

Abstract
The Gram-negative bacterium Helicobacter pylori is both a normal constituent of the human gastric microbiota as well as a pathogen tightly associated with severe gastric disorders. The ability of H. pylori to activate the inflammasome and caspase-1 in antigen-presenting and other cells, and the resulting processing and release of caspase-1-dependent cytokines, impacts both the immunomodulatory and pathogenic activities of H. pylori. This article summarizes recent insights by us and others on the bacterial and host prerequisites of inflammasome activation. H. pylori predominantly activates the NLRP3 inflammasome through a process that requires TLR2-dependent licensing. We identified the urease enzyme, a colonization determinant known to be required for acid adaptation, as critically required for activation of the TLR2/NLRP3/caspase-1 axis. The phenotypes of urease mutants, as well as mouse strains defective for TLR2 or NLRP3, are discussed with respect to their ability to support persistent colonization, immune tolerance and immunity to H. pylori.
Keywords: allergic diseases, gastrointestinal microbiota, microbial immunomodulation, Nod-like receptors, pro-inflammatory cytokines, regulatory T-cells, toll-like receptors, transposon mutagenesis
H. pylori has pathogenic as well as immunomodulatory properties
Two major trends have dominated public health in developed countries since the second half of the 20th century. The incidence of infectious diseases has declined sharply in that time frame, whereas immunological disorders such as multiple sclerosis (MS), inflammatory bowel disease (IBD), allergies, and type I diabetes have become much more common over the same time period.1 The incidence of infections with the gastric bacterial pathogen Helicobacter pylori has followed those of other infectious diseases, with childhood acquisition rates dropping in the US and other developed countries from >50% to 10% between the beginning and the end of the 20th century.2 In line with the known carcinogenic properties of H. pylori infection,3 a beneficial effect of this trend has been the steady decline in gastric cancer rates and gastric cancer-associated mortality in countries from which H. pylori has largely disappeared.4 The loss of H. pylori in developed countries has been paralleled by an increase in esophageal diseases such as esophagitis, Barrett's esophagus and esophageal cancer, with the latter having increased in incidence by over 6 fold in the years from 1975 to 2000 alone;5 however, a potential causal link between these 2 trends remains to be proven. Epidemiological studies have further shown an inverse association of H. pylori infection with asthma and other allergies with respiratory tract manifestations, which was particularly strong in children and adolescents and in individuals with early onset allergies and asthma.6-10 The chronic inflammatory skin disease atopic dermatitis/eczema has also been inversely linked to H. pylori infection in studies including over 3000 German school children and almost 2000 Japanese university students.11,12 Similarly, according to several meta-analyses, H. pylori infection is inversely associated with IBD. 13,14 Only 27% of IBD patients had evidence of infection with H. pylori compared to 41% of patients in the control group.13 Epidemiological studies conducted in Japan and the UK have concluded that H. pylori may further be protective in multiple sclerosis (MS).15,16
Following up on the various observational studies in human populations, we and others have begun to examine a possible protective effect of experimental H. pylori infection in animal models of allergic asthma, IBD and MS. In a murine model of allergic asthma induced by ovalbumin or house dust mite antigen sensitization and challenge, H. pylori infection confers almost complete protection against the airway hyper-responsiveness, broncho-alveolar eosinophilia, lung inflammation and goblet cell metaplasia that are hallmarks of asthma in humans and mice.17 The protective effects are particularly pronounced in animals that have been experimentally infected early in life,17 i.e. at an age when humans typically contract the infection from their mothers. 18 Asthma protection conferred by H. pylori is abolished by antibiotic eradication therapy prior to allergen challenge, and depends critically on regulatory T-cells (Tregs).17 The systemic depletion of Tregs abrogates asthma protection, and conversely, pure populations of CD4+CD25+ Tregs are sufficient to transfer protection from neonatally infected donors to naive recipients.17 These results are in line with our earlier demonstration that neonatal infection with H. pylori induces Treg-mediated immune tolerance to the bacteria.19 In addition to the beneficial effects on allergen-induced asthma, we and others have shown that H. pylori protects against chronic intestinal inflammation in models of IBD;20-22 a recent study has reported protection against experimental autoimmune encephalomyelitis (EAE), a mouse model of MS.16
The immunomodulatory effects of H. pylori require IL-18 signaling
Interestingly, the suppressive activity of Tregs in the asthma model depends critically on interleukin-18.23 In the absence of IL-18 signaling, neonatal tolerance to the infection cannot be established; CD4+CD25+ T-cells derived from IL-18−/− or IL-18R−/− donors are not protective against asthma.23 Additional experiments have revealed that IL-18 is produced by dendritic cells (DCs) upon exposure to H. pylori infection.23 The lack of IL-18 production by DCs appears to be responsible for the failure of IL-18−/− or IL-18R−/− mice to develop H. pylori-specific tolerance. IL-18 proficiency is required both in DCs derived from bone marrow and DCs isolated immunomagnetically from mesenteric lymph nodes for the conversion of naive CD4+ T-cells into CD25+FoxP3+ Tregs and IL-18R signaling in T-cell is critical for their differentiation into FoxP3+ Tregs.23 In line with these observations, FoxP3+ Treg numbers in the MLNs of both infected IL-18−/− and IL-18R−/− mice are significantly lower than those of infected wild type mice.23 The other caspase-1-dependent cytokine, IL-1β, exerts entirely different functions in the H. pylori/host interaction, and in many ways opposes the activity of IL-18.24 IL-1 receptor-deficient mice fail to control Helicobacter and are colonized as heavily as T-cell-deficient mice; Th1 and Th17 responses are not generated in the absence of IL-1 signaling, and as a consequence, the mice are protected against infection-associated immunopathology.24 Furthermore, IL-1β levels are elevated in the gastric mucosa of symptomatic H. pylori-infected individuals25 and polymorphisms associated with increased steady state levels of IL-1β predispose carriers to gastric cancer.26 Indeed, the stomach-specific expression of human IL-1β is sufficient to induce gastric inflammation and gastric cancer in transgenic mice.27 Based on the described findings on the impact of caspase-1-dependent IL-18 and IL-1β on the outcome of the H. pylori/host interaction, we hypothesized that H. pylori must possess inflammasome ligands that trigger caspase-1 activation in gastric lamina propria DCs or other antigen-presenting cells sampling the gastric lumen. In this addendum, we summarize our results showing that H. pylori activates the TLR2/NLRP3/caspase-1 axis to promote cytokine processing and secretion, which in turn is required for Treg generation, persistence and protection against allergic disease.28
H. pylori activates the NLRP3 inflammasome in a TLR2-dependent manner
We initially asked which pattern recognition receptors might contribute to inflammasome activation by H. pylori in (bone marrow-derived, BM) DCs. We compared the ability of BM-DCs from various knock-out mouse strains to activate caspase-1 and to process and secrete IL-1β and IL-18. NLRP3 and the adaptor protein ASC, but not the Nod-like receptors NLRC4 or NLRP6, or the cytoplasmic DNA sensor AIM2, turned out to be absolutely required for these processes (Fig. 1). Our results were well in line with other publications that had also identified the NLRP3 inflammasome as the predominant type of inflammasome to become activated upon H. pylori exposure of murine DCs.29,30 We then turned our attention to TLRs and their contribution to inflammasome activation; quite unexpectedly, TLR2 proficiency was found to be a clear prerequisite of NLRP3 inflammasome activation, as TLR2−/− DCs failed to activate caspase-1 and secrete caspase-1-dependent cytokines. In contrast, all other examined TLRs (TLR4,5,9) and the IL-1 receptor were dispensable for NLRP3 inflammasome activation. Further work revealed that H. pylori-induced TLR2 signaling led to the transcriptional activation of NLRP3, which in both bone-marrow-derived and splenic DCs is a rate-limiting step for H. pylori-induced inflammasome activation (Fig. 1). NLRP3 expression was dependent on NF-κB, as an inhibitor of NF-κB nuclear translocation blocked both NLRP3 transcriptional activation and caspase-1-dependent cytokine processing and secretion. We also examined a putative role for a recently identified non-canonical inflammasome activation pathway involving TRIF, IRF3/7, and type I IFN production and signaling as well as caspase-11,31 but found this pathway to be dispensable for H. pylori–induced caspase-1 activation and cytokine production. Having thus uncovered a critical contribution of NLRP3 and TLR2 to H. pylori-specific inflammasome activation, we experimentally infected the respective knockout strains with a virulent isolate of H. pylori that readily recapitulates important features of human H. pylori-associated gastric disorders.19 Interestingly, H. pylori infection of both TLR2−/− and NLRP3−/− mice phenocopied the effects of caspase-1 or IL-18 gene deletion,24 i.e., these mice were able to control the bacteria more efficiently and exhibited more pronounced Th1 (and Th17, data not shown) responses upon infection. In contrast, FoxP3+ Treg frequencies in the stomach-draining mesenteric lymph nodes were lower in TLR2−/− mice, presumably due to their inability to produce bioactive IL-18. Indeed, a defect in any of the factors of the TLR2/NLRP3/caspase-1/IL-18 axis produces a phenotype that is reminiscent of Treg or DC depletion, which leads to better infection control and more pronounced chronic inflammation and immunopathology.19,23,32 Although we weren't able to attribute the NLRP3 and TLR2 phenotypes to DCs due to the lack of available floxed alleles, the fact that DCs are required for immune tolerance rather than immunity to H. pylori23,32 makes it seem likely that inflammasome activation in DCs and the DC-intrinsic production of IL-18 contribute critically to Treg conversion and the tolerogenic phenotype of infected mice.
Figure 1.
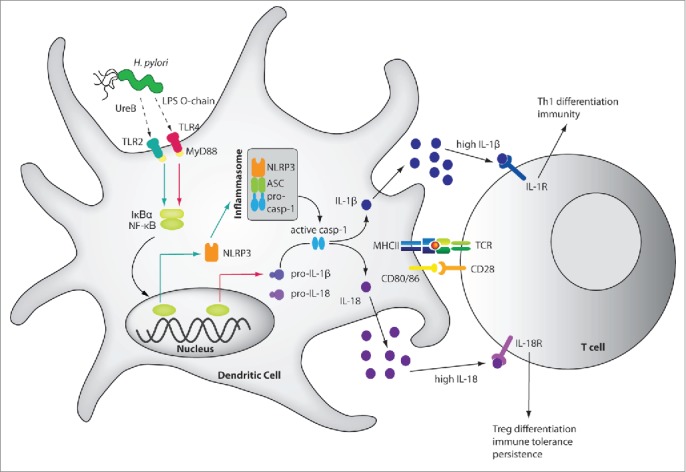
Schematic of H. pylori-induced inflammasome activation, cytokine processing and downstream effects. H. pylori LPS and the urease B subunit (UreB) promote NLRP3 inflammasome and caspase-1 activation as well as IL-1β and IL-18 processing and secretion. H. pylori LPS activates IL-1β expression via TLR4, MyD88 and NF-κB (indicated by red arrows), whereas UreB signals via TLR2, MyD88 and NF-κB to activate NLRP3 transcription (green arrows). The assembly of NLRP3, ASC and pro-caspase-1 is triggered through an as yet unknown mechanism, leading to caspase-1 activation and to the processing of pro-IL-1β and pro-IL-18. The mature cytokines are released, bind to their receptors on naive T-cells and promote Th1 differentiation and H. pylori control in the case of IL-1β, and Treg differentiation, immune tolerance and persistence in the case of IL-18. The pro-form of IL-18 is constitutively expressed in DCs.
H. pylori LPS and urease prime IL-1β and NLRP3 expression
Having identified the TLR2/NLRP3/IL-18 axis as an important modulator of the H. pylori/host interaction, we next asked which H. pylori factors drive inflammasome activation. As the Cag pathogenicity island and certain well-known H. pylori immunomodulators (e.g. the γ-glutamyl-transpeptidase and the vacuolating cytotoxin A33) could be ruled out as contributing significantly to H. pylori-induced caspase-1 activation and IL-1β secretion, we performed a genome-wide screen for mutants lacking the ability to induce IL-1β secretion by BM-DCs. We opted for an ELISA-based readout because it allowed us to screen thousands of mutants at reasonable cost. IL-1β secretion was assayed because we had determined earlier that the processing and secretion of this cytokine by BM-DCs is entirely dependent on caspase-124 (and on NLRP3, see above). We took advantage of a transposon library that had been used previously to characterize the essential genes of H. pylori for survival in vitro34 and for the colonization of the murine stomach.35 We applied a rather stringent cut-off (>75% reduction in IL-1β secretion relative to BM-DCs infected with the parental wild type strain on the same 96 well plate). Only mutants that could be validated in a second round of screening were subjected to sequencing of transposon-flanking regions to identify the transposon insertion site. The screen was saturated, i.e. no new insertion sites could be identified, after approximately 2000 individual clones had been assessed. The 64 identified transposon mutants could be mapped to 32 different gene loci and grouped into distinct classes with defects in LPS synthesis, restriction/modification and chemotaxis; a fourth predominant group had insertions in the urease gene cluster. Mechanistic follow up studies were conducted on mutants that lack LPS synthesis genes, as well as mutants with defects in urease production, because genes in these categories were recurrently identified in the screen (at least 3 and up to 5 times). Many of the transposon mutant phenotypes could be confirmed by gene-specific, isogenic deletion mutants in various strain backgrounds. To address the functional contribution of LPS to IL-1β secretion, we constructed a gene-specific deletion mutant in the gene HPG27_146, which encodes LPS 1,2-glycosyltransferase; as shown previously,36 mutants lacking this enzyme produce LPS without an O-chain and without Lewis antigen. We were able to attribute the defect of the LPS mutants in inducing IL-1β secretion to their deficiency in triggering the transcriptional activation of pro-IL-1β (Fig. 1). Further analyses with purified wild type LPS confirmed previous studies showing that H. pylori LPS is comparatively bio-inactive (i.e., 1000 times more is needed to trigger a response similar to that of E. coli LPS);37,38 however, in contrast to a previous study,38 we found the LPS to signal exclusively through TLR4 and MyD88, but not TLR2. The transcriptional activation of pro-IL-1β thus depends on a functional LPS/TLR4/MyD88 axis. The second category we chose to follow up mechanistically had defects in urease production. The H. pylori enzyme urease consists of 2 subunits, UreA and UreB, and catalyzes the hydrolysis of urea into carbon dioxide and ammonia. Urease is active in the bacterial cytosol and is widely believed to serve as the most important adaptation of H. pylori to its acidic gastric niche. Urease mutants generally fail to colonize mice.39 Interestingly, we found that mutants lacking the entire ureA/B operon (ure mutants) were defective for caspase-1 activation as judged by its auto-proteolytic processing (Fig. 1); this phenotype could be rescued by addition of recombinant UreB, but not UreA, suggesting that a structural component of this urease subunit rather than the enzymatic activity of the holoenzyme is required for inflammasome activation. Interestingly, the biochemical complementation of the ure mutant defect with UreB was restricted to wild type DCs, and was not seen in TLR2−/− DCs. This observation led us to examine whether the TLR2-dependent licensing of the NLRP3 inflammasome by H. pylori requires urease. Indeed, ure mutants failed to induce NLRP3 expression, which mechanistically explains their defect in caspase-1 activation and IL-1β/IL-18 processing and secretion. In summary, performing a genome-wide transposon library screen allowed us to identify 2 distinct, non-redundant H. pylori factors contributing to inflammasome activation and caspase-1-dependent cytokine production, with the LPS/TLR4/MyD88 axis promoting pro-IL-1β expression and the urease/TLR2/MyD88 axis promoting NLRP3 expression; the latter represents a critical step in inflammasome licensing and activation by H. pylori, at least in the examined DCs of bone-marrow and splenic origin. Independent studies on human blood-derived DCs confirmed that both classes of mutants also exhibit a clear defect in inducing IL-1β secretion in human cells, suggesting that the priming of pro-IL-1β expression and NLRP3 inflammasome licensing represent the rate-limiting steps of inflammasome activation and cytokine processing in humans as well as mice.
H. pylori urease is required for Treg induction and immune tolerance and for the suppression of allergen-specific T-cell responses
Mice lacking caspase-1, or IL-18, or its receptor all control experimental H. pylori infections more effectively than wild type mice, generate more pronounced Th1 and Th17 responses to the infection and develop excessive infection-induced gastric immunopathology upon H. pylori exposure.23,24 IL-18−/− and IL-18R−/− animals are further also known to exhibit defects in Treg differentiation and function, which explains these strains' above-mentioned traits.23 As discussed above, NLRP3−/− and TLR2−/− mice phenocopy the defects of caspase-1- and IL-18 signaling-deficient strains as they also control H. pylori infections better, and develop stronger Th1 and Th17 responses. To address whether the H. pylori factors identified in our screen contribute to NLRP3 inflammasome activation, cytokine secretion and Treg differentiation in vivo, we deleted the respective genes in the mouse-colonizing, highly immunogenic strain of H. pylori described above (termed PMSS1) that we and others have used extensively to study immunomodulation, persistence and gastric pathology.19 Whereas the LPS mutant unfortunately failed to colonize mice under any of the conditions tested and therefore could not be analyzed in vivo, the urease mutant in the PMSS1 background had no defect in initial colonization. This result was surprising given that urease mutants in 2 other strain backgrounds, M6 and SS1, have either long been known to be entirely incapable of colonizing mice or piglets (M6)39,40 or were found to be defective for colonization in our current study (SS1). The discrepancy among strains is especially puzzling as PMSS1 is a derivative of SS1 and differs from its parent mainly in the expression of a functional type IV secretion system.41 Interestingly, the urease mutant in the PMSS1 background (which colonized adult animals at wild type levels and neonatally infected animals at significantly reduced levels) induced less gastric production of IL-1β, IL-18, less active caspase-1 and less NLRP3 expression than the parental wild type strain as judged by ELISA, Western blotting and qRT-PCR of gastric mucosal extracts, thereby confirming the in vitro phenotype of the urease mutant. The lower bacterial counts of the PMSS1 urease mutant in neonatally infected mice coincided with higher Th1 and lower Treg frequencies in the mesenteric lymph nodes, and higher IFN-γ expression in the gastric mucosa. As all parameters thus pointed to a defect of the urease mutant in inducing tolerogenic immune responses, we subjected wild type and urease mutant-infected mice to house dust mite allergen sensitization and challenge to induce asthma-like symptoms. Whereas wild type-infected animals were protected against the eosinophilia, lung inflammation, goblet cell metaplasia and Th2 cytokine production that are all hallmarks of allergic asthma in this model, the urease mutant-infected mice developed normal disease. Similarly, CD4+CD25+ Tregs derived from the mesenteric lymph nodes of wild type-infected, but not mutant-infected donors transferred protection to naive recipients, suggesting that the protection induced by the wild type bacteria was Treg-mediated. Indeed, TLR2−/− mice were not protected after wild type H. pylori infection, and CD4+CD25+ Tregs derived from TLR2−/− or NLRP3−/− did not confer protection to naive recipients; moreover, treatment with an IL-18 neutralizing antibody abrogated the protection conferred by wild type bacteria. All data thus point to a critical role of the urease/TLR2/NLRP3/IL-18 axis in promoting tolerogenic (Treg-driven) responses to allergens and in suppressing allergen-specific Th2 responses and the associated pulmonary pathology (see schematic in Fig. 2). In conclusion, the contribution of urease to NLRP3 inflammasome activation and cytokine processing (especially of IL-18) that was identified in our screen translates into a critical function of this protein in high-level persistent infection and immunomodulation in the mammalian host (Fig. 2). Our results suggest a role for H. pylori urease that is independent of its role in acid adaptation; our interpretation of the urease mutant phenotype is in agreement with previous data from gnotobiotic piglets, in which urease was required for colonization even if the stomach pH had been neutralized by treatment with the proton pump inhibitor omeprazole.40
Figure 2.
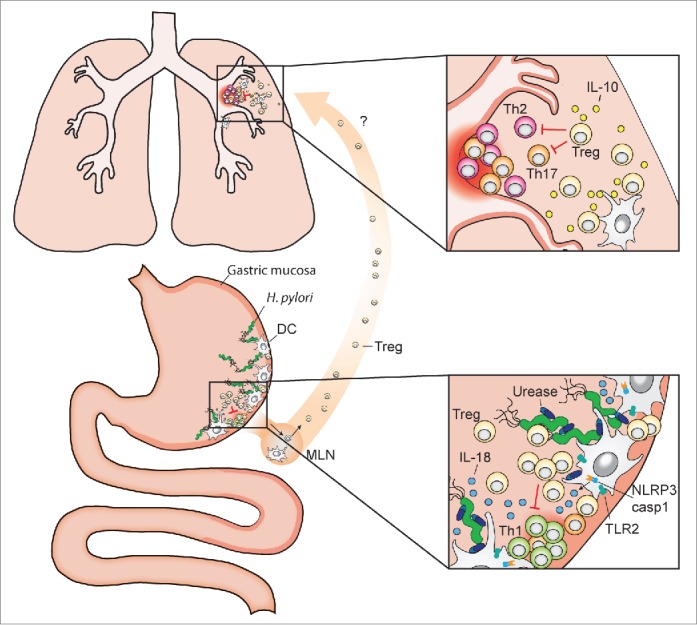
Systemic effects of gastric H. pylori colonization and inflammasome-dependent IL-18 production. The pathobiont H. pylori colonizes the gastric mucosa of roughly one half of the human population. Dendritic cells (DCs) and potentially other resident antigen-presenting cells sample H. pylori antigens and prime T-cell responses, either locally in the gastric mucosa and/or upon their migration to the draining mesenteric lymph nodes (MLN). DCs that have encountered urease-proficient H. pylori activate TLR2 signaling, the NLRP3 inflammasome and caspase-1 (lower inset), and process and secrete IL-18 (as well as IL-1β, not shown here), which promotes Treg differentiation. H. pylori-induced Tregs are required for the local suppression of anti-H. pylori T-effector cell responses and persistent H. pylori infection, and are also believed to migrate to the lung, where they prevent allergen-specific immune responses (upper inset). Treg- and DC-derived IL-10 contributes to H. pylori-specific allergy prevention in the lung.
Several questions remain to be addressed in the future: most importantly, it will be of special interest to examine the putative (direct) interaction of UreB with TLR2, the essential first step for NLRP3 expression. As TLR2 is better known for its ability to detect lipoproteins and -peptides, lipoteichoic acid, lipoarabinomannan and phosphatidylinositol-anchored lipids, UreB might be among the first of a new class of non-lipid agonists to activate TLR2 signaling. In this context, it will also be interesting to investigate the subcellular localization of the urease enzyme. Whereas the bulk of the enzyme is clearly cytoplasmic, periplasmic, surface and extracellular pools have been documented and/or proposed.42,43 As several other pathogens, including extragastric Helicobacter species and mycobacteria, are known to produce ureases, it will be worthwhile to address whether these enzymes also exhibit immunomodulatory properties in the respective hosts. Finally, the ultimate trigger for H. pylori-driven inflammasome assembly remains to be identified. Although our screen has revealed priming and licensing factors, we failed to identify factors affecting the aggregation of pre-formed NLRP3, ASC and pro-caspase-1. In conclusion, more work remains to be done to elucidate all aspects of H. pylori-induced inflammasome activation and cytokine secretion, and the various biological implications of this highly regulated process for the outcome (health vs. disease) of the Helicobacter/host interaction.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Work in the laboratory of A.M. is funded by the Swiss National Science foundation (310030-143609 and BSCGIO 157841/1).
References
- 1.Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med 2002; 347:911-20; PMID:12239261; http://dx.doi.org/ 10.1056/NEJMra020100 [DOI] [PubMed] [Google Scholar]
- 2.Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nat Rev Microbiol 2009; 7:887-94; PMID:19898491; http://dx.doi.org/ 10.1038/nrmicro2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 1991; 325:1127-31; PMID:1891020; http://dx.doi.org/ 10.1056/NEJM199110173251603 [DOI] [PubMed] [Google Scholar]
- 4.Forman D. Re: The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst 2005; 97:1013-4; author reply 4; PMID:15998956; http://dx.doi.org/ 10.1093/jnci/dji180 [DOI] [PubMed] [Google Scholar]
- 5.Pohl H, Welch HG. The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst 2005; 97:142-6; PMID:15657344; http://dx.doi.org/ 10.1093/jnci/dji024 [DOI] [PubMed] [Google Scholar]
- 6.Blaser MJ, Chen Y, Reibman J. Does Helicobacter pylori protect against asthma and allergy? Gut 2008; 57:561-7; PMID:18194986; http://dx.doi.org/ 10.1136/gut.2007.133462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y, Blaser MJ. Inverse associations of Helicobacter pylori with asthma and allergy. Arch Intern Med 2007; 167:821-7; PMID:17452546; http://dx.doi.org/ 10.1001/archinte.167.8.821 [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, Blaser MJ. Helicobacter pylori colonization is inversely associated with childhood asthma. J Infect Dis 2008; 198:553-60; PMID:18598192; http://dx.doi.org/ 10.1086/590158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reibman J, Marmor M, Filner J, Fernandez-Beros ME, Rogers L, Perez-Perez GI, Blaser MJ. Asthma is inversely associated with Helicobacter pylori status in an urban population. PLoS One 2008; 3:e4060; PMID:19112508; http://dx.doi.org/ 10.1371/journal.pone.0004060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amberbir A, Medhin G, Erku W, Alem A, Simms R, Robinson K, Fogarty A, Britton J, Venn A, Davey G. Effects of Helicobacter pylori, geohelminth infection and selected commensal bacteria on the risk of allergic disease and sensitization in 3-year-old Ethiopian children. Clin Exp Allergy 2011; 41(10):1422-30; PMID:21831135 [DOI] [PubMed] [Google Scholar]
- 11.Herbarth O, Bauer M, Fritz GJ, Herbarth P, Rolle-Kampczyk U, Krumbiegel P, Richter M, Richter T. Helicobacter pylori colonisation and eczema. J Epidemiol Community Health 2007; 61:638-40; PMID:17568058; http://dx.doi.org/ 10.1136/jech.2006.046706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shiotani A, Miyanishi T, Kamada T, Haruma K. Helicobacter pylori infection and allergic diseases: epidemiological study in Japanese university students. J Gastroenterol Hepatol 2008; 23:e29-33; PMID:17725593; http://dx.doi.org/ 10.1111/j.1440-1746.2007.05107.x [DOI] [PubMed] [Google Scholar]
- 13.Luther J, Dave M, Higgins PD, Kao JY. Association between Helicobacter pylori infection and inflammatory bowel disease: a meta-analysis and systematic review of the literature. Inflamm Bowel Dis 2010; 16:1077-84; PMID:19760778; http://dx.doi.org/ 10.1002/ibd.21116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sonnenberg A, Genta RM. Low prevalence of Helicobacter pylori infection among patients with inflammatory bowel disease. Aliment Pharmacol Ther 2012; 35:469-76; PMID:22221289; http://dx.doi.org/ 10.1111/j.1365-2036.2011.04969.x [DOI] [PubMed] [Google Scholar]
- 15.Li W, Minohara M, Su JJ, Matsuoka T, Osoegawa M, Ishizu T, Kira J. Helicobacter pylori infection is a potential protective factor against conventional multiple sclerosis in the Japanese population. J Neuroimmunol 2007; 184:227-31; PMID:17296235; http://dx.doi.org/ 10.1016/j.jneuroim.2006.12.010 [DOI] [PubMed] [Google Scholar]
- 16.Cook KW, Crooks J, Hussain K, O'Brien K, Braitch M, Kareem H, Constantinescu CS, Robinson K, Gran B. Helicobacter pylori infection reduces disease severity in an experimental model of multiple sclerosis. Front Microbiol 2015; 6:52; PMID:25762984; http://dx.doi.org/ 10.3389/fmicb.2015.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arnold IC, Dehzad N, Reuter S, Martin H, Becher B, Taube C, Müller A. Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. J Clin Invest 2011; 121:3088-93; PMID:21737881; http://dx.doi.org/ 10.1172/JCI45041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weyermann M, Rothenbacher D, Brenner H. Acquisition of Helicobacter pylori infection in early childhood: independent contributions of infected mothers, fathers, and siblings. Am J Gastroenterol 2009; 104:182-9; PMID:19098867; http://dx.doi.org/ 10.1038/ajg.2008.61 [DOI] [PubMed] [Google Scholar]
- 19.Arnold IC, Lee JY, Amieva MR, Roers A, Flavell RA, Sparwasser T, Muller A. Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology 2011; 140:199-209; PMID:20600031; http://dx.doi.org/ 10.1053/j.gastro.2010.06.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Higgins PD, Johnson LA, Luther J, Zhang M, Sauder KL, Blanco LP, Kao JY. Prior Helicobacter pylori infection ameliorates Salmonella typhimurium-induced colitis: Mucosal crosstalk between stomach and distal intestine. Inflamm Bowel Dis 2011; 17(6):1398-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luther J, Owyang SY, Takeuchi T, Cole TS, Zhang M, Liu M, Erb-Downward J, Rubenstein JH, Chen CC, Pierzchala AV, et al.. Helicobacter pylori DNA decreases pro-inflammatory cytokine production by dendritic cells and attenuates dextran sodium sulphate-induced colitis. Gut 2011; 60:1479-86; PMID:21471567; http://dx.doi.org/ 10.1136/gut.2010.220087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engler DB, Leonardi I, Hartung ML, Kyburz A, Spath S, Becher B, Rogler G, Muller A. Helicobacter pylori-specific protection against inflammatory bowel disease requires the NLRP3 inflammasome and IL-18. Inflamm Bowel Dis 2015; 21:854-61; PMID:25742401; http://dx.doi.org/ 10.1097/MIB.0000000000000318 [DOI] [PubMed] [Google Scholar]
- 23.Oertli M, Sundquist M, Hitzler I, Engler DB, Arnold IC, Reuter S, Maxeiner J, Hansson M, Taube C, Quiding-Jarbrink M, et al.. DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest 2012; 122:1082-96; PMID:22307326; http://dx.doi.org/ 10.1172/JCI61029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hitzler I, Sayi A, Kohler E, Engler DB, Koch KN, Hardt WD, Muller A. Caspase-1 Has Both Proinflammatory and Regulatory Properties in Helicobacter Infections, Which Are Differentially Mediated by Its Substrates IL-1beta and IL-18. J Immunol 2012; 188:3594-602; PMID:22403439; http://dx.doi.org/ 10.4049/jimmunol.1103212 [DOI] [PubMed] [Google Scholar]
- 25.Yamaoka Y, Kita M, Kodama T, Sawai N, Kashima K, Imanishi J. Induction of various cytokines and development of severe mucosal inflammation by cagA gene positive Helicobacter pylori strains. Gut 1997; 41:442-51; PMID:9391240; http://dx.doi.org/ 10.1136/gut.41.4.442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, et al.. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 2000; 404:398-402; PMID:10746728; http://dx.doi.org/ 10.1038/35006081 [DOI] [PubMed] [Google Scholar]
- 27.Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, Betz KS, Penz-Oesterreicher M, Bjorkdahl O, Fox JG, et al.. Overexpression of Interleukin-1 beta Induces Gastric Inflammation and Cancer and Mobilizes Myeloid-Derived Suppressor Cells in Mice. Cancer Cell 2008; 14:408-19; PMID:18977329; http://dx.doi.org/ 10.1016/j.ccr.2008.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koch KN, Hartung ML, Urban S, Kyburz A, Bahlmann AS, Lind J, Backert S, Taube C, Muller A. Helicobacter urease-induced activation of the TLR2/NLRP3/IL-18 axis protects against asthma. J Clin Invest 2015; 125:3297-302; PMID:26214524; http://dx.doi.org/ 10.1172/JCI79337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Semper RP, Mejias-Luque R, Gross C, Anderl F, Muller A, Vieth M, Busch DH, Prazeres da Costa C, Ruland J, Gross O, et al.. Helicobacter pylori-Induced IL-1beta Secretion in Innate Immune Cells Is Regulated by the NLRP3 Inflammasome and Requires the Cag Pathogenicity Island. J Immunol 2014; 193:3566-76; PMID:25172489; http://dx.doi.org/ 10.4049/jimmunol.1400362 [DOI] [PubMed] [Google Scholar]
- 30.Kim DJ, Park JH, Franchi L, Backert S, Nunez G. The Cag pathogenicity island and interaction between TLR2/NOD2 and NLRP3 regulate IL-1beta production in Helicobacter pylori-infected dendritic cells. Eur J Immunol 2013; 43:2650-8; PMID:23818043; http://dx.doi.org/ 10.1002/eji.201243281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 2012; 150:606-19; PMID:22819539; http://dx.doi.org/ 10.1016/j.cell.2012.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hitzler I, Oertli M, Becher B, Agger EM, Müller A. Dendritic Cells Prevent Rather Than Promote Immunity Conferred by a Helicobacter Vaccine Using a Mycobacterial Adjuvant. Gastroenterology 2011; 141:186-96; PMID:21569773; http://dx.doi.org/ 10.1053/j.gastro.2011.04.009 [DOI] [PubMed] [Google Scholar]
- 33.Oertli M, Noben M, Engler DB, Semper RP, Reuter S, Maxeiner J, Gerhard M, Taube C, Muller A. Helicobacter pylori gamma-glutamyl transpeptidase and vacuolating cytotoxin promote gastric persistence and immune tolerance. Proc Natl Acad Sci U S A 2013; 110:3047-52; PMID:23382221; http://dx.doi.org/ 10.1073/pnas.1211248110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salama NR, Shepherd B, Falkow S. Global transposon mutagenesis and essential gene analysis of Helicobacter pylori. J Bacteriol 2004; 186:7926-35; PMID:15547264; http://dx.doi.org/ 10.1128/JB.186.23.7926-7935.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baldwin DN, Shepherd B, Kraemer P, Hall MK, Sycuro LK, Pinto-Santini DM, Salama NR. Identification of Helicobacter pylori genes that contribute to stomach colonization. Infect Immun 2007; 75:1005-16; PMID:17101654; http://dx.doi.org/ 10.1128/IAI.01176-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moran AP, Shiberu B, Ferris JA, Knirel YA, Senchenkova SN, Perepelov AV, Jansson PE, Goldberg JB. Role of Helicobacter pylori rfaJ genes (HP0159 and HP1416) in lipopolysaccharide synthesis. FEMS Microbiol Lett 2004; 241:57-65; PMID:15556710; http://dx.doi.org/ 10.1016/j.femsle.2004.10.004 [DOI] [PubMed] [Google Scholar]
- 37.Moran AP, Lindner B, Walsh EJ. Structural characterization of the lipid A component of Helicobacter pylori rough- and smooth-form lipopolysaccharides. J Bacteriol 1997; 179:6453-63; PMID:9335296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yokota S, Ohnishi T, Muroi M, Tanamoto K, Fujii N, Amano K. Highly-purified Helicobacter pylori LPS preparations induce weak inflammatory reactions and utilize Toll-like receptor 2 complex but not Toll-like receptor 4 complex. FEMS Immunol Med Microbiol 2007; 51:140-8; PMID:17645528; http://dx.doi.org/ 10.1111/j.1574-695X.2007.00288.x [DOI] [PubMed] [Google Scholar]
- 39.Eaton KA, Gilbert JV, Joyce EA, Wanken AE, Thevenot T, Baker P, Plaut A, Wright A. In vivo complementation of ureB restores the ability of Helicobacter pylori to colonize. Infect Immun 2002; 70:771-8; PMID:11796610; http://dx.doi.org/ 10.1128/IAI.70.2.771-778.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eaton KA, Krakowka S. Effect of gastric pH on urease-dependent colonization of gnotobiotic piglets by Helicobacter pylori. Infect Immun 1994; 62:3604-7; PMID:8063376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barrozo RM, Cooke CL, Hansen LM, Lam AM, Gaddy JA, Johnson EM, Cariaga TA, Suarez G, Peek J, Richard M, et al.. Functional plasticity in the type IV secretion system of Helicobacter pylori. PLoS Pathog 2013; 9(2):e1003189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schoep TD, Fulurija A, Good F, Lu W, Himbeck RP, Schwan C, Choi SS, Berg DE, Mittl PR, Benghezal M, et al.. Surface properties of Helicobacter pylori urease complex are essential for persistence. PLoS One; 5:e15042; PMID:21124783; http://dx.doi.org/ 10.1371/journal.pone.0015042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phadnis SH, Parlow MH, Levy M, Ilver D, Caulkins CM, Connors JB, Dunn BE. Surface localization of Helicobacter pylori urease and a heat shock protein homolog requires bacterial autolysis. Infect Immun 1996; 64:905-12; PMID:8641799 [DOI] [PMC free article] [PubMed] [Google Scholar]