

Abstract
Toll-like receptor 4 (TLR4) is important in promoting the immune response in various cancers. Recently, TLR4 is highly expressed in a stage-dependent manner in gastric cancer, but the regulatory mechanism of TLR4 expression has been not elucidated it. Here, we investigated the mechanism underlying regulation of TLR4 expression through promoter methylation and histone modification between transcriptional regulation and silencing of the TLR4 gene in gastric cancer cells. Chromatin immunoprecipitation was carried out to screen for factors related to TLR4 methylation such as MeCP2, HDAC1, and Sp1 on the TLR4 promoter. Moreover, DNA methyltransferase inhibitor 5-aza-deoxycytidine (5-aza-dC) induced demethylation of the TLR4 promoter and increased H3K4 trimethylation and Sp1 binding to reactivate silenced TLR4. In contrast, although the silence of TLR4 activated H3K9 trimethylation and MeCP2 complex, combined treatment with TLR4 agonist and 5-aza-dC upregulated H3K4 trimethylation and activated with transcription factors as Sp1 and NF-κB. This study demonstrates that recruitment of the MeCP2/HDAC1 repressor complex increases the low levels of TLR4 expression through epigenetic modification of DNA and histones on the TLR4 promoter, but Sp1 activates TLR4 high expression by hypomethylation and NF-κB signaling in gastric cancer cells.
Keywords: toll-like receptor 4, gastric cancer, Sp1, methyl-CpG-binding domain protein 2, methylation
INTRODUCTION
Toll-like receptor 4 (TLR4) is an important member of type I transmembrane protein family. TLR4 is expressed not only on immune cells but also on cancer cells such as cervical cancer [1–3] and lung cancer [4]. Although the TLR4 profile varies in different tumor cells, current evidence indicates that the expression of TLR4 and signaling cascade are involved in tumor growth, progression and invasion [5]. For example, TLR4 signaling increased COX-2 and PGE2 signaling and early colorectal carcinogenesis, inhibited apoptosis and promoted angiogenesis [6]. TLR4-mediated cancer growth involved in breast tumor progression and downregulation of TLR4 prevented breast cancer progression and survival [7]. TLR4 expressed on human lung cancer cells is functionally active, and may play important roles in promoting immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance [8]. TLR4 acts as a functional receptor in the pre-metastatic phase in pulmonary metastasis [9]. These reports implied that TLR4 are expressed on human tumor cells and may play important roles in the progression of cancer.
A recent study suggests that TLR4 expression is suppressed by epigenetic events via methylation of DNA and histone, which has been recognized as a critical mechanism modulating TLR4 expression [10]. Epigenetic modification of CpG islands within gene promoters plays a major role in cancer [11,12]. Moreover, methylation of the TLR4 promoter is associated with TLR4 silencing in a variety of cells, including intestinal epithelial and stem-cell–derived vascular cells [13,14], but the precise epigenetic mechanism underlying progression of gastric cancer is not fully understood. Silencing by DNA methylation consists of two main mechanisms, de novo methylation and methylation mediated by transcription factor binding; the latter inhibits gene expression via repressor binding to methylated CpG sites [15]. Following methylation, methyl-CpG-binding domain (MBD) proteins, such as MBD1, MBD2, MBD3, and methyl-CpG binding protein 2 (MeCP2) are commonly recruited to the CpG site, repress transcription by recruiting Sin3A, which interacts with histone deacetylases (HDACs), and form a corepressor complex [16]. Transcription factor Sp1 binds to the TLR4 promoter [17]. Sp1 binding to the promoters of target genes is often associated with prevention of CpG methylation and acts as an activator [18]. Consistent with this, we hypothesize that methylation of cytosine in CpG islands within Sp1 binding sites may directly suppress to Sp1 binding ability. In addition, we suggest that the differential DNA methylation of the TLR4 promoter in gastric cancer cell lines expressing TLR4 at high and low levels is an important mechanism underlying TLR4 transcription, with TLR4-silenced cells showing increased MeCP2 binding and TLR4-upregulated cells showing enhanced Sp1 binding, to the TLR4 promoter.
RESULTS
TLR4 mRNA and protein expression in gastric cancer cell lines and tissues
To explore potential TLR4 expression in gastric cancer, immunohistochemical analysis of TLR4 on a tissue array consisting of 55 sets of gastric cancer tissues and adjacent normal tissues was carried out each of tumor stages I and II, 24 cases of stage III, and 3 cases of stage IV. We observed that TLR4 was expressed at a high level in cancer tissue compared to normal tissue, and level was dependent on tumor stage (Figure 1A). Quantification of staining intensity using by Image J (http://openwetware.org/wiki/Sean_Lauber:ImageJ Threshold_Analysis) demonstrated that 20% of the area of tumor stage I samples, 55% in tumor stage II, 80% in tumor stage III, and 90% in tumor stage IV were TLR4-positive, compared to 10% in normal tissue (Figure 1B). Further quantification of TLR4 staining intensity in these tissues revealed that TLR4 expression was elevated in gastric cancer tissues (70%), but not normal tissues (10%) (data not shown). Furthermore, we confirmed TLR4 mRNA level in 15 pairs of gastric cancer tissue and adjacent normal tissue using quantitative PCR (qPCR) and found 5-fold enhancement in gastric cancer tissues (p < 0.0016) (Figure 1C). We next examined TLR4 mRNA level in eight gastric cancer cell lines using qPCR and reverse transcription-PCR. Interestingly, TLR4 mRNA expression was overwhelmingly low levels in all gastric cancer cell lines and high level in SNU-668, AGS, and SNU-216 (Figure 1D). The MethHC database (http://MethHC.mbc.nctu.edu.tw) shows that TLR4 is expressed through promoter demethylation, but is occasionally downregulated by hypermethylation in gastric cancer tissues. This in silico data suggest that TLR4 expression may be regulated by an epigenetic mechanism, and we hypothesized that TLR4 expression may be silenced by promoter methylation in gastric cancer cell lines. Taken together, these data indicate that TLR4 is overexpressed in gastric cancer and suggest the possibility that TLR4 may be a useful biomarker and therapeutic target for gastric cancer, but the precise molecular mechanism underlying regulation of TLR4 transcription is unclear.
Figure 1. TLR4 expression pattern in gastric tumors.
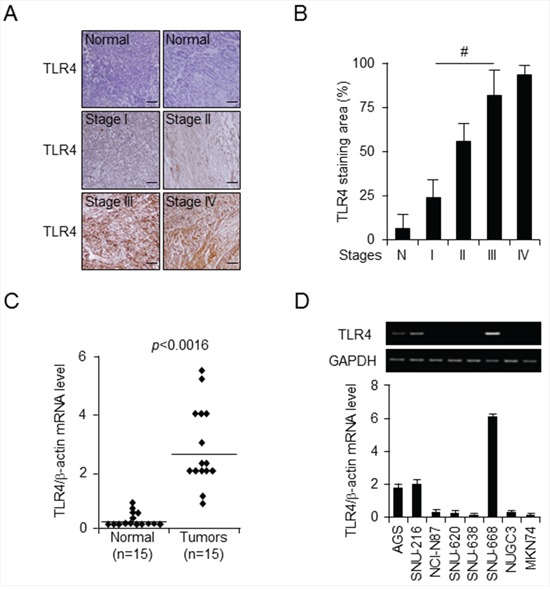
A. and B. Representative images (A) and quantitation (B) of immunohistochemical analysis of TLR4 expression in tissue array of pairs (n = 55) of normal and gastric cancer tissues at indicated stages (stage I, n = 14; stage II, n = 14; stage III, n = 24; and stage IV, n = 3). Original magnification, 200×; scale bars, 50 μm. Statistical significance was calculated using a t-test; #, p < 0.01. C. TLR4 mRNA expression was monitored by quantitative PCR in 15 paired gastric normal and tumor tissues from gastric cancer patients. D. qPCR and reverse transcription-PCR analysis of TLR4 mRNA expression level was carried out for the indicated gastric cancer cell lines.
TLR4 silencing by promoter methylation in gastric cancer cell lines
Following promoter analysis using the program TF Search (http://www.cbrc.jp/research/db/TFSEARCH.html) to identify candidate regulators on the TLR4 gene, binding sites of transcription factor Sp1 were identified on the TLR4 promoter region (Figure 2A). It is well known that promoter methylation generally interferes with Sp1 binding activity [19]. Demethylation of promoter CpG sites is a key component of epigenetic regulation, and transcription factors, such as Sp1 and Sp3, are required for demethylation and gene expression [20]. We further investigated the methylation status of putative Sp1 binding sites (CCCGCC), including a CpG site on the TLR4 promoter using quantitative methylation-specific PCR (qMSP) in gastric normal and cancer tissues. The Sp1 binding site was generally hypomethylated in 15 gastric cancer tissues (38%) compared to normal tissues (80%) (Figure 2B) and in gastric cancer cell lines (Figure 2C), and these results were associated with a high level of TLR4 mRNA expression (Figure 2D). We used qMSP to assess the extent of methylation of the TLR4 promoter in gastric cancer cell lines expressing low levels of TLR4 and observed that these cells showed high levels of DNA methylation compared with SNU-668 cells, which express a high level of TLR4 (Figure 2C and 2D). However, no change in methylation level of the TLR4 promoter was observed upon treatment of SNU-668 cells with DNA methyltransferase inhibitor 5-aza-deoxycytidine (5-aza-dC), indicating that the TLR4 promoter is hypomethylated. Epigenetic events, such as DNA methylation and histone modification, frequently induce silencing of gene transcription [21]. Both 5-aza-dC and trichostatin A (TSA) are often used to reverse silencing by promoter methylation. Therefore, we investigated the effect of 5-aza-dC on promoter methylation (Figure 2C) and of 5-aza-dC and TSA on expression of TLR4 (Figure 2D and Supplementary Figure S1A) in gastric cancer. Consistent with the basal methylation level in gastric cancer cell lines, the CpG island in the Sp1 binding site of the TLR4 promoter was dramatically demethylated by treatment with 1 μM 5-aza-dC of gastric cancer cell lines expressing low levels of TLR4 in AGS, SNU-216, SNU-668, NUGC-3, and MKN-74 cells (Figure 2C), accompanied by a dramatic increase in TLR4 mRNA levels in these cells (Figure 2D). However, TLR4 mRNA level was increased by treatment with 100 nM TSA only in SNU-638 and NUGC-3 cells (Supplementary Figure S1A). Another toll-like receptor, TLR2, also induces NF-κB signaling, similar to TLR4 [22]. Moreover, although TLR4 is associated with LPS-induced signaling, recent reports suggest that TLR2 is located upstream of TLR4 in this pathway [23]. We additionally confirmed basal expression level of TLR2 and effect of 5-aza-dC in gastric cancer cell lines using qPCR, but TLR2 expression level was not affected by 5-aza-dC except SNU-668 cells (Supplementary Figure S1B). These results imply that TLR4 expression is strongly associated with promoter methylation status, which may be one of the most important factors to regulate TLR4 expression in gastric cancer cell lines.
Figure 2. Analysis of relationship between TLR4 expression and promoter methylation in gastric cancer.
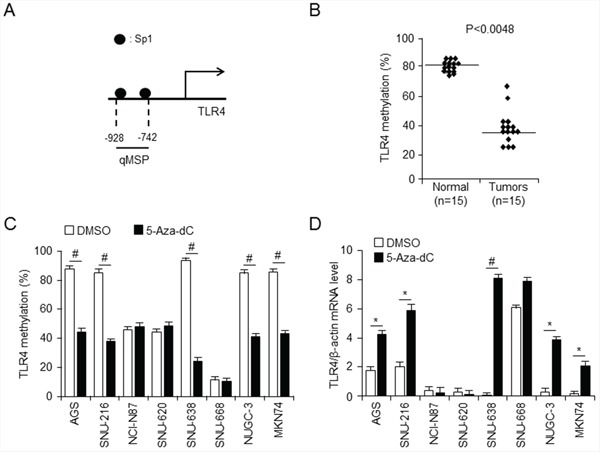
A. Schematic diagram of TLR4 promoter, showing positions of Sp1 binding sites. quantitative methylation-specific PCR (qMSP) were analyzied for methylation pattern of CpG sites on TLR4 promoter in normal and gastric cancer tissues (p < 0.0048) B. and the indicated gastric cancer cell lines C. treated with 5-aza-dC or DMSO (control) (#p < 0.001). The TLR4 promoter was methylated at a much higher level in low level of TLR4 mRNA expressing gastric cancer cell line SNU638, but not in SNU668. D. qPCR analysis effect of 5-aza-dC on TLR4 expression in the indicated gastric cancer cells. * p < 0.05, # p < 0.01. Low level of TLR4 mRNA expressing SNU638 gastric cancer cells was demethylated by 5-aza-dC.
TLR4 expression is regulated through DNA and histone methylation at the Sp1 binding site
Based on the results shown in Figure 2, we proposed that silencing of TLR4 is correlated with hypermethylation. Transcription factors, such as Sp1 and CTCF, are recruited to and suppressed CpG sites in promoters from DNA methylation, activating gene expression, whereas HDAC activity and MBD proteins are transcriptional repressors [24,25]. To determine whether methylation or lack of methylation at the CpG site on the TLR4 promoter influences Sp1 binding, we first carried out quantitative chromatin immunoprecipitation (qChIP) to identify the Sp1 binding site on the TLR4 promoter in SNU-638, SNU-216, and SNU-668 gastric cancer cells which show differential expression of TLR4 mRNA as shown Figure 2D (Figure 3A). This experiment suggested that Sp1, a transcriptional activator, was able to bind the TLR4 promoter in SNU-668 cells, which express TLR4 at a high level, compared with the hypermethylated TLR4 promoter in the SNU-638 cells. Treatment of SNU-638 cells with 5-aza-dC demethylated the TLR4 promoter and led to increased binding of Sp1. Subsequently, to investigate whether HDACs, known to be repressors of transcription, were related to binding of Sp1, qChIP was performed with antibodies targeting the HDACs such as HDAC1, 2, and 6 and SIRT 1, 2, and 3 in SNU-216, SNU-638, and SNU-668 cells. qChIP analysis demonstrated that HDAC1 and HDAC2 participate in strong binding of Sp1 in SNU-216 and SNU-638 cells, in contrast to SNU-668 cells, and 5-aza-dC–induced demethylation of the Sp1 site blocks binding of HDAC1 and HDAC2 (Figure 3B and Supplementary Figure S2A). Moreover, the effect of treatment of gastric cancer cells with HDAC inhibitor TSA was similar to that of 5-aza-dC with regard to differential HDAC1 and HDAC2 binding (Supplementary Figure S2B and S2C). The recruitment of HDACs is associated with MBD proteins, which are known to be transcriptional co-repressors [26]. MBD proteins known to be transcription repressors, MBD1 and MeCP2 recruit HDACs to methylated DNA [27]. Binding of MBD1 and MeCP2 to the TLR4 promoter was also analyzed using qChIP with primers for the Sp1 binding site in gastric cancer cells (Figure 3C). qChIP analysis using antibodies targeting MBD1 and MeCP2 indicated that MeCP2 binds to the Sp1 binding site on the TLR4 promotor in low level of TLR4 expressing SNU-638 cells, but not to the hypomethylated promoter in SNU-668 cells, and 5-aza-dC treatment did not lead to MeCP2 binding to the demethylated TLR4 promoter in gastric cancer cells. MeCP2 binding to the methylated CpG site in the TLR4 promoter results in recruitment of corepressors, such as HDAC1 and HDAC2, and transcriptional repression. Moreover, MeCP2 cooperates in maintenance of a repressive chromatin state by acting as a link between DNA and histone methylation [28]. When we analyzed ChIP-seq profiles for methylated histones H3K4me3, H3K9me3, and H3K27me3 in gastric primary tissues (in the NCBI Gene Expression Omnibus repository under accession numbers GSM910579, GSM910585, and GSM910561), the level of trimethylated H3K4 in the proximal promoter region of TLR4 was elevated compared to the levels of trimethylated H3K9 and H3K27. (Epigenome: ChIP-Seq Analysis of H3K9me3 in Human Gastric Tissue; renlab.H3K9me3.STL001GA.01.01). To test whether trimethylation of H3K4, H3K9, and H3K27 is affected by TLR4 mRNA expression, we carried out qChIP of the Sp1 binding site using histone methylation-related antibodies in SNU-638 and SNU-668 cells, which differentially express TLR4. We found that 5-aza-dC and TSA treatments dramatically increased binding of H3K4me3 in SNU-638 cells, which express TLR4 at a low level, and profoundly decreased levels of H3K9me3 and H3K27me3 binding (Figure 3D and Supplementary Figure S3). In contrast, H3K4me3 bound more strongly to the hypomethylated Sp1 binding site in SNU-668 cells than trimethylated H3K9 and H3K27, and this binding was not affected by 5-aza-dC or TSA. Taken together, these data demonstrate a complex epigenetic mechanism associated with differential expression of TLR4. These findings also suggest that Sp1 and binding of trimethylated H3K4 to the TLR4 promoter act as activators of hypomethylated TLR4 expression, but binding of MeCP2, H3K9me3, and H3K27me3 form a repressor complex and silence TLR4.
Figure 3. Relative relationship between Sp1 and MeCP2 binding to TLR4 promoter.
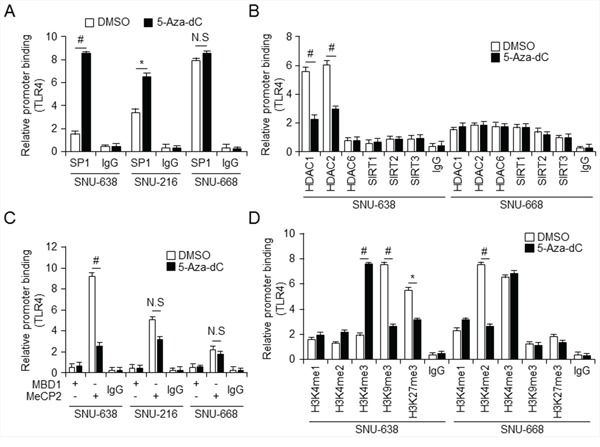
A–D. Chromatin immunoprecipitation (ChIP) was performed with lysates of control (DMSO-) and 5-aza-dC–treated SNU638, SNU216, and SNU668 gastric cancer cells and (control) IgG and antibodies against Sp1 (A); HDAC1, HDAC2, HDAC6, SIRT1, SIRT2, SIRT3 (B); MBD1, MeCP2 (C); and H3K4me1, H3K4me2, H3K4me3, H3K9me3, and H3K27me3 (D). * p < 0.05, # p < 0.01.
Epigenetic regulation of TLR4 is mediated by MeCP2 and Sp1 via HDACs
MBD proteins, such as MeCP2, MBD1, and MBD2, together with histone modifications, can repress transcription from methylated promoters [29]. To further explore the mechanism of transcriptional regulation at the TLR4 promoter, we performed short interfering RNA (siRNA)-mediated knockdown of MeCP2 and Sp1 in SNU638 and SNU668 cells. Cells were then treated with LPS and/or 5-aza-dC alone or together, followed by assessment of cell viability, and qMSP and Western analysis. No difference in cell viability was observed between MeCP2 and Sp1 knockdown and control siRNA-treated cells or between any cells treated with 5-aza-dC and/or LPS (Supplementary Figure S4). In SNU-638 cells, MeCP2 level, decreased by MeCP2 knockdown compared to control siRNA-treated cells, was also downregulated by treatment with LPS and 5-aza-dC alone and together, whereas H3K9me3 level increased with LPS and 5-aza-dC treatment (Figure 4B). Moreover, Sp1 knockdown also resulted in a decrease in H3K4me3, whereas MeCP2 and H3K9me3 levels increased and were not so much different effects with LPS and 5-aza-dC treatment. Interestingly, MeCP2 downregulation contributes to TLR4 activation and upregulation with LPS and 5-aza-dC treatment in SNU-638 cells, but SNU-668 Sp1 knockdown cells downregulate TLR4 expression, which was also reduced by LPS and 5-aza-dC treatment, compared to control siRNA-transfected cells (Figure 4C). Together, these findings suggest that MeCP2 regulates H3K9 trimethylation with TLR4 activation, but Sp1-mediated TLR4 activation contributes to H3K4 trimethylation. Interestingly, MeCP2 binding following treatment with 5-aza-dC or LPS was associated with activator Sp1 and H3K4me3. MeCP2 regulates H3K9 trimethylation with TLR4 activation, but Sp1-mediated TLR4 activation contributes to H3K4 trimethylation.
Figure 4. Knockdown of Sp1 and MeCP2 regulate TLR4 expression through modification of DNA and histone methylation.
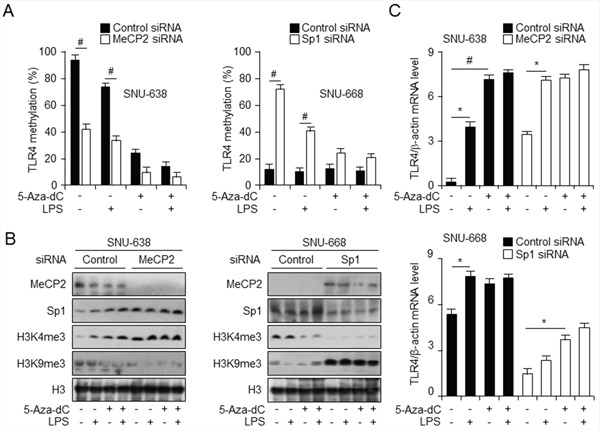
A. qMSP analysis of CpG sites in TLR4 promoter in gastric cancer cells transfected with control short interfering RNA (siRNA) and siRNA targeting MeCP2 (left) or Sp1 (right) and treated with lipopolysaccharide (LPS) and 5-aza-dC. # p < 0.01. B. Western analysis of the indicated proteins in nuclear fraction of cells described in (A) to monitor modifications in methylation of histones. C. qPCR analysis of TLR4 expression level, normalized to that of β-actin, in cells described in (A). *p < 0.05, #p < 0.01.
Combination 5-aza-dC and LPS treatment enhances binding activity of Sp1 and H3K4me3 in gastric cancer cells via activation of TLR4 signaling
To identify whether combination treatment with 5-aza-dC and LPS mediated NF-κB signaling and triggered cell death in TLR4 (Figure 5A) and TLR2 (Figure 5B) knockdown cells treated with BCL2 inhibitor ABT199, we first carried out siRNA-mediated knockdown of TLR4 and TLR2 in SNU-668 cells, which express TLR4 at a high level, treated the cells with 5-aza-dC and LPS and then with ABT-199, and monitored cell viability. Interestingly, viability was significantly reduced only in cells treated with ABT-199 alone, but viability of ABT-199–treated control and TLR4 knockdown cells increased following 5-aza-dC and LPS pretreatment. However, silencing effect of TLR2 was similar with the cell viability of TLR4 knockdown in SNU-668. Although TLR2 gene was silenced by TLR2 siRNA, these results might be regulated by TLR4 promoter methylation and activated TLR4 mRNA level by 5-aza-dc in SNU-668 cells. TLR4 expression, like cell viability, is also upregulated in cells expressing TLR4 at a high level and in TLR4 knockdown cells. These finding suggest that NF-κB signaling is blocked with BCL2 inhibition, triggering apoptosis, but the effect of combined treatment with 5-aza-dC and LPS also relatively repressed cell death with ABT-199. Our results suggest that epigenetic regulation of TLR4 expression regulates NF-κB signaling via TLR4 (Figure 5C).
Figure 5. ABT-199 treatment decreased, and combined treatment with LPS and 5-aza-dC restored, TLR4 expression and cell viability.
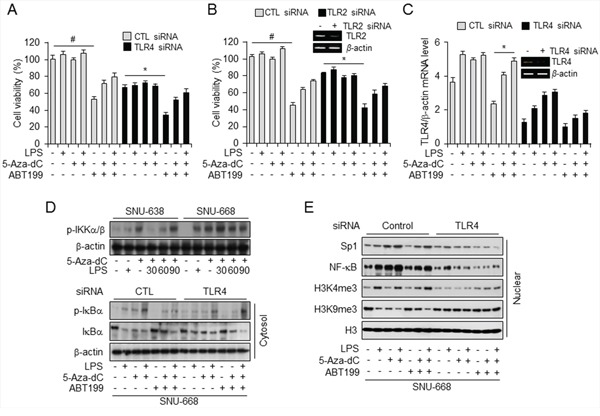
A–C. Analysis of cell viability (A and B) and TLR4 mRNA level (C) in SNU668 cells transfected with control siRNA and siRNA targeting TLR4 and TLR2, and treated with LPS, 5-aza-dC, and ABT-199 alone and in the indicated combinations. *p < 0.05, #p < 0.01. D. Western analysis of p-IKKα/β in SNU638 and SNU668 cells, untreated or pretreated with 5-aza-dC and incubated with LPS for the indicated time (minutes) (top) and of total and phosphorylated IκBα in the cytosolic fraction of SNU668 cells transfected with control siRNA or siRNA targeting TLR4 and treated with LPS, 5-aza-dC, and ABT-199 alone and in the indicated combinations. β-actin served as internal control. E. Western analysis of Sp1, NF-κB, H3K4me3, H3K4me9, and H3 in the nuclear fraction of SNU668 cells transfected with control siRNA or siRNA targeting TLR4 and treated with LPS, 5-aza-dC, and ABT-199 alone and in the indicated combinations.
Next, to elucidate whether TLR4 activation induced by 5-aza-dC and LPS affects NF-κB signaling, we investigated the effect of combined pretreatment with 5-aza-dC and LPS in gastric cancer cell lines expressing low and high levels of TLR4. We found significant time-dependent enhancement of p-IKKα/β level with LPS treatment of 5-aza-dC–pretreated SNU-638 and SNU-668 cells (Figure 5D). Then, to demonstrate the occurrence of epigenetic events at the TLR4 promoter and NF-κB signaling through TLR4 knockdown, we analyzed nuclear and cytosolic fractions of TLR4 knockdown and control cells by western blotting (Figure 5D and 5E). First, to demonstrate NF-κB signaling induced by treatment with 5-aza-dC and LPS, proteins in the cytosolic fractions were quantified together with β-actin, We observed increased phosphorylation of IκBα in treated control siRNA-transfected cells In addition, ABT-199 treatment following LPS and 5-aza-dC pretreatment phosphorylated IκBα to a greater extent than treatment with ABT-199 alone, but to a lesser extent than combined 5-aza-dC and LPS treatment. Conversely, levels of phosphorylated IκBα were lower in TLR4 knockdown cells than in control transfected cells. Moreover, Western analysis of nuclear fractions showed that H3, Sp1, and NF-κB were more inactivated in TLR4 knockdown cells than in control transfected cells, but H3K4me3 and H3K9me3 levels were unchanged. On the basis of these results, we suggest that combined treatment with 5-aza-dC and LPS activates TLR4 signaling, which leads to phosphorylation of IKK α/β and IκBα and promotes nuclear translocation of NF-κB.
TLR4 expression induced by combined treatment with 5-aza-dC and LPS activates TLR4/NF-κB signaling
Previous studies suggest that co-expression of TLR4 with its ligand, LPS, induce activation of NF-κB in the tumor microenvironment, and also induce tumor growth and development [30,31]. Moreover, LPS-induced TLR4/NF-κB signaling allows nuclear translocation of NF-κB [32]. Our immunofluorescence data indicate that combined treatment with LPS and 5-aza-dC stimulates NF-κB in the cytosol and recruits it to the nucleus in SNU-638 and SNU-668 cells (Figure 6A). In addition, NF-κB translocates faster and to a greater extent in cells treated with LPS following pretreatment with 5-aza-dC than in cells treated with LPS alone. LPS and 5-aza-dC induction of NF-κB activity was investigated in gastric cell lines expressing TLR4 at low and high levels and transfected with the luciferase reporter construct pGL4-NFκB-luc (Figure 6B). NF-κB expression, demonstrated by luciferase activity, was increased in cells treated with LPS and 5-aza-dC compared with cells treated with DMSO alone. Although there was a strong change with LPS treatment, combination treatment with LPS and 5-aza-dC highly increased NF-κB activity and nuclear translocation in SNU-638 and SNU-216 which is low level of TLR4 expression rather than SNU-668 cells which is high TLR4 expression. These results are indicated that the low level of TLR4 expression which is hypermethylated condition may increase the demethylated effect of 5-aza-dC and NF-κB signal pathway than high level of TLR4 expression.
Figure 6. Combined treatment with LPS and 5-aza-dC activates TLR4/NF-κB signaling.
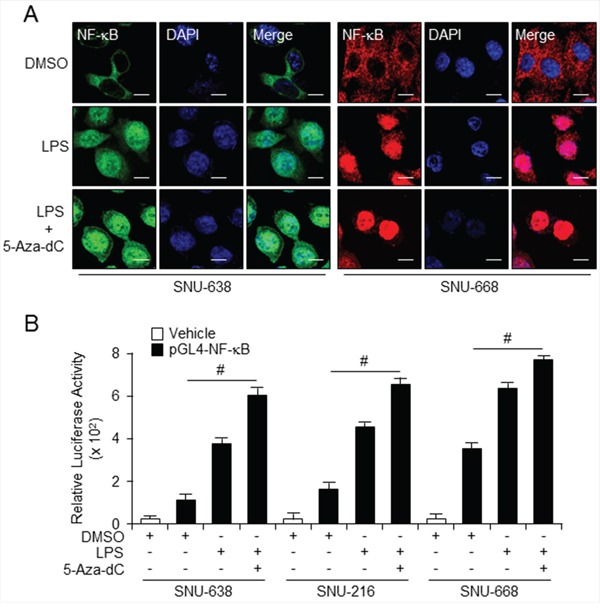
A. Immunofluorescence analysis using confocal microscopy of NF-κB in SNU638 cells stained with secondary antibody labeled with Alexa Fluor 488 (green) and SNU668 cells stained with secondary antibody labeled with Alexa Fluor 555 (red) after 3-d pretreatment with 5-aza-dC followed by 30-min incubation in LPS alone. Scale bar, 20 mm. B. Relative NF-κB expression in SNU638, SNU216, and SNU668 cells transfected with pGL4 (control) or pGL4 NF-κB-luc vector using Lipofectamine 2000, assessed by luciferase assay after 3-d incubation with LPS and 5-aza-dC alone and in the indicated combinations. Each samples were assayed in triplicate (# p < 0.01).
DISCUSSION
In this study, our results suggested that TLR4 expression in gastric cancer correlated with tumor stages and difference of TLR4 expression related to epigenetic modification via DNA methylation. TLR4 is not only important in the regulation of immune responses to infection [33], but also is involved in noninfectious inflammatory diseases, such as tumor invasion and survival [34]. High level of TLR4 expression was found to be associated with increased incidence of metastasis and poor prognosis in breast carcinoma, colorectal cancer and hepatocellular carcinoma [35–37]. Although the expression of TLR4 in gastric cancer cells has been examined, the mechanisms and the molecular pathways mediated by TLR4 signaling in gastric tumorigenesis are still controversial. On the one hand, TLR4, as a negative regulator of cancer, inhibits tumor growth and progression. TLR4 expressed in cancer can prevent infection by microbial pathogens such as hepatitis B and C viruses [38,39], Helicobacter pylori [40,41], and human papilloma virus [42], which may cause cancer. High dose of LPS, TLR4 ligand, can directly kill tumor cells so that it has been used to treat colorectal and lung cancer in Phase II clinical trials [43]. TLR4 activation may also lead to tumor regression by increasing vascular permeability [44]. On the other hand, TLR4 stimulation promotes tumorigenesis. TLR4 upregulate the NF-κB signaling and produce anti-apoptotic proteins that promote carcinogenesis and cancer cell growth and proliferation [45,46]. When the function of TLR4 signaling pathways is gradually unveiled and confirmed in cancer cells, their regulation attracts much attention. In the past few decades, many regulators of TLR4 signaling pathways, including both positive and negative ones, have been found [47,48]. Understanding the precise mechanism of TLR4 regulation is important to understanding the development and progression of gastric cancer. In these results, we found that TLR4 expression was frequently upregulated in human gastric cancer tissues and cells such as AGS, SNU-216 and SNU-668, but was often downregulated in several gastric cancer cell lines such as NCI-N87, SNU-620, SNU-638, NUGC3 and MKN74. Different levels of TLR4 expression commonly occurs by promoter methylation, and we also found that both MeCP2 and H3K9me3 contribute as repressors of TLR4 expression, whereas transcription factor Sp1 and H3K4me3 participate as activators of TLR4 transcription.
To further investigate the signal transduction and mechanism of TLR4 expression in various gastric cancer cells, the epigenetic modification of TLR4 has been characterized using 5-aza-dc in vitro. Epigenetic modifications, such as DNA methylation and histone modification, have been studied in various events in the cancer environment [49]. Recent reports demonstrate promoter methylation of tumor-related genes, including MGMT, CDKN2B and RASSF1 [50]. DNA methyltransferase DNMT1 is important in regulation of gene expression, and DNMT1 inhibitor 5-aza-dC demethylates CpG sites via DNMT1 degradation [51]. In this study, we demonstrate that the methylation status of CpG sites on the TLR4 promoter regulates transcription, and combination treatment of cells with 5-aza-dC and LPS induced TLR4/NF-κB signaling events related to critical regulators, including transcriptional repressor MeCP2 and transcriptional activator Sp1. MeCP2 expression was silenced through the interaction of transcriptional repression domain (TRD) with a corepressor, the Sin3A complex, which includes HDAC1 and HDAC2, and it was recently reported that MeCP2 is involved in histone modification in vitro and in vivo [52]. Moreover, Fuks et al. suggest that MeCP2-mediated methylation of the H19 gene is specific for H3K9 [53]. TLR4-induced proinflammatory cytokine IL-6 is silenced by promoter methylation and binding of MeCP2 and H3K9me, and 5-aza-dC–induced reactivation of IL-6 induced NF-κB signaling [54].
Gastric cancer is a highly heterogeneous disease and necessary to figure out the alterations involved in individual gastric cancer so as to increase the chance to predict prognosis and establish effective treatment options. Gastric adenocarcinoma accounting for 90-95% of gastric cancers has two histological types- intestinal and diffuse types based on microscopic observation and growth patterns. They are widely different in their molecular pathogeneses [55]. Nonetheless, epigenetic modifications may play important role in the development of both types of gastric carcinomas. Activation of TLR4/NF-κB signaling in gastric cancer cell lines expressing TLR4 at low and high levels by the TLR4 ligand LPS and DNA methyltransferase inhibitor 5-aza-dC provides a new direction for research on TLR4 signaling in cells silenced by promoter methylation, and promote cell survival and proliferation [56]. Moreover, BCL2 inhibitor ABT-199 induced cell death in gastric cancer cell lines, but 5-aza-dC/LPS and ABT199 cotreatment relatively decreased cell death.
Recent studies have demonstrated that transcription factor Sp1 may bind the TLR4 promoter [57]. Sp1 binding is commonly involved in the demethylation of CpG sites on various gene promoters [58]. When we carried out ChIP with a Sp1 antibody, Sp1 binding increased to a greater extent in SNU-668 cells, which express TLR4 at a high level, compared to SNU-638 cells which express TLR4 at a low level, and also decreased Sp1 levels of nuclear fraction in TLR4 knockdown cells. Here, Sp1 binding suppressed CpG methylation, and Sp1-binding–induced demethylation plays a role as an activator of TLR4 transcription. MeCP2 was first shown to bind methylated DNA and is an epigenetic transcriptional repressor that inhibits gene expression through the interpretation of DNA methylation and histone modification [59]. DNA methylation is frequently associated with gene silencing in various tumors, and MeCP2 may also be a potential regulator of tumorigenesis [60]. MBD proteins bind to methylated cytosines and interact with corepressors, including HDACs [61]. This complex of MeCP2, HDACs, and methylated DNA represents an important mechanism of gene silencing and chromatin remodeling [62]. Moreover, methylation at lysine 9 of histone H3 (H3K9) and DNA methylation are associated with MeCP2, and highlight MeCP2-associated histone modification for transcriptional repression [63].
In summary, by promoting rounds of histone and DNA methylation, MeCP2 binding of the TLR4 promoter maintains the hypermethylation status of CpG sites and repressor-mediated silencing of TLR4 expression, but Sp1 binding induced TLR4 activation from a hypomethylated promoter in cells expressing TLR4 at a high level, and LPS and 5-aza-dC also induced TLR4/NF-κB signaling. Our findings provide basic information for the different expression of TLR4 via epigenetic modification of TLR4 promoter in various gastric cancer cells and the relationship between epigenetic modification and TLR4 expression may contribute to this regulation of these alterations results in enhanced levels of TLR4 that can confer responsiveness to anti-tumor drug.
MATERIALS AND METHODS
Genomic DNA extraction from gastric tissues
Post-surgical samples of gastric carcinoma and nearby normal tissues were obtained from Samsung medical hospital in Seoul, Korea. A few milligrams of tumor and adjacent normal tissue were taken and genomic DNA and RNA was isolated using a genomic DNA purification kit (Promega, Madison, WI, USA) and Trizol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocols.
Cell culture
Human gastric cancer cell lines, AGS, SNU-216, NCI-N87, SNU-620, SNU-638, MKN74 and SNU-668, were obtained from the Korean Cell Line Bank (Seoul, Korea) and grown in Dulbecco modified Eagle medium (Gibco-BRL, Grand Island, NY) supplemented with heat-inactivated 10% fetal bovine serum (Gibco-BRL) and antibiotics (100 U/mL Penicillin and 100 mg/mL streptomycin, Gibco-BRL). Cells were maintained at 37°C in a humidified, 5% CO2/air atmosphere. To identify regulation by methyltransferase, 5′-Aza-2′-deoxycytidine (Sigma, St. Louis MO, USA), a methyltransferase inhibitor, was added to the culture medium 1 μM for 72 hrs, respectively, to induce demethylation of the cytosine residues.
Sodium bisulfite modification and methylation-specific PCR
Chromosomal DNA was isolated from the cell cultures in a 75 cm2 culture flask using a genomic DNA purification kit (Promega, Madison, WI) according to the manufacturer's protocol. The extracted DNA was eluted with 250 μl of distilled water. Sodium bisulfite modification of genomic DNA was carried out using an EZ DNA Methylation Kit (Zymo Research, USA) according to the manufacturer's protocol using 0.1 mg of purified DNA. Primers used in this study were as follows: methylated TLR4 (sense, 5′-GTT TAG CGG TTT ACA TGA TTT GAT-3′, antisense, 5′-CCC GCC CCT TCT TAT AAA AAA ACT-3′); unmethylated TLR4 (sense, 5′-GTT TAG TGG TTT ATA TGA TTT GAT-3′, antisense, 5′-CCC ACC CCT TCT TAT AAA AAA ACT-3′). Real-time Methylation-Specific PCR (MSP) was performed using a Power SYBR Green Kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's protocol. A methylation index was calculated for each sample using the following formula: methylation index = 1 / [1 + 2−(CTu − CTme)] × 100%, where CTu is the average cycle threshold (CT) obtained from duplicate quantitative PCR analyses using the unmethylated primer pair, and CTme is the average CT obtained using the methylated primer pair.
Quantitative and reverse transcription PCR
Total RNA from cell culture was prepared using Trizol reagent according to the manufacturer's protocols (Invitrogen, Carlsbad, CA, USA). Reverse transcription was conducted using 10 μg of total RNA with a reverse transcription kit (Promega). Expression levels of selected genes were measured by quantitative PCR (qPCR) analysis in order to confirm consistency with those from the microarray data. Primers used in this study were as follows: TLR4 for qPCR (sense, 5′-GCT TAT CTG AAG GTG TTG CA-3′, antisense, 5′-CAG AGT TTC CTG CAA TGG AT-3′); TLR2 for qPCR (sense, 5′-GGC CAG CAA ATT ACC TGT GT-3′, antisense, 5′-AGG CGG ACA TCC TGA ACC T-3′); TLR4 for reverse transcription PCR (sense, 5′-TTG GGA CAA CCA GCC TAA AG-3′, antisense, 5′-TGC CAT TGA AAG CAA CTC TG-3′); TLR2 for reverse transcription PCR (sense, 5′-GGC CAG CAA ATT ACC TGT GT-3′, antisense, 5′-ATA CCA CAG GCC ATG GAA AC-3′); ACTB (sense, 5′-GAG ACC TTC AAC ACC CCA GCC-3′, antisense, 5′-GGA TCT TCA TGA GGT AGT CAG-3′); GAPDH (sense, 5′-CAT GAC CAC AGT CCA TGC CAT-3′, antisense, 5′-AAG GCC ATG CCA CTG AGC TTC-3′). ACTB and GAPDH were used as reaction controls. 1 ml of cDNA was used for the PCR, and triplicate reactions were performed for each sample using a Power SYBR Green Kit (Applied Biosystems, Foster City, CA) with gene-specific primers on an ABI StepOnePlus instrument (Applied Biosystems). RNA quantity was normalized to β-actin and GAPDH content, and gene expression was quantified according to the 2−ΔCt method.
Transfection
SNU-638 and SNU-668 cells (2 × 105 or 1 × 106 cell/well) in 24- or 6-well plate were transfected with these double-stranded siRNAs (30 nmol/ml) of MeCP2, Sp1, TLR2 and TLR4 (Bioneer) for 24 hr by the Lipofectamine 2000 (Invitrogen) method and recovered in RPMI1640 medium (Welgene) containing 10% fetal bovine serum for 24 hrs.
Western blot analysis
Cells resuspended RIPA lysis buffer (50 mmol/L Tris-HCl, pH7.4, 150 mmol/L NaCl, 1% NP40, 0.25% Na-deoxycholate,1 mmol/L phenylmethylsulfonylfluoride (PMSF), 1 mmol/L sodium orthovanadate, 1X sigma protease inhibitor cocktail). Protein was measured using a BCA Protein Assay Kit (Pierce, Rockford, USA). Proteins were size-fractionated by PAGE and transferred to PVDF membrane. Nonspecific binding was blocked by incubation with PBS-T including 5% powdered milk and 1% Triton X-100. Membranes were incubated overnight at 4°C with primary antibodies (1:1,000) including Sp-1, MeCP2, IκBa, p-IκBa (Santa cruz) and H3K4me3, H3K9me3, H3 (Millipore) and p-IKKα/β (Cellsignaling) followed by incubation with polyclonal HRP-conjugated secondary antibodies (Sigma, 1:6,000) for 1 hour at room temperature. Membranes with any HRP-conjugate secondary antibody provided by Clarity Western ECL substrate (Bio-rad, USA) and the protein bands were visualized by exposure to X-ray film.
Chromatin immunoprecipitation(ChIP) assay
ChIP assays were performed using an EZ ChIP Chromatin Immunoprecipitation kit (Millipore, Billerica, MA, USA) as described in the supplier's protocol. Briefly, the cross-linked chromatin was sonicated after cell lysis and then incubated with antibodies against HDAC1, HDAC2, HDAC6, SIRT1, SIRT2, SIRT3, MBD1, MeCP2, Sp-1 (Santa Cruz Biotechnology) and H3K4monomethylation (H3K4me1), H3K4dimethylation (H3K4me2), H3K4trimethylation (H3K4me3), H3K9trimethylation (H3K9me3), H3K27trimethylation (H3K27me3) (Millipore) at 4°C overnight. The immunocomplex was precipitated with Protein A-agarose (Millipore), and the beads were washed, sequentially treated with 10 μl of RNase A (37°C for 30 min) and 75 μl of Proteinase K (45°C for 4 h), and incubated at 65°C overnight to reverse cross-link the chromatin. The DNA was recovered by phenol-chloroform extraction and coprecipitation with glycogen, and dissolved in 50 μl of Tris-EDTA (TE) buffer. DNA was amplified by PCR using 1 μl of the precipitated DNA. PCR primers as follows: TLR4 (sense, 5′-GTG AGT TTC TTC ACA AGA AGG G-3′, antisense, 5′-GGA GAG AGA GCC TTG AAA GAG G-3′) were designed to amplify the putative Sp-1 binding sites at the TLR4 gene promoter. Quantitative PCR conditions were 40 cycles at 94°C for 40 s, 60°C for 1 min, and 72°C for 40s.
Nuclear and cytoplasmic fractionation
Fractionation of cytoplasmic and nuclear extracts was carried out using Nuclear Extract kit (Active motif) according to manufacturer's instructions. Supernatants used as cytoplasmic fraction, whereas pellets were resuspended in 50 ml of complete lysis buffer and supernatants were used as nuclear fractions by centrifuging at 14,000 × g for 10 minutes at 4°C.
Tissue microarray
Tissue microarray slides for human gastric normal and tumor were purchased from SuperBioChips (SuperBioChips Laboratories, Seoul, Korea). Tissue sections were deparaffinized by xylene and blocked by normal serum. Primary antibody was diluted with anti-TLR4 (Santa Cruz Biotechnology, Inc.) antibody with CAS blocking solution (invitrogen) for overnight. And slides incubated with biotinylated secondary antibody for overnight. Using DAB substrate kit (DAB substrate kit, vector laboratories), tissue slides stained with substrate at room temperature until suitable staining develops immunohisto- chemistry staining was performed as the manufacturer recommended (VECTASTAIN ABC KIT, Vector laboratories).
Immunofluorescence
After 5′-Aza-dC and LPS combination treatment, SNU-638 and SNU-668 were fixed for 15 min with 4% paraformaldehyde and stained by cell staining for immunofluorescence microscopy method according to the manufacturer's protocol (Life technology). The slides were incubated with blocking buffer (1% BSA in PBS) for 30 min at 37°C and exposed overnight to anti-NF-κB (Santa Cruz) antibody (1:200 dilution in blocking buffer) at 4°C. Secondary antibody was stained using red-fluorescent Alexa Fluor 555 goat anti-rabbit and -mouse, and green-fluorescent Alexa Fluor 488 goat anti-rabbit and -mouse. Nuclear DNA was stained with DAPI (Sigma). Samples were analyzed with a ZEISS LSM 510 META confocal microscope.
Cell viability assay
WST-1 assay was performed according to the manufacturer's instructions (Roche, Mannheim, Germany) with 30 μl of WST-1 reagent was added to each well of a 24-well plate (1 × 104 cell/well). After 1h of incubation using CO2 incubator, the conversion of WST-1 reagent into chromogenic formazan was monitored with a spectrophotometer. On day 1 after plating, cells were treated with methyltransferase inhibitor, 5′-Aza-2′-deoxycytidine (Sigma, St. Louis MO, USA), 1 μM for 72 hr to induce demethylation of the cytosine residues, respectively and also added Bcl2 inhibitor, ABT-199 (Abmole BioScience), 1 μM for 24 hr. LPS (Sigma, St. Louis MO, USA) was applied to the gastric cancer cells at concentrations of 10 mg/ml.
Luciferase assay
pGL4.32 (luc2P/NF-κB-RE/Hygro) vector (pGL4NFkB-luc) and pGL4.74 (hRluc/TK) vector (pGL4-Renilla) were purchased from Promega (Madison, WI, USA). SNI-638, SNI-216 and SNU-668 cells at 70% confluence in 6-well plates were transfected with reporter luciferase plasmid, pGL4 basic, pGL4-Renilla and pGL4NFkB-luc, using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions, respectively. Luciferase activity was measured 36 hours after transfection in three independent cultures using a dual-luciferase reporter assay kit (Promega) on Molecular Devices Filter Max F3 (Sunnyvale, CA, USA).
Statistical analysis
Student's t-test was used to detect differences in the methylation and expression level between normal and cancerous tissues. P-values < 0.05 were considered significant.
SUPPLEMENTARY FIGURES
Footnotes
CONFLICTS OF INTEREST
The authors have declared no competing interest.
GRANT SUPPORT
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Ministry of Science, ICT & Future Planning (NRF-2014R1A2A2A09052492) and KRIBB Initiative of the Korea Research Council of Fundamental Science and Technology.
REFERENCES
- 1.Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol Pharm Bull. 2005;28:886–892. doi: 10.1248/bpb.28.886. [DOI] [PubMed] [Google Scholar]
- 2.Wang J, Lin D, Peng H, Shao J, Gu J. Cancer-derived immunoglobulin G promotes LPS-induced proinflammatory cytokine production via binding to TLR4 in cervical cancer cells. Oncotarget. 2014;5:9727–9743. doi: 10.18632/oncotarget.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu L, Wang L, Li M, Zhong J, Wang Z, Chen S. Expression of toll-like receptor 4 is downregulated during progression of cervical neoplasia. Cancer Immunol Immunother. 2010;59:1021–1028. doi: 10.1007/s00262-010-0825-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhan Z, Xie X, Cao H, Zhou X, Zhang XD, Fan H, Liu Z. Autophagy facilitates TLR4- and TLR3-triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy. 2013;10:257–268. doi: 10.4161/auto.27162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu L, Chen S. Toll-like receptors expressed in tumor cells: targets for therapy. Cancer Immunol Immunother. 2008;57:1271–1278. doi: 10.1007/s00262-008-0459-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Federico Feroze Tak. The PGE2-EP receptor axis in cololectal cancer and angiogenesis. Journal of Tumor. 2014;2:208–218. [Google Scholar]
- 7.Liao SJ, Zhou YH, Yuan Y, Li D, Wu FH, Wang Q, Zhu JH, Yan B, Wei JJ, Zhang GM, Feng ZH. Triggering of Toll-like receptor 4 on metastatic breast cancer cells promotes αvβ3-mediated adhesion and invasive migration. Breast Cancer Res Treat. 2012;133:853–863. doi: 10.1007/s10549-011-1844-0. [DOI] [PubMed] [Google Scholar]
- 8.He W, Liu Q, Wang L, Chen W, Li N, Cao X. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol Immunol. 2007;44:2850–2859. doi: 10.1016/j.molimm.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 9.Hiratsuka S, Watanabe A, Sakurai Y, Akashi-Takamura S, Ishibashi S, Miyake K, Shibuya M, Akira S, Aburatani H, Maru Y. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008;10:1349–1355. doi: 10.1038/ncb1794. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi K, Sugi Y, Hosono A, Kaminogawa S. Epigenetic regulation of TLR4 gene expression in intestinal epithelial cells for the maintenance of intestinal homeostasis. Journal of Immunology. 2009;183:6522–6529. doi: 10.4049/jimmunol.0901271. [DOI] [PubMed] [Google Scholar]
- 11.Nephew KP, Huang TH. Epigenetic gene silencing in cancer initiation and progression. Cancer Letter. 2003;190:125–133. doi: 10.1016/s0304-3835(02)00511-6. [DOI] [PubMed] [Google Scholar]
- 12.Nagy E, Gajjar KB, Patel ll, Taylor S, Martin-Hirsch PL, Stringfellow HF, Martin FL, Phillips DH. MGMT promoter hypermethylation and K-RAS, PTEN and TP53 mutations in tamoxifen-exposed and non-exposed endometrial cancer cases. Br J Cancer. 2014;110:2874–2880. doi: 10.1038/bjc.2014.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zampetaki A, Xiao Q, Zeng L, Hu Y, Xu Q. TLR4 expression in mouse embryonic stem cells and in stem cell-derived vascular cells is regulated by epigenetic modifications. Biochem Biophys Res Commun. 2006;347:89–99. doi: 10.1016/j.bbrc.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi K, Sugi Y, Nakano K, Tsuda M, Kurihara K, Hosono A, Kaminogawa S. Epigenetic control of the host gene by commensal bacteria in large intestinal epithelial cells. J Biol Chem. 2011;286:35755–35762. doi: 10.1074/jbc.M111.271007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Comb M, Goodman HM. CpG methylation inhibits proenkephalin gene expression and binding of the transcription factor AP-2. Nucleic Acids Res. 1990;18:3975–3982. doi: 10.1093/nar/18.13.3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 17.Wasiluk KR, McCulloch KA, Banton KL, Dunn DL. Sp1 elements regulate transcriptional activity within the murine Toll-like receptor 4 promoter. Surg Infect (Larchmt) 2006;7:489–499. doi: 10.1089/sur.2006.7.489. [DOI] [PubMed] [Google Scholar]
- 18.Holler M, Westin G, Jirincy J, Schaffner W. Sp1 transcription factor binds DNA and activates transcription even when the binding site is CpG methylated. Genes Dev. 1998;2:1127–1135. doi: 10.1101/gad.2.9.1127. [DOI] [PubMed] [Google Scholar]
- 19.Cao YX, Jean JC, Williams MC. Cytosine methylation of an Sp1 site contributes to organ-specific and cell-specific regulation of expression of the lung epithelial gene t1alpha. Biochem J. 2000;350:883–890. [PMC free article] [PubMed] [Google Scholar]
- 20.Simonsson S, Gurdon J. DNA demethylation is necessary for the epigenetic reprogramming of somatic cell nuclei. Nat Cell Biol. 2004;6:984–990. doi: 10.1038/ncb1176. [DOI] [PubMed] [Google Scholar]
- 21.Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. 2005;2:S4–S11. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]
- 22.Chen H, Cowan MJ, Hasday JD, Vogel SN, Medvedev AE. Tobacco smoking inhibits expression of proinflammatory cytokines and activation of IL-1R-associated kinase, p38, and NF-kappaB in alveolar macrophages stimulated with TLR2 and TLR4 agonists. J Immunol. 2007;179:6097–6106. doi: 10.4049/jimmunol.179.9.6097. [DOI] [PubMed] [Google Scholar]
- 23.Dobrovolskaia MA, Medvedev AE, Thomas KE, Cuesta N, Toshchakov V, Ren T, Cody MJ, Michalek SM, Rice NR, Vogel SN. Induction of in vitro reprogramming by Toll-like receptor (TLR)2 and TLR4 agonists in murine macrophage: effects of TLR “homotolerance” versus “heterotolerance” on NF-kappa B signaling pathway components. J Immunol. 2003;170:508–519. doi: 10.4049/jimmunol.170.1.508. [DOI] [PubMed] [Google Scholar]
- 24.Bird AP, Wolffe AP. Methylation-induced repression - belts, braces, and chromatin. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- 25.Kemp CJ, Moore JM, Moser R, Bernard B, Teater M, smith LE, Rabaia NA, Gurley KE, Guinney J, Busch SE, Shaknovich R, Lobanenkov VV, Lighitt D, Shmulevich I, Melnick A, Filippova GN. CTCF haploinsufficiency destabilizes DNA methylation and predisposes to cancer. Cell Rep. 2014;7:1020–1029. doi: 10.1016/j.celrep.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 27.Cassel S, Carouge D, Gensburger C, Anglard P, Burgun C, Dietrich JB, Aunis D, Zwiller J. Fluoxetine and cocaine induce the epigenetic factors MeCP2 and MBD1 in adult rat brain. Mol Pharmacol. 2006;70:487–492. doi: 10.1124/mol.106.022301. [DOI] [PubMed] [Google Scholar]
- 28.Abuhatzira L, Shemer R, Razin A. MeCP2 involvement in the regulation of neuronal alpha-tubulin production. Hum Mol Genet. 2009;18:1415–1423. doi: 10.1093/hmg/ddp048. [DOI] [PubMed] [Google Scholar]
- 29.Hendrich B, Tweedie S. The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet. 2003;19:269–277. doi: 10.1016/S0168-9525(03)00080-5. [DOI] [PubMed] [Google Scholar]
- 30.Liu L, Li YH, Niu YB, Sun Y, Guo ZJ, Li Q, Li C, Feng J, Cao SS, Mei QB. An apple oligogalactan prevents against inflammation and carcinogenesis by targeting LPS/TLR4/NF-kB pathway in a mouse model of colitis-associated colon cancer. Carcinogenesis. 2010;31:1822–1832. doi: 10.1093/carcin/bgq070. [DOI] [PubMed] [Google Scholar]
- 31.Szajnik M, Szczepanski MJ, Czystowska M, Elishaev E, Mandapathil M, Nowak-Markwitz E, Spaczynski M, Whiteside TL. TLR4 signaling induced by lipopolysaccharide or paclitaxel regulates tumor survival and chemoresistance in ovarian cancer. Oncogene. 2009;28:4353–4363. doi: 10.1038/onc.2009.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang Q, Akashi S, Miyake K, Petty HR. Lipopolysac-charide induces physical proximity between CD14 and toll-like receptor 4 (TLR4) prior to nuclear translocation of NF-kappa B. J Immunol. 2000;165:3541–3544. doi: 10.4049/jimmunol.165.7.3541. [DOI] [PubMed] [Google Scholar]
- 33.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 34.Hua D, Liu MY, Cheng ZD, Qin XJ, Zhang HM, Chen Y, Qin GJ, Li JN, Han XF, Liu DX. Small interfering RNA-directed targeting of Toll-like receptor 4 inhibits human prostate cancer cell invasion, survival, and tumorigenicity. Mol. Immunol. 2009;46:2876–2884. doi: 10.1016/j.molimm.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 35.Gonzalez-Reyes S, Marin L, Gonzalez L, Gonzalez LO, del Casar JM, Lamelas ML, Gonzalez-Q uintana JM, Vizoso FJ. Study of TLR3, TLR4 and TLR9 in breast carcinomas and their association with metastasis. BMC Cancer. 2010;10:665. doi: 10.1186/1471-2407-10-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cammarota R, Bertolini V, Pennesi G, Bucci EO, Gottardi O, Garlanda C, Laghi L, Barberis MC, Sessa F, Noonan DM, Albini A. The tumor microenvironment of colorectal cancer: stromal TLR-4 expression as a potential prognostic marker. J Transl Med. 2010;8:112. doi: 10.1186/1479-5876-8-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Velazquez RF, Rodriguez M, Navascues CA, Linares A, Perez R, Sotorrios NG, Martinez I, Rodrigo L. Prospective analysis of risk factors for hepatocellular carcinoma in patients with liver cirrhosis. Hepatology. 2003;37:520–527. doi: 10.1053/jhep.2003.50093. [DOI] [PubMed] [Google Scholar]
- 38.Broering R, Wu J, Meng Z, Hilgard P, Lu M, Trippler M, Szczeponek A, Gerken G, Schlaak JF. Toll-like receptor-stimulated non-parenchymal liver cells can regulate hepatitis C virus replication. J Hepatol. 2008;48:914–922. doi: 10.1016/j.jhep.2008.01.028. [DOI] [PubMed] [Google Scholar]
- 39.Wu J, Lu M, Meng Z, Trippler M, Broering R, Szczeponek A, Krux F, Dittmer U, Roggendorf M, Gerken G, Schlaak JF. Toll-like receptor-mediated control of HBV replication by nonparenchymal liver cells in mice. Hepatology. 2007;46:1769–1778. doi: 10.1002/hep.21897. [DOI] [PubMed] [Google Scholar]
- 40.Ferrero RL. Innate immune recognition of the extracellular mucosal pathogen, Helicobacter pylori. Mol Immunol. 2005;42:879–885. doi: 10.1016/j.molimm.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 41.Uno K, Kato K, Atsumi T, Suzuki T, Yoshitake J, Morita H, Ohara S, Kotake Y, Shimosegawa T, Yoshimura T. Toll-like receptor (TLR) 2 induced through TLR4 signaling initiated by Helicobacter pylori cooperatively amplifies iNOS induction in gastric epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1004–G1012. doi: 10.1152/ajpgi.00096.2007. [DOI] [PubMed] [Google Scholar]
- 42.Yang R, Murillo FM, Cui H, Blosser R, Uematsu S, Takeda K, Akira S, Viscidi RP, Roden RB. Papillomavirus-like particles stimulate murine bone marrow-derived dendritic cells to produce α interferon and Th1 immune responses via MyD88. J Virol. 2004;78:11152–11160. doi: 10.1128/JVI.78.20.11152-11160.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Otto F, Schmid P, Mackensen A, Wehr U, Seiz A, Braun M, Galanos C, Mertelsmann R, Engelhardt R. Phase II trial of intravenous endotoxin in patients with colorectal and non-small cell lung cancer. Eur J Cancer. 1996;32A:1712–1718. doi: 10.1016/0959-8049(96)00186-4. [DOI] [PubMed] [Google Scholar]
- 44.Garay RP, Viens P, Bauer J, Normier G, Bardou M, Jeannin JF, Chiavaroli C. Cancer relapse under chemotherapy: why TLR2/4 receptor agonists can help. Eur J Pharmacol. 2007;563:1–17. doi: 10.1016/j.ejphar.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 45.Goto Y, Arigami T, Kitago M, Nguyen SL, Narita N, Ferrone S, Morton DL, Irie RF, Hoon DS. Activation of Toll-like receptors 2, 3, and 4 on human melanoma cells induces inflammatory factors. Mol Cancer Ther. 2008;7:3642–3653. doi: 10.1158/1535-7163.MCT-08-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee CH, Wu CL, Shiau AL. Toll-like receptor 4 signaling promotes tumor growth. J Immunother. 2010;33:73–82. doi: 10.1097/CJI.0b013e3181b7a0a4. [DOI] [PubMed] [Google Scholar]
- 47.Ling GS, Bennett J, Woollard KJ, Szajna M, Fossati-Jimack L, Taylor PR, Scott D, Franzoso G, Cook HT, Botto M. Integrin CD11b Positively regulates TLR4-induced signalling pathways in dendritic cells but not inmacrophages. Nat Commun. 2014 doi: 10.1038/ncomms.4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, Yang X, Hong J, Songyang Z, Chen ZJ, Wang RF. NLRX1 Negatively Regulates TLR-Induced NF-kB Signaling by Targeting TRAF6 and IKK. Immunity. 2011;34:843–853. doi: 10.1016/j.immuni.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shukla S, Meeran SM, Katiyar SK. Epigenetic regulation by selected dietary phytochemicals in cancer chemoprevention. Cancer Lett. 2014;355:9–17. doi: 10.1016/j.canlet.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 51.Ghoshal K, Datta J, Majumder S, Bai S, Kutay H, Motiwala T, Jacob ST. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol. 2005;25:4727–4741. doi: 10.1128/MCB.25.11.4727-4741.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Drewell RA, Goddard CJ, Thomas JO, Surani MA. Methylation-dependent silencing at the H19 imprinting control region by MeCP2. Nucleic Acids Res. 2002;30:1139–1144. doi: 10.1093/nar/30.5.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fuks F, Hurd PJ, Deplus R, Kouzarides T. The DNA methyltransferase associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003;31:2305–2312. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dandrea M, Donadell M, Costanzo C, Scarpa A, Palmieri M. MeCP2/H3meK9 are involved in IL-6 gene silencing in pancreatic adenocarcinoma cell lines. Nucleic Acids Res. 2009;37:6681–6690. doi: 10.1093/nar/gkp723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jinawath N, Furukawa Y, Hasegawa S, Li M, Tsunoda T, Yamaguchi T, Imamura H, Inoue M, Shiozaki H, Nakamura Y. Comparison of gene-expression profiles between between diffuse- and intestinal-type gastric cancers using a genome-wide cDNA microarray. Oncogene. 2004;23:6830–6844. doi: 10.1038/sj.onc.1207886. [DOI] [PubMed] [Google Scholar]
- 56.Yang H, Zhou H, Feng P, Zhou X, Wen H, Xie X, Shen H, Zhu X. Reduced expression of Toll-like receptor 4 inhibits human breast cancer cells proliferation and inflammatory cytokines secretion. J Exp Clin Cancer Res. 2010;29:92. doi: 10.1186/1756-9966-29-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De Oliveira NF, Andia DC, Planello AC, Pasetto S, Marques MR, Nociti FH, Jr, Line SR, De Souza AP. TLR2 and TLR4 gene promoter methylation status during chronic periodontitis. J Clin Periodontol. 2011;38:975–983. doi: 10.1111/j.1600-051X.2011.01765.x. [DOI] [PubMed] [Google Scholar]
- 58.Brandeis M, Frank D, Keshet I, Siegfried Z, Mendelsohn M, Nemes A, Temper V, Razin A, Cedar H. Sp1 elements protect a CpG island from de novo methylation. Nature. 1994;371:435–438. doi: 10.1038/371435a0. [DOI] [PubMed] [Google Scholar]
- 59.Meehan RR, Lewis JD, Bird AP. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 1992;20:5085–5092. doi: 10.1093/nar/20.19.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 61.Ballestar E, Wolffe AP. Methyl-CpG-binding proteins. Targeting specific gene repression. Eur J Biochem. 2001;268:1–6. doi: 10.1046/j.1432-1327.2001.01869.x. [DOI] [PubMed] [Google Scholar]
- 62.Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet. 2006;7:415–426. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- 63.Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 2003;278:4035–4040. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.