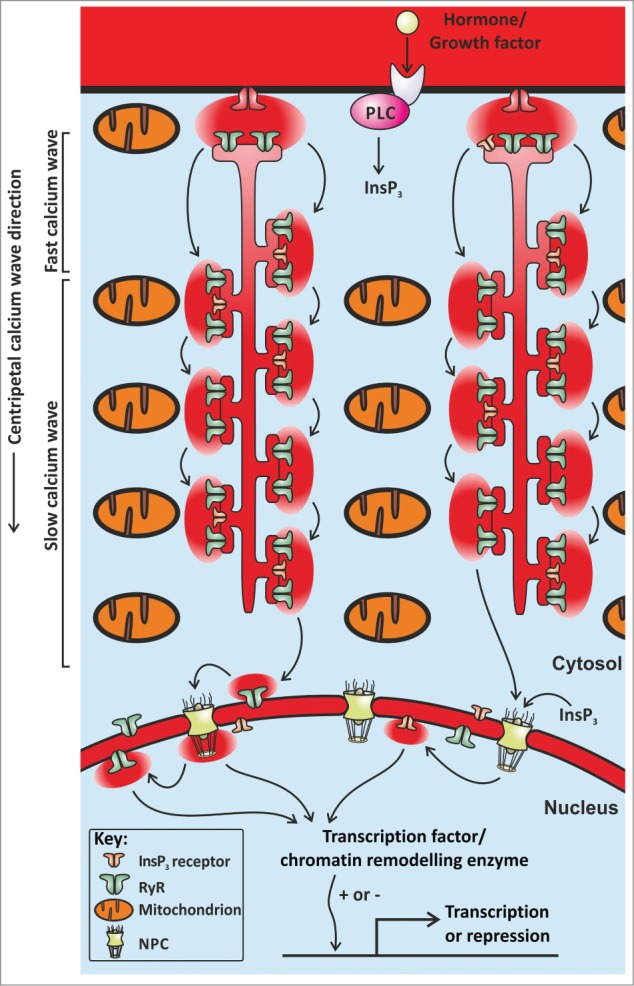
The Figure shows an illustration of part of an atrial myocyte. The arrows denote the movement of Ca2+ ions as a centripetal wave. The Ca2+ wave propagates via CICR between neighbouring clusters of RyRs. In the presence of InsP3, Ca2+ wave propagation is boosted by the opening of InsP3Rs. Both Ca2+ and InsP3 can pass through nuclear pore complexes (NPC), and trigger nucleoplasmic Ca2+ signals that influence gene transcription. Mitochondria sequester Ca2+ ions, and thereby retard Ca2+ wave propagation.
References
- 1.Hohendanner F, et al.. Channels 2015; 9:129-38; PMID:25891132; http://dx.doi.org/ 10.1080/19336950.2015.1040966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bers DM. Nature 2002; 415:198-205; PMID:11805843; http://dx.doi.org/ 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 3.Bootman MD, et al.. Biochimica et biophysica acta 2011; 1813:922-34; PMID:21295621; http://dx.doi.org/ 10.1016/j.bbamcr.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 4.Orchard CH, et al.. Experimental physiology 2009; 94:509-19; PMID:19297389; http://dx.doi.org/ 10.1113/expphysiol.2008.043984. [DOI] [PubMed] [Google Scholar]
- 5.Dibb KM, et al.. Journal of molecular and cellular cardiology 2013; 58:84-91; PMID:23147188; http://dx.doi.org/ 10.1016/j.yjmcc.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Rietdorf K, et al.. PloS one 2014; 9:e88649; PMID:24586364; http://dx.doi.org/ 10.1371/journal.pone.0088649. [DOI] [PMC free article] [PubMed] [Google Scholar]