

Abstract
Two metallosynthons, namely (Et4N)2[Ni(NpPepS)] (1) and (Et4N)2[Ni(PhPepS)] (2) containing carboxamido-N and thiolato-S as donors have been used to model the bimetallic Mp-Nid subsite of the A-cluster of the enzyme ACS/CODH. A series of sulfurbridged Ni/Cu dinuclear and trinuclear complexes (3-10) have been synthesized to explore their redox properties and affinity of the metal centers toward CO. The structures of (Et4N)2[Ni(PhPepS)] (2), (Et4N)[Cu(neo)Ni(NpPepS)]•0.5Et2O•0.5H2O (3•0.5Et2O•0.5H2O), (Et4N)[Cu(neo)Ni(PhPepS)]•H2O (4•H2O), (Et4N)2[Ni{Ni(NpPepS)}2]•DMF (5•DMF), (Et4N)2[Ni(DMF)2{Ni(NpPepS)}2]•3DMF (6•3DMF), (Et4N)2[Ni(DMF)2{Ni(PhPepS)}2] (8), and [Ni(dppe)Ni(PhPepS)]•CH2Cl2 (10•CH2Cl2) have been determined by crystallography. The Nid mimics 1 and 2 resist reduction and exhibit no affinity toward CO. In contrast, the sulfur-bridged Ni center (designated NiC) in the trinuclear models 5–8 are amenable to reduction and binds CO in the Ni(I) state. Also, the sulfur-bridged NiC center can be removed from the trimers (5–8) by treatment with 1,10-phenanthroline much like the “labile Ni” from the enzyme. The dinuclear Ni-Ni models 9 and 10 resemble the Nip-Nid subsite of the A-cluster more closely and only the modeled Nip site of the dimers can be reduced. The Ni(I)-Ni(II) species display EPR spectra typical of a Ni(I) center in distorted trigonal bipyramidal and distorted tetrahedral geometries for 9red and 10red, respectively. Both species bind CO and the CO-adducts 9red-CO and 10red-CO display strong νco at 2044 and 1997 cm-1, respectively. The reduction of 10 is reversible. The CO-affinity of 10 in the reduced state and the νco value of 10red-CO closely resemble the CO-bound reduced A-cluster (νco = 1996 cm-1).
Introduction
Acetyl coenzyme A synthase/carbon monoxide dehydrogenase (ACS/CODH) is a bifunctional metalloenzyme that catalyzes two important biological reactions, namely the reversible reduction of CO2 into CO (CODH activity) and the synthesis of acetyl coenzyme A (acetyl CoA) from CO, CH3 from a methylated corrinoid iron-sulfur protein, and the thiol CoA (ACS activity).1,2 Once formed, acetyl CoA is converted into cell carbon or respired as acetate depending on the metabolic needs of the cell. This enzyme is present in a number of anaerobic bacteria including acetogens, methanogens, and sulfate-reducers that utilize the Wood-Ljungdahl pathway for autotrophic carbon fixation.3 Indeed, ACS/CODH has been implicated in the chemoautotrophic origin of life in which primitive organisms consumed CO or CO2 from volcanic or hydrothermal sites for the formation of carbon-carbon bonds.4 Interest in ACS/CODH stems from its unusual metallocluster active sites, role in the global carbon cycle (reducing the levels of gaseous pollutants like CO2), and the reactions it catalyzes utilizing well known organometallic ligands like CO and CH3, analogous to the industrial Monsanto acetic acid process.
Although a large volume of spectroscopic and biochemical data were available for the enzyme for some time,1,2 the site of acetyl CoA synthesis (A-cluster) has come under intense scrutiny following the publication of three high-resolution crystal structures.5-7 The A-cluster of ACS/CODH has been shown to exist in two different oxidation states, Aox (S = 0) and Ared-CO (S = ½), the one electron reduced CO-adduct formed under CO atmosphere. The paramagnetic Ared-CO state is readily identified by a strong rhombic EPR signal (g = 2.08, 2.07, and 2.03) that exhibits dipolar broadening when isotopically substituted with 61Ni, 57Fe, or 13CO (hence referred to as the NiFeC signal).1,8 In addition, Ared-CO displays a strong band in its IR spectrum at 1996 cm-1 (νco) consistent with a terminally bound CO molecule.9 Addition of CH3+ to CO-treated ACS/CODH affords a diamagnetic state, although the order of CO and CH3+ binding is still debatable.10 Titration of ACS/CODH with the bidentate chelator 1,10-phenanthroline (phen) results in the removal of ~ 30 % of the Ni present in the enzyme.11 Such removal of “labile Ni” abolishes the NiFeC signal and also shuts down acetyl CoA synthesis with no effect on CODH activity. Reconstitution of phen-treated enzyme with NiCl2 replenishes both the NiFeC signal and synthase activity.11 These results strongly suggest that “labile Ni” is a requirement for ACS activity.
Crystallographic studies on ACS/CODH from the acetogenic bacterium Moorella thermoacetica (formerly known as Clostridium thermoaceticum) have revealed some interesting and unexpected features never seen before in any metalloenzyme. The overall α2β2 protein comprises several metalloclusters in the different subunits.5,6 The β subunits at the center of the protein contain the B, C, and D clusters, the site of CODH activity (C-cluster). The α-subunits, located at the terminal ends of the protein, contain the A-cluster, the site responsible for acetyl CoA formation. Interestingly, the structure of the A-cluster reported by two separate groups turned out to be quite different even though the enzyme was isolated from the same organism (M. thermoacetica). The first structure of ACS/CODH reported by Drennan and coworkers revealed a homodimeric α2β2 protein whose active site is relatively unexposed to solvent that contain a trio of different metals present at the A-cluster.5 The A-cluster in the Drennan structure consists of an Fe4S4 cubane bridged through one Cys-S residue to a bimetallic site that contains a square-planar Ni(II) (Nid, distal to Fe4S4) ion with N2S2 coordination derived from two backbone carboxamido nitrogens and two Cys-S residues. The Nid site is further bridged through two Cys-S donors to a tetrahedral Cu(I) (Mp, proximal to Fe4S4) center. A fourth nonprotein ligand is also bound to Cu(I) to complete the coordination sphere (Figure 1). The second structure by Fontecilla-Camps and coworkers, however, revealed a very different A-cluster site.6 First, the α2β2 quaternary structure is no longer symmetric and consists of an αcββαo structure in which one of the α-subunits (αo) is more open and exposed to solvent. In the Ao-cluster of αo, a square-planar Ni(II) ion occupies the Mp site that is ~ 20 Å removed from a closed CO tunnel. Quite in contrast, the other α subunit (αc) is closed off to solvent and a tetrahedral Zn(II) ion resides at the Mp site (Figure 1). This Ac-cluster is located at the end of an open hydrophobic CO tunnel from the C-cluster. The discrepancy in the structures of the A-cluster from the same enzyme triggered an intense debate regarding the identity of the true catalytic metal ion at the Mp site. The debate has recently subsided after careful biochemical11a,12,13 and computational studies.14,15 It is now evident that Ni at the Mp site is required for the ACS activity. A very recent structural study on an ACS/CODH from the hydrogenogenic bacterium Carboxydothermus hydrogenoformans has also revealed the presence of Ni at the Mp site.7
Figure 1.
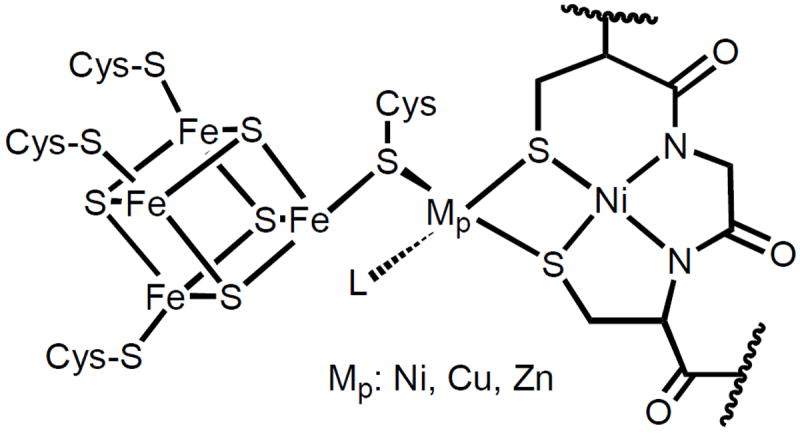
Schematic of the A-cluster site of ACS/CODH from Moorella thermoacetica. Mp: Ni(II), Cu(I), or Zn(II); L is a non-protein ligand.
The extent to which the unusual coordination architecture of the A-cluster dictates its ability to assemble acetyl CoA is still an unanswered question. The presence of carboxamido-N donors at the Nid site is quite uncommon in biology. At this time, coordination of deprotonated carboxamido-N has only been observed in the oxidized P-cluster of nitrogenase16, nitrile hydratase17 (which contains a similar Cys-X-Cys metal binding motif as the Nid site), and most recently in Ni-SOD.18 Since the publication of the first structure of ACS/CODH, several low molecular weight models of the ACS site have been synthesized and studied to determine the intrinsic properties of the metal-containing active site(s).19-25 In these pursuits, researchers have focused mostly on the bimetallic Mp—Nid site of ACS and ignored the Fe4S4 portion (unlikely site of substrate binding). For example, Rauchfuss and coworkers have synthesized the model complex [{Ni(CO)2}{NiS2N’2}]2− (structure a, Figure 2) with two terminal CO ligands bound to the bridged Ni center in the 0 oxidation state.24 This complex was the first example of a structurally characterized, mixed-valence, sulfur-bridged, dinuclear Ni model containing carboxamido-N coordination that resembles the CO-bound form of the enzyme. More recently, Riordan and coworkers have employed the metallopeptide unit [Ni(CGC)]2- to synthesize biologically relevant Ni-Ni models like [Ni(CGC)Ni(dRpe)] (structure b, Figure 2).20a Although phosphine donors are not present in the enzyme, they do allow for more favorable reduction of the Nip site of the model to the Ni(I) state and binding of CO (as shown by electrochemistry). However, none of the models from this study has been characterized further by structural and spectroscopic (such as EPR, FTIR) studies. Clearly, more model complexes are required to establish the properties of the active site(s) of the ACS A-cluster and what implications these have on the overall mechanism of acetyl CoA synthesis.
Figure 2.

Structures of (a) [{Ni(CO)2}{NiS2N’2}]2- and (b) [Ni(CGC)Ni(dRpe)]
As part of our continuing efforts on modeling the bimetallic Mp—Nid portion of the A-cluster of ACS, we describe herein the syntheses and properties of several di/trinuclear metal complexes utilizing the dicarboxamido-dithiolato Ni(II) metallosynthons (Et4N)2[Ni(NpPepS)] (1)26 and (Et4N)2[Ni(PhPepS)] (2) reported by us in preliminary accounts.27 The redox behavior of two Cu(I)-Ni(II) models, namely (Et4N)[Cu(neo)Ni(NpPepS)] (3) and (Et4N)[Cu(neo)Ni(PhPepS)] (4) clearly demonstrate that catalytically active A-cluster cannot utilize Cu(I) at the Mp site. The four trinuclear models (Et4N)2[Ni{Ni(NpPepS)}2] (5),27b (Et4N)2[Ni(DMF)2{Ni(NpPepS)}2] (6),27b (Et4N)2[Ni{Ni(PhPepS)}2] (7), and (Et4N)2[Ni(DMF)2{Ni(PhPepS)}2] (8) and the two dinuclear models [Ni(terpy)Ni(NpPepS)] (9)27a and [Ni(dppe)Ni(PhPepS)] (10)27a provide the opportunity to study, for the first time, a series of complexes with variable donor sets and coordination geometries at the bridged Ni(II) center (Nip mimics). In this paper, we report the results of redox studies and CO binding at the Mp (M = Cu, Ni) centers of these models and compare the spectral data of the reduced CO-adducts with that of the Ared-CO (S = ½) signal of the enzyme in order to establish the mode of CO binding by the A-cluster of ACS/CODH.

Experimental Section
Thiosalicylic acid, 1,2-phenylenediamine, 1,8-diaminonaphthalene, triethylamine, [Ni(dppe)Cl2], CuCl, [Cu(MeCN)4]PF6, 2,9-dimethyl-1,10-phenanthroline (neocuproine, abbreviated as neo), 2,2′:6′,2″-terpyridine (terpy), 1,2-ethanedithiol (edt), and 1,10-phenanthroline (phen) were purchased from Aldrich Chemical Co. and used without further purification. NpPepSH4 (N,N′-naphthalenebis(o-mercaptobenzamide)26, 2,2’-dithiosalicyl chloride26, (Et4N)2[Ni(NpPepS)]26 (1), (Et4N)2[NiCl4]28, [Cu(neo)Cl]29a, (Na)3[Cu3(edt)3]29b, and [Ni(terpy)Cl2]30 were synthesized by following published procedures. All manipulations were carried out under N2 (where needed) using Schlenk lines or dry box techniques. The solvents were purified by standard techniques and distilled prior to use.
Synthesis of Compounds. PhPepSH4 (N,N′-phenylenebis(o-mercaptobenzamide)
The synthesis of the ligand comprises the following steps.
Step 1. (PhPepS-)2
A solution of 1,2-phenylenediamine (0.23 g, 2.13 mmol) and triethylamine (0.67 g, 6.63 mmol) dissolved in 10 mL of CH2Cl2 was quickly added to a solution of 2,2’-dithiosalicyl chloride (0.72 g, 2.10 mmol) in 10 mL CH2Cl2. The resulting pale yellow-brown solution was stirred for 48 h at room temperature. This solution was then washed with aqueous NaHCO3 and NaCl. The CH2Cl2 layer was dried with MgSO4, filtered, and the solvent was removed by rotary evaporation to yield an oily yellow residue. The oil was triturated three times with Et2O (10 mL) to afford a pale yellow solid. Yield: 0.32 g (41 %). 1H NMR (298 K, CDCl3, 500 MHz): δ (ppm from TMS) 8.59 (s, 1H, NH), 7.67 (d, 1H), 7.53 (t, 1H), 7.39 (m, 2H), 7.21 (m, 2H). Selected IR bands (KBr matrix, cm-1): 3217 (m, νNH), 1652 (vs, νco).
Step 2. PhPepSH4
To a degassed THF solution (10 mL) of (PhPepS-)2 (0.32 g, 0.85 mmol) was slowly added solid NaBH4 (0.17 g, 4.49 mmol) in small portions at 4 °C. The bright yellow-orange solution was then allowed to stir at room temperature for 16 h. Next, the THF solution was concentrated to ~10 % of the original volume via short-path vacuum distillation and 6 M acetic acid was added dropwise at 4 °C until the pH was 3. The light cream colored solid that precipitated almost immediately upon addition of acid was collected by filtration, washed with degassed water, and dried on a high-vacuum line for 2 h. Yield: 0.30 g (90 %). 1H NMR (298 K, CDCl3, 500 MHz): δ (ppm from TMS) 8.73 (s, 1H, NH), 7.69 (d, 1H), 7.56 (t, 1H), 7.36 (d, 1H), 7.31 (t, 1H), 7.22 (m, 2H), 4.53 (br s, 1H, SH). Selected IR bands (KBr plates, cm-1): 3300 (m, νNH), 2552 (w, νSH), 1652 (vs, νco).
(Et4N)2[Ni(PhPepS)] (2)
A batch of NaH (0.054 g, 2.24 mmol) was added to a solution of PhPepSH4 (0.17 g, 0.45 mmol) in 10 mL of degassed DMF and the pale yellow-brown solution was allowed to mix for ~ 20 min to ensure that all NaH had reacted. To this solution was then added a batch of (Et4N)2[NiCl4] (0.21 g, 0.44 mmol) dissolved in 2 mL of DMF. The dark red-brown solution thus obtained was stirred for 4 h at room temperature. Next, DMF was removed via short-path vacuum distillation and the residue was dissolved in 25 mL of MeCN and filtered. The resulting red solution was concentrated to ~ 50 % of the original volume and the desired product was isolated as a red microcrystalline solid. Yield: 0.25 g (82 %). Anal calcd. for C36H52N4O2S2Ni (2): C, 62.16; H, 7.53; N, 8.05; found: 62.11; H, 7.61; N, 8.02. Selected IR bands (KBr plates, cm-1): 1521 (vs, νco). Electronic absorption spectrum in MeCN, λmax nm (ε, M-1 cm-1): 545 (sh 610), 384 (8 845), 331 (19 510). 1H NMR (298 K, CD3CN, 500 MHz): δ (ppm from TMS) 8.06 (d, 1H), 7.95 (d, 1H), 7.22 (d, 1H), 6.78 (m, 2H), 6.51 (t, 1H), 3.14 (q, 8H), 1.11 (t, 12H).
(Et4N)[Cu(neo)Ni(NpPepS)] (3)
A batch of 0.04 g (0.14 mmol) of [Cu(neo)Cl] was added to a red-orange solution of 0.10 g (0.14 mmol) of 1 in 10 mL of DMF. The burgundy red homogeneous solution thus obtained was stirred for 12 h at room temperature. Next, DMF was removed in vacuo and 10 mL of MeCN was added to the residue. The pale red solution with a dark precipitate was filtered and the dark red solid was washed with 10 mL of MeCN and dried in vacuo. Yield: 0.087 g (71 %). Anal calcd. for C46H46N5O2S2CuNi (3): C, 62.27; H, 5.23; N, 7.89; found: 62.14; H, 5.31; N, 7.92. Selected IR bands (KBr plates, cm-1): 1516 (vs, νco). Absorption spectrum in MeCN, λmax nm (ε, M-1 cm-1): 495 (12 250), 420 (12 130), 326 (32 000). 1H NMR (298 K, CD3CN, 500 MHz): δ (ppm from TMS) 8.43 (d, 1H), 7.96 (s, 1H), 7.93 (d, 1H), 7.68 (d, 1H), 7.50 (t, 2H), 7.17 (t, 1H), 6.93 (t, 1H), 6.50 (d, 1H), 6.43 (t, 1H), 3.11 (q, 4H), 2.74 (s, 3H), 1.16 (t, 6H).
(Et4N)[Cu(neo)Ni(PhPepS)] (4)
A batch of 0.05 g (0.16 mmol) of [Cu(neo)Cl] was slowly added to a solution of 0.11 g (0.15 mmol) of 2 in 15 mL of MeCN. The deep burgundy red solution was then stirred at room temperature for 2 h. Next, the solvent was removed via short path vacuum distillation and the red oily residue was triturated with 20 mL of degassed MeCN/Et2O (1:1) mixture. The red solid thus obtained was collected by filtration, washed with cold Et2O, and dried on a high-vacuum line for 1 h. Yield: 0.086 g (66 %). Anal calcd. for C42H44N5O2S2CuNi (4): C, 60.25; H, 5.30; N, 8.37; found: 60.34; H, 5.23; N, 8.39. Selected IR bands (KBr plates, cm-1): 1524 (vs, νco). Absorption spectrum in MeCN, λmax nm (ε, M-1 cm-1): 495 (2 940), 363 sh (10 650), 312 sh (19 600). 1H NMR (298 K, CD3CN, 500 MHz): δ (ppm from TMS) 8.40 (d, 1H), 8.11 (s, 1H), 7.94 (d, 1H), 7.84 (d, 1H), 7.67 (t, 1H), 6.75 (t, 1H), 6.62 (t, 1H), 6.50 (d, 1H), 6.31 (t, 1H), 3.34 (s, 3H), 3.12 (q, 4H), 1.17 (t, 6H).
(Et4N)2[Ni{Ni(NpPepS)}2] (5)
A batch of 0.027 g of (Et4N)2[NiCl4] (0.059 mmol) dissolved in 2 mL MeCN was added to a solution containing 0.087 g (0.117 mmol) of 1 in 10 mL of degassed MeCN. The initial red-orange color of the solution turned pale orange and a dark red microcrystalline precipitate appeared within minutes. This heterogeneous solution was stirred for 1 h and then the mixture was filtered. The microcrystalline dark solid was collected, washed with dry Et2O, and dried in vacuo. Yield: 0.065 g (85 %). Anal calcd. for C64H68N6O4S4Ni3 (5): C, 59.61; H, 5.31; N, 6.52; found: 59.70; H, 5.27; N, 6.51. Selected IR bands (KBr plates, cm-1): 1538 (vs, νco).
(Et4N)2[Ni(DMF)2{Ni(NpPepS)}2] (6)
A batch of 0.053 g (0.04 mmol) of 5 was allowed to stir for 2 h at room temperature in 10 mL of degassed DMF. The initial heterogeneous solution slowly became homogeneous and turned bright red in color. The DMF was removed via vacuum distillation and the resulting oily red material was triturated with 10 mL of dry Et2O to obtain the desired product as a fine red solid. Yield: 0.053 g (94 %). Anal calcd. for C70H82N8O6S4Ni3 (6): C, 58.56; H, 5.76; N, 7.80; found: 58.64; H, 5.73; N, 7.79. Selected IR bands (KBr plates, cm-1): 1645 (s, νco DMF), 1532 (vs, νco). Absorption spectrum in DMF, λmax nm (ε, M-1 cm-1): 525 (sh), 437 (12 410).
(Et4N)2[Ni{Ni(PhPepS)}2] (7)
A solution of 0.035 g of (Et4N)2[NiCl4] (0.075 mmol) dissolved in 3 mL of MeCN was added to a red-orange solution of 0.102 g (0.147 mmol) of 2 in 20 mL of degassed MeCN. The color of the reaction mixture rapidly turned to dark red. The deep red solution was stirred for 1 h at room temperature and then its volume was reduced to half by short path vacuum distillation. Storage of this solution at −20 °C for 16 h resulted in the precipitation of a red crystalline solid. This microcrystalline red solid was collected, washed with dry Et2O, and dried in vacuo. Yield: 0.080 g (92 %). Anal calcd. for C56H64N6O4S4Ni3 (7): C, 56.55; H, 5.42; N, 7.07; found: 56.64; H, 5.37; N, 7.10. Selected IR bands (KBr plates, cm-1): 3052 (w), 2983 (w), 1591 (s), 1566 (s), 1538 (vs, νco), 1467 (s), 1442 (s), 1393 (w), 1345 (s), 1295 (w), 1268 (m), 1220 (w), 1182 (w), 1172 (w), 1140 (w), 1118 (w), 1059 (w), 1040 (w), 1002 (w), 956 (w), 791 (w), 755 (m), 691 (w), 601 (w), 515 (w). Absorption spectrum in MeCN, λmax nm (ε, M-1 cm-1): 850 sh (800), 660 (2500), 560 sh (3030), 470 (3850). 1H NMR (298 K, CD3CN, 500 MHz): δ (ppm from TMS) 8.07 (d, 1H), 7.95 (d, 1H), 7.21 (d, 1H), 6.78. (m, 2H), 6.50 (t, 1H), 3.14 (q, 4H), 1.13 (t, 6H).
(Et4N)2[Ni(DMF)2{Ni(PhPepS)}2] (8)
A batch of 0.027 g (0.022 mmol) of 7 was allowed to stir for 2 h at room temperature in 5 mL of degassed DMF. Next, the solvent was removed via vacuum distillation and the resulting oily red material was triturated with 5 mL of dry Et2O to obtain the desired product as a fine red solid. Yield: 0.028 g (97 %). Anal calcd. for C62H78N8O6S4Ni3 (8): C, 55.75; H, 5.89; N, 8.39; found: 55.74; H, 5.83; N, 8.41. Selected IR bands (KBr plates, cm-1): 1646 (vs, νco DMF), 1538 (vs, νco). Absorption spectrum in DMF, λmax nm (ε, M-1 cm-1): 527 (1080), 380 sh (15 300), 335 (38 130).
[Ni(terpy)Ni(NpPepS)] (9)
A slurry of 0.048 g (0.13 mmol) of [Ni(terpy)Cl2] in 1 mL of MeCN was added to a solution containing 0.100 g (0.13 mmol) of 1 in 10 mL MeCN. Within 5 min, the initial red-orange solution turned pale and a red-brown precipitate appeared. The precipitate was filtered, washed with cold MeCN, and dried in vacuo. Yield: 0.076 g (75 %). Anal calcd. for C39H25N5O2S2Ni2 (9): C, 60.27; H, 3.24; N, 9.01; found: 60.21; H, 3.30; N, 8.97. Selected IR bands (KBr plates, cm-1): 3051 (w), 1585 (s), 1568 (s), 1531 (vs, νco), 1474 (m), 1451 (m), 1429 (w), 1385 (s), 1355 (m), 1321 (w), 1265 (w), 1248 (w), 1184 (w), 1163 (w), 1112 (w), 1095 (w), 1054 (w), 1035 (w), 1016 (w), 958 (w), 820 (w), 771 (m), 742 (m), 680 (w), 650 (w), 642 (w), 619 (w), 592 (w). Absorption spectrum in DMF, λmax nm (ε, M-1 cm-1): 450 sh (8500), 427 (10 200), 337 (34 680).
[Ni(dppe)Ni(PhPepS)] (10)
A batch of 0.154 g (0.292 mmol) of [Ni(dppe)Cl2] was slowly added to a solution of 0.204 g (0.293 mmol) of 2 in 10 mL of MeCN. Almost immediately, the red-brown solution turned pale and a blue-green microcrystalline solid appeared in the reaction mixture. The solid was filtered, washed with cold MeCN, and dried in vacuo. Yield: 0.223 g (85 %). Anal calcd. for C46H46N5O2S2CuNi (10): C, 61.92; H, 4.07; N, 3.14; found: 62.03; H, 4.16; N, 3.12. Selected IR bands (KBr plates, cm-1): 1538 (vs, νco). Electronic absorption spectrum in CH2Cl2, λmax nm (ε, M-1 cm-1): 594 (3 040), 506 (sh 1 890), 345 (21 910). 1H NMR (298 K, CDCl3, 500 MHz): δ (ppm from TMS) 8.03 (m, 2H), 7.90 (s, 2H), 7.61 (s, 5H), 7.39 (s, 1H), 7.31 (s, 2H), 6.96 (t, 1H), 6.74 (t, 1H), 6.51 (t, 1H), 6.43 (d, 1H), 1.87 (t, 2H).
Physical Measurements
Electronic absorption spectra were recorded on a Perkin-Elmer Lambda 9 UV/Vis/NIR Spectrophotometer. A Perkin-Elmer Spectrum One or Nicolet Nexus 870 FTIR was used to monitor the infrared spectra. The 1H NMR spectra were recorded at 298 K on a Varian Unity Plus 500 MHz spectrometer. EPR spectra were collected on a Bruker EleXsys E500 spectrometer at X-band frequencies at liquid N2 temperature. A Johnson-Matthey magnetic susceptibility balance was used to determine the room-temperature magnetic susceptibility values of the solid complexes. Electrochemical measurements were performed with Princeton Applied Research instrumentation (model 273A) at 298 K in DMF or CH2Cl2 using either 0.1 M (Et4N)(ClO4) or (nBu4N)(PF6) as the supporting electrolyte. The working electrode was a Beckman Pt-inlay working electrode and the potentials were measured versus a saturated calomel electrode (SCE).
X-ray Data Collection and Structure Solution and Refinement
Red needles of (Et4N)2[Ni(PhPepS)] (2) were grown by slow diffusion of Et2O into a dilute solution of 2 in MeCN at 4 °C. Red plates of (Et4N)[Cu(neo)Ni(NpPepS)]•0.5Et2O•0.5H2O (3•0.5Et2O•0.5H2O) were obtained from slow diffusion of Et2O into a saturated solution of 3 in MeCN/DMF (1/1). Red needles of (Et4N)[Cu(neo)Ni(PhPepS)]•H2O (4•H2O) were obtained upon slow cooling of an MeCN/Et2O (1/1) solution of the complex at −20 °C. Black blocks of (Et4N)2[Ni{Ni(NpPepS)}2]•DMF (5•DMF) were obtained by slow diffusion of Et2O into a saturated solution of the complex in MeCN/DMF (9/1). Red parallelepipeds of (Et4N)2[Ni(DMF)2{Ni(NpPepS)}2]•3DMF (6•3DMF) were grown by diffusion of Et2O into a dilute solution of the complex in DMF. Red blocks of (Et4N)2[Ni(DMF)2{Ni(PhPepS)}2] (8) were grown by slow diffusion of Et2O into a dilute solution of the complex in DMF. Black prisms of [Ni(dppe)Ni(PhPepS)]•CH2Cl2 (10•CH2Cl2) were obtained from slow evaporation of a concentrated solution of 10 in a CH2Cl2/toluene (3/1) mixture at room temperature. Diffraction data for 2, 4•H2O, and 10•CH2Cl2 were collected at 91 K on a Bruker APEX system. Diffraction data for 3•0.5Et2O•0.5H2O, 5•DMF, and 8 were collected at 91 K on a Bruker SMART 1000 system. For these structures Mo Kα (0.71073 Å) radiation was used and the data were corrected for absorption. Diffraction data for 6•3DMF were collected at 130 K on a Siemens P4 system. Cu Kα (1.54178 Å) radiation was used and the data were corrected for absorption. The structures were solved using the standard SHELXS-97 package. Machine parameters, crystal data, and data collection parameters for all the complexes are summarized in Table S1 while selected bond distances and angles are reported in Table S2. Both these tables have been submitted as Supporting Information along with other crystallographic data for all of the complexes.
Results and Discussion
Synthesis
The Nid mimics (Et4N)2[Ni(NpPepS)] (1)26 and (Et4N)2[Ni(PhPepS)] (2)27a served as the metallosynthons in the construction of the higher nuclearity analogues reported in this account. To date, several Ni(II)-dicarboxamido-dithiolato complexes have been reported in the literature.19,21,31-33 The synthetic route that we have followed to obtain the Ni(II) complexes 1 and 2 was originally developed in this laboratory for the synthesis of Fe(III)-carboxamide complexes.34 Reaction of (Et4N)2[NiCl4] with the deprotonated (with the aid of NaH) ligands NpPepS4- or PhPepS4- in solvents like DMF readily afford complexes 1 and 2 in high yields. These Ni(II) complexes with N2S2 chromophores are inherently stable in the solid state.
Failures in the initial attempts to synthesize Ni-Cu dinuclear model complexes in the present work provide some synthetic tips for isolation of such systems. Addition of simple Cu(I) salts like CuCl or [Cu(MeCN)4]PF6 to DMF solutions of the Nid mimic 1 invariably results in the formation of the trinuclear species (Et4N)[Cu{Ni(NpPepS)}2] regardless of the choice of solvent, Cu(I) salt, or reagent stoichiometry.27b This observation suggests that in order to synthesize discrete dinuclear species, one must employ Cu(I) starting complexes with less labile ligands. Use of the trinuclear Cu(I) complex, [Cu3(edt)3]3- (edt = 1,2-ethanedithiol)29b, however, leads to no reaction with 1 or 2 in solvents like DMF, MeCN, or MeOH at room temperature. When reaction mixtures with 1 is heated to 65 °C in DMF for 24 h, one obtains the trinuclear complex (Et4N)[Cu{Ni(NpPepS)}2] instead of the desired S-bridged Ni-Cu complex [Cu(edt)Ni(NpPepS)]3-. In contrast, when one employs [Cu(neo)(Cl)]29a (neo = neocuproine = 2,9-dimethyl-1,10-phenanthroline) as the starting Cu(I) complex, the reaction indeed takes the desired course. For instance, reaction of [Cu(neo)Cl] with 1 or 2 in MeCN at room temperature cleanly affords the Ni-Cu dinuclear complexes (Et4N)[Cu(neo)Ni(NpPepS)] (3) and (Et4N)[Cu(neo)Ni(PhPepS)] (4), respectively in high yields. Clearly, the strong preference of the neocuproine ligand for Cu(I) centers as well as the tetrahedral geometric preference of Cu(I) allows for the formation of 3 and 4. Complexes 3 and 4 were also obtained when [Cu(neo)(SR)] (R = C6H5, p-C6H4-Cl)29c are used as the starting Cu(I) complexes. In the solid state, 3 and 4 are stable for months and no oxidation of the Cu(I) center is observed (as verified by 1H NMR and electronic absorption spectroscopy).
Similar reactivity has also been noted in our attempts to synthesize dinuclear Ni(II) species. Reactions of Ni(II) salts like (Et4N)2[NiCl4] with 1 or 2 in MeCN afford the corresponding trinuclear complexes (Et4N)2[Ni{Ni(NpPepS)}2] (5) and (Et4N)2[Ni{Ni(PhPepS)}2] (7) in high yield. These trinuclear complexes form regardless of the Ni(II) salt/metallosynthon stoichiometry. Isolation of these complexes is aided by the high insolubility of 5 and 7 in MeCN; the trinuclear complexes precipitate out of solution within minutes. Interestingly, dissolution of 5 and 7 in DMF (a slow process) results in coordination of two molecules of DMF to the central Ni(II) ion (designated as NiC) affording the DMF adducts (Et4N)2[Ni(DMF)2{Ni(NpPepS)}2] (6) and (Et4N)2[Ni(DMF)2{Ni(PhPepS)}2] (8), respectively. This binding is also reversible. When the DMF adducts 6 and 8 are treated with solvents like MeCN or THF, the respective trinuclear species 5 and 7 form almost immediately (verified by IR and 1H NMR).
The dinuclear Ni-Ni model complexes have been synthesized by following the strategy used to isolate the Ni-Cu complexes 3 and 4. Reaction of one equiv of [Ni(terpy)Cl2] (terpy = 2,2′:6′,2″-terpyridine) with 1 in MeCN affords [Ni(terpy)Ni(NpPepS)] (9), a Ni-Ni dimer in which the modeled Nip site is five-coordinate. The isolation of 9 is aided by its lack of solubility in a variety of polar solvents like MeOH and MeCN due to the overall neutral charge of the complex. Since the as isolated Nip site of ACS is four-coordinate in the crystal structure, we employed a second Nip synthon, [Ni(dppe)Cl2] (dppe = 1,2-bis(diphenylphosphino)ethane), in such reaction. This Nip synthon is expected to favor an easier reduction to the Ni(I) oxidation state and further facilitate binding of CO. However, all attempts to synthesize a Ni-Ni dimer with 1 and [Ni(dppe)Cl2] led to the formation of the trinuclear species 5. In contrast, reaction of 2 with 1 equiv of [Ni(dppe)Cl2] in MeCN affords [Ni(dppe)Ni(PhPepS)] (10) as a blue-green microcrystalline solid in 85 % yield. Steric interactions between the folded structure of the {Ni(NpPepS)} moiety26 and the dppe ligand frame presumably prevent the formation of the dinuclear complex in the first case while the more rigid ligand frame of 2 allows formation of the desired dimer 10 without such hindrance. It is therefore evident that proper design of the ligand frame of the Nid metallosynthon is crucial for the successful isolation of Nip-Nid ACS models.
Structure and Properties. (Et4N)2[Ni(PhPepS)] (2)
The structure of [Ni(PhPepS)]2- (anion of 2) is shown in Figure 3. The coordination geometry around nickel is square-planar arising from two carboxamido nitrogens and two thiolato sulfurs in cis geometry. Although the overall disposition of the PhPepS4- ligand frame is similar to that of NpPepS4- in (Et4N)2[Ni(NpPepS)] (1),26 there are some noted differences. For example, the extent of the ‘butterfly’ folding of the aryl sulfur rings observed in 1 is not as extreme in the case of 2. This is reflected in the more rigid fivemember chelate ring of the phenylenediamido portion of 2 (nearly coplanar with the NiN2S2 plane) compared to the six-member chelate ring of the naphthalenediamido portion of 1 (almost perpendicular to the NiN2S2 plane). Interestingly, [Ni(tsalphen)], an analogous Ni(II) complex derived from a ligand frame similar to 2 but with imine nitrogen coordination, is completely planar.35 This suggests that the bending of the aryl rings are most likely due to the presence of the carboxamido nitrogens in 2. The angles at the Ni(II) center are slightly distorted from the ideal 90° value due to the short bite of the phenylenediamido portion of the ligand frame (N1–Ni–N2, 85.52(11)°). The average Ni–Namido bond length (1.907(3) Å) compares well with other Ni(II)-carboxamido complexes.21,26,31-33,36 The average Ni–S bond distance (2.158(10) Å) is also within the range of Ni-S distances found in analogous Ni(II)-dicarboxamido-dithiolato species.21,26,31-33
Figure 3.

ORTEP diagram of the anion of [Ni(PhPepS)]2− (anion of 2) (50 % probability) with the atom-labeling scheme. H atoms are omitted for the sake of clarity.
Coordination of deprotonated carboxamido nitrogen to metal centers is readily indicated by the red-shift of the carbonyl stretching frequency (νco) of the complex with respect to the νco of the free ligand.34 Complex 2 exhibits its νco at 1521 cm-1 compared to 1652 cm-1 for the free ligand PhPepSH4. In MeCN, 2 exhibits a dark-red color arising from strong ligand-to-metal charge transfer absorption at 384 nm and a d-d band at 545 nm typical for Ni(II) complexes with dicarboxamido-dithiolato (N2S2) coordination.21,26,31-33 The clean 1H NMR spectrum of 2 in CD3CN confirms that the square-planar geometry of 2 is retained in solution. Ligation of deprotonated carboxamido nitrogen to nickel in general provides exceptional stability to the 2+ and 3+ oxidation states.21,31-33 In DMF, 2 displays an irreversible metal centered oxidation wave at 0.22 V (vs SCE). This value is more positive than the oxidation potential of other Ni(II)N2S2 complexes possibly due to the weaker donor strength of the aryl thiolate donors in 2 compared to the alkyl thiolates present in analogous complexes. Not surprisingly, 2 exhibits no reduction wave down to a potential of −1.8 V (vs SCE, DMF) due to the presence of strong σ-donors (carboxamido-N and thiolato-S) around the Ni(II) center.
(Et4N)[Cu(neo)Ni(NpPepS)] (3)
The structure of 3 consists of a square-planar Ni(II) ion bridged to a distorted tetrahedral Cu(I) center via the thiolato-S donors of the NpPepS4- ligand frame (Figure 4). Close scrutiny of the Ni–Namido and Ni–S bond distances in 3 (av: 1.899(4) and 2.1950(15) Å, respectively) reveals little changes from those noted for the Ni(II) monomer 1. The Cu---Ni distance of 3 (3.074 Å) is somewhat longer than that of the Cu form of ACS/CODH (2.792 Å).5 Steric constraints imposed by the NpPepS4- ligand frame are presumably responsible for the longer metal-metal distance compared with the enzyme value. The average Cu–N distance in 3 (2.047(4) Å) is typical of Cu(I)-neocuproine type complexes.37 However, the two Cu–S distances are quite different with Cu–S1: 2.2670(16) and Cu–S2: 2.3198(16) Å. The distorted arrangement of the donor atoms around the Cu(I) center is further supported in the large deviation of the bond angles (N3–Cu–N4, 82.66(18)° and N3–Cu–S1, 129.79(14)°) from the ideal tetrahedral value of 109.5°. Similar distortion has also been observed in other sulfur-bridged Ni(II)-Cu(I) complexes.25,27b
Figure 4.
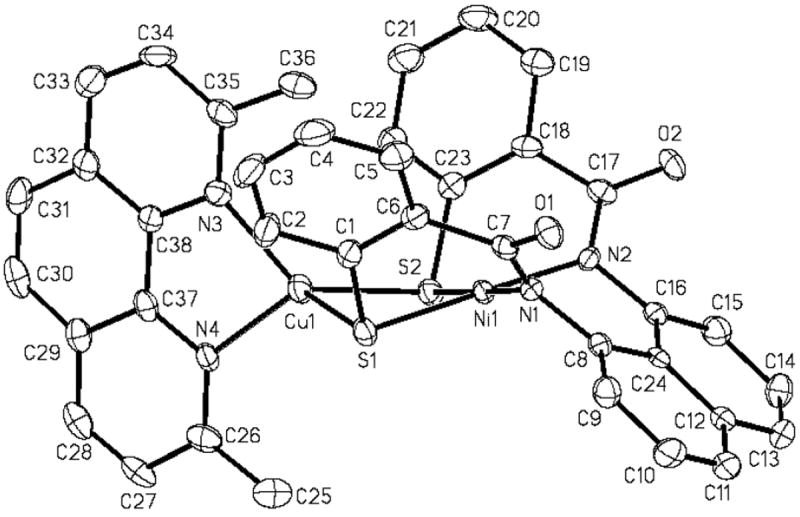
ORTEP diagram of [Cu(neo)Ni(NpPepS)]− (anion of 3) (50 % probability) with the atom-labeling scheme. H atoms are omitted for the sake of clarity.
Metallation of the sulfur ligands of the {Ni(NpPepS)} moiety in 3 is readily indicated by the blue shift of the νco value of the complex (1516 cm-1 compared to 1510 cm-1 in monomeric 1). Enhanced charge donation by the carboxamido-N to compensate for the decreased donor strength of sulfur (following metallation by Cu) is presumably responsible for this blue-shift. Complex 3 is diamagnetic and exhibits a clean 1H NMR spectrum. Although metallation of thiolate-S donors can alter the charge density at the Ni(II) center making the Ni(I) oxidation state more accessible,23 3 does not exhibit any observable redox wave down to –1.8 V (vs SCE, DMF). Clearly, the Ni(I) state is not supported in this complex even with sulfur-metallation. This lends further support toward a non-redox role for the Nid site in the enzyme.
(Et4N)[Cu(neo)Ni(PhPepS)] (4)
The structure of the anion of the dinuclear Ni(II)-Cu(I) complex 4 is shown in Figure 5. Much like 3, this complex also consists of a square-planar Ni(II) ion sulfur-bridged to a distorted tetrahedral Cu(I) center. The metric parameters of the NiN2S2 portion of 4 (av: Ni–Namido, 1.9037(11) Å) are similar to that of monomer 2 except for slight elongation of the Ni–S bond distance (av: 2.1872(4) Å). Also, the bending of the aryl thiolate rings away from the NiN2S2 coordination plane is more noticeable in 4 compared to 2 as a result of the steric clash between the methyl groups of the neocuproine unit with the PhPepS4- ligand frame. This is reflected in the change of the C1–S1–Ni angle which is more compressed in 4 (99.06(5)°) than in 2 (106.53(11)°) and the highly distorted tetrahedral geometry about the Cu(I) center. For example, there is a significant difference in the two Cu–N bond lengths (Cu–N3: 2.0194(14) Å; Cu–N4: 2.0894(14) Å), the two Cu–S distances (Cu–S1: 2.2496(4) Å; Cu–S2: 2.3508(4) Å), and the bond angles range from 82.08(5)° (N3–Cu–N4) to 140.13(4)° (N3–Cu–S1). As a result the Cu---Ni bond distance of 4 (3.1435(2) Å) is longer than that observed in Cu-ACS. Like 3, complex 4 exhibits a clean diamagnetic 1H NMR spectrum and exhibits no Ni(II)/Ni(I) reduction wave in its cyclic voltammogram.
Figure 5.
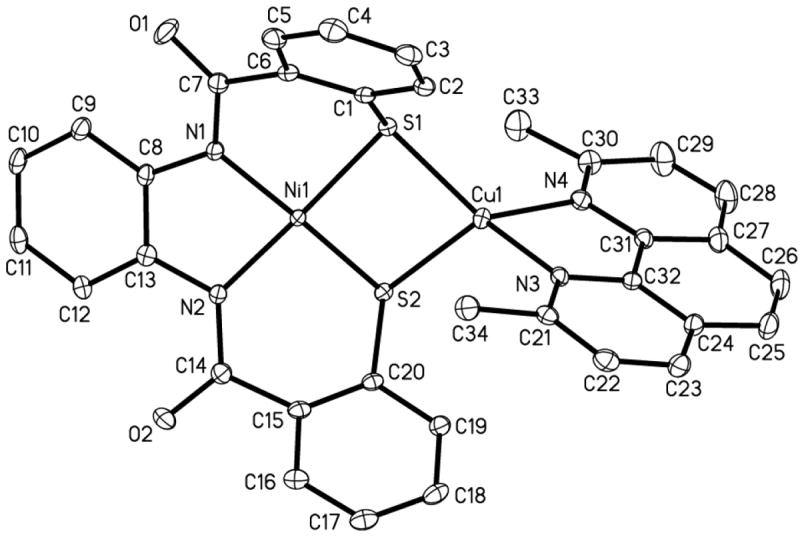
ORTEP diagram of [Cu(neo)Ni(PhPepS)]− (anion of 4) (50 % probability) with the atom-labeling scheme. H atoms are omitted for the sake of clarity.
(Et4N)2[Ni{Ni(NpPepS)}2] (5)
An ORTEP drawing of the anion of the trinuclear Ni(II) complex (Et4N)2[Ni{Ni(NpPepS)}2] (5) is shown in Figure 6. The two terminal Ni(II) centers (designated as NiT; Ni1 and Ni3 in Figure 5) and the central Ni(II) center (designated as NiC; Ni2 in Figure 5) of 5 all lie in one extended plane in a slant chair configuration resulting from the orientation of the naphthalene rings. The two aryl sulfur rings of the NpPepS4- ligand frame fold in an up-down sequence and appear as railings for the N2NiTS2–NiC–S2NiTN2 platform. The NiT–Namido and NiT–S bond distances in 5 (av: 1.880(3) and 2.1769 Å, respectively) are similar to those observed in 1 while the average NiC–S bond length (2.2368(10) Å) is slightly longer than those distances observed in similar Ni(II) trinuclear species.21,38 Unique folding of the NpPepS4- ligand around the NiT centers in 5 is presumably responsible for this lengthening of the NiC–S bonds. Further comparison of the structural parameters of 5 with those of 1 reveal several differences in the coordination structure and ligand folding about the Ni(II) centers. For example, in 5, the S1–Ni1–S2 bite angle is more compressed (86.85(4)°) compared to 1 (91.91(10)°) as a result of NiC coordination. Most notably, the ‘butterfly’ folding of the aryl sulfur rings in 5 allow the two adjacent rings on each NpPepS4- ligand frame to become much closer to each other compared to 1. Finally, the C1–S1–Ni1 bond angle is considerably more closed in 5 (87.72(13)°) than in 1 (101.2(2)°).
Figure 6.
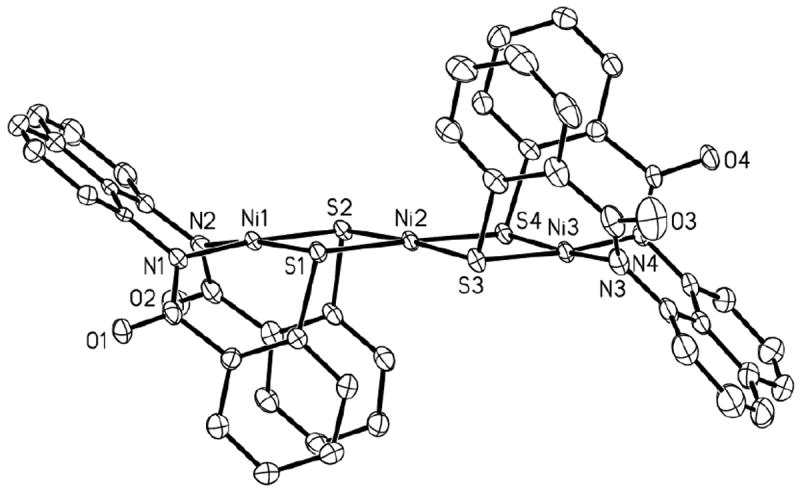
ORTEP diagram of [Ni{Ni(NpPepS)}2]2− (anion of 5) (50 % probability) with the atom-labeling scheme. H atoms are omitted for the sake of clarity.
The extremely low solubility of 5 in CD3OD, CD3CN and CD2Cl2 prevented the recording of its 1H NMR spectrum although magnetic moment measurement (polycrystalline sample) confirmed its S = 0 spin state. As discussed below, dissolution of 5 in deuterated DMF or DMSO affords paramagnetic species due to solvent binding to NiC and such solutions exhibit broad NMR signals. Metallation of the sulfurs in 5 was also confirmed by the blue-shift of its νco band (1538 cm-1) compared to 1 (1510 cm-1).
(Et4N)2[Ni(DMF)2{Ni(NpPepS)}2] (6)
The structure of the anion of the DMF adduct 6 is shown in Figure 7. The binding of two molecules of DMF to NiC in 5 results in significant structural rearrangement. Major rearrangement occurs to accommodate the two additional DMF ligands coordinated to the NiC center in cis fashion by rotation of one of the NiTN2S2 units by ~ 90° to afford a distorted octahedral coordination unit around NiC. Although the average Ni–Namido and Ni–S bond lengths of the NiT sites in 6 (1.893(3) and 2.1758(12) Å, respectively) remain unchanged (compared to 1 and 5), the NiC–S bond distances (av: 2.4447(12) Å) increase significantly compared with 5 (av: 2.2368(10) Å) upon expansion of the coordination sphere. Additionally, this results in a longer Ni---Ni separation in 6 (3.374 Å) compared to 5 (3.2269(7) Å). The bending of the aryl sulfur rings of the NiTN2S2 units in 6 is also less pronounced (C1–S1–Ni1, 97.67(14)° compared to 87.72(13)° in 5). A small trans influence of the coordinated DMF molecules in 6 is reflected in the two longer NiC–S bond distances (Ni2–S1, 2.4877(13) Å; Ni2–S3, 2.4495(12) Å). The increase in NiC–S could also arise from the increased metal-ligand electron repulsion in going from low-spin planar 5 (S = 0) to high-spin octahedral 6 (S = 1).
Figure 7.
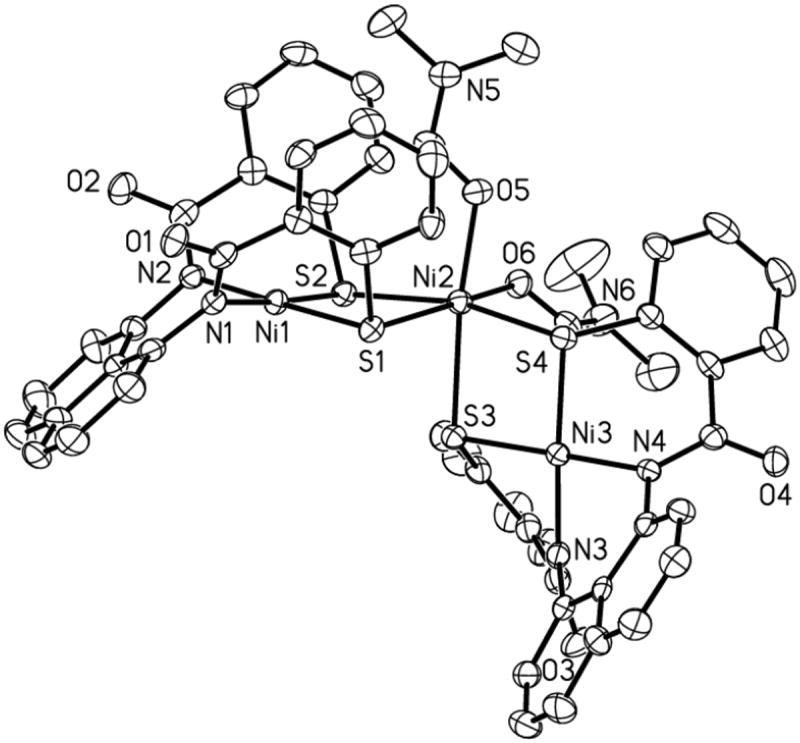
ORTEP diagram of [Ni(DMF)2{Ni(NpPepS)}2]2- (anion of 6) (50 % probability) with the atom-labeling scheme. H atoms are omitted for the sake of clarity.
Complex 6 exhibits an additional νco stretch at 1645 cm-1 due to the O-coordinated DMF molecules at the NiC center. The S = 1 spin state of 6 has been confirmed by measurement of the magnetic moment of the complex in the polycrystalline state (μeff = 2.87 μB at 298 K).
(Et4N)2[Ni{Ni(PhPepS)}2] (7)
At present we have not been able to grow good quality crystals of this complex for diffraction studies. However, microanalytical and spectroscopic data confirm an extended planar structure similar to that of complex 5. This diamagnetic complex is more soluble in a variety of solvents than its analogue 5 and its clean 1H NMR spectrum in CD3CN is consistent with square-planar coordination around the three Ni(II) centers. The electronic absorption spectrum of 7 consists of several peaks in the 400-500 nm range arising from multiple ligand-to-metal charge transfer (LMCT) bands arising from the NiS4 and NiN2S2 chromophores. As expected, the νco band of 7 is shifted to higher energy (1538 cm-1) compared to the monomeric complex 2 (1521 cm-1).
(Et4N)2[Ni(DMF)2{Ni(PhPepS)}2] (8)
The structure of the anion of the trinuclear complex 8 is shown in Figure 8. Much like the analogous trimer 6, complex 8 also comprises two NiTN2S2 units coordinated to an octahedral NiC center (Ni2 in Figure 8). However, the NiTN2S2 moieties prefer to stay coplanar due to the decreased steric demands of the PhPepS4- ligand frame in 8. This results in coordination of the two DMF molecules in a trans position at the NiC center. The average NiC–S and NiC–O bond distances of 8 (2.4475(16) and 2.063(5) Å, respectively) are very similar to those observed in 6 and other related complexes. When compared with 2, the overall folding and metric parameters of the PhPepS4- ligand frame in 8 appear relatively unchanged with only a slight increase in the NiT–S bond length (av: 2.1723(18) Å) presumably due to metallation of sulfur by NiC. Collectively, the structures of 6 and 8 demonstrate that minor changes in the ligand design can alter the binding mode (e.g. cis vs trans coordination) of ligands and reactivity of metal centers in these model complexes. Complex 8 displays a strong νco band at 1646 cm-1 due to the O-coordinated DMF ligands and its magnetic moment value (μeff = 2.89 μB at 298 K, polycrystalline sample) is consistent with an S = 1 ground state.
Figure 8.
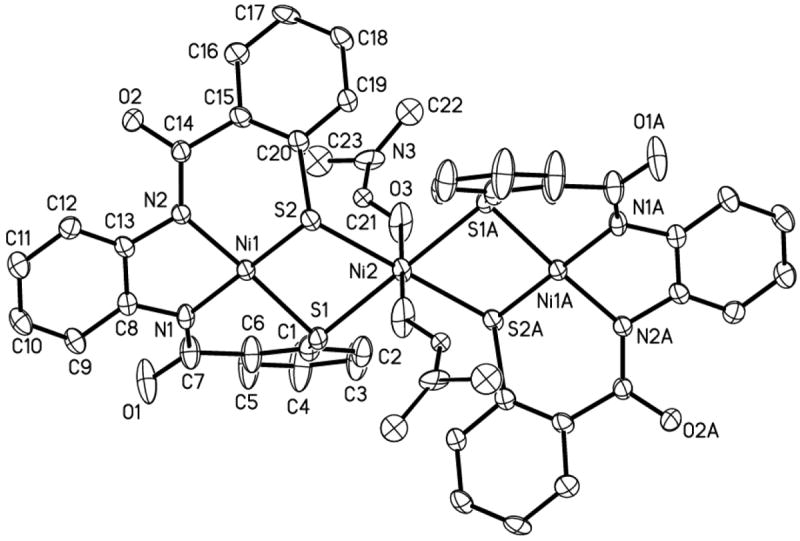
ORTEP diagram of [Ni(DMF)2{Ni(PhPepS)}2]2− (anion of 8) (50 % probability) with the atom-labeling scheme. H atoms are omitted for the sake of clarity.
[Ni(terpy)Ni(NpPepS)] (9)
Although we have not completed structural studies on this model complex due to lack of suitable crystals, it has been thoroughly characterized by various spectroscopic techniques and its composition has been confirmed by microanalytical data. The blue-shift in the νco band of complex 9 (1531 cm-1) strongly supports the proposed dinuclear structure and the ESI-MS data support the presence of a [Ni(terpy)]2+ moiety bound to a [Ni(NpPepS)]2- unit. Additional support for a five-coordinate Ni(II) center in 9 comes from the magnetic susceptibility studies (μeff = 3.02 μB at 298 K, polycrystalline sample). Since 9 exhibits an irreversible reduction at −1.13 V (vs SCE) in DMF, it is evident that the Ni(I) state is accessible in this model complex. Although the potential is still outside the biological range, this property of 9 allows one the opportunity to study the possibility of CO binding in the reduced state (Ared-CO mimic). The results of such studies are discussed in detail in a forthcoming section.
[Ni(dppe)Ni(PhPepS)] (10)
The structure of the neutral dinuclear complex 10 is shown in Figure 9. In this Ni-Ni model, the two Ni(II) centers are connected via the two thiolato-S donors of the PhPepS4- ligand frame and both Ni centers exist in square-planar geometry. The spatial arrangement of the donor atoms of the NiN2S2 portion of this complex does not change significantly from that in the monomeric complex 2 and hence similar Ni–Namido and Ni–S bond distances (av: 1.890(17) and 2.150(5) Å, respectively) are noted for 10. The dihedral angle between the two Ni(II) square-planes in 10 is 111.4° and the Ni---Ni separation is 2.8255(4) Å (Ni---Ni in ACS/CODH = 3.00 Å).6 Coordination of the Nip mimic to the two S-donors of the PhPepS4- results in a decrease bend of the aryl thiolate rings due to the shorter bite angle of the cis-dithiolato portion of the ligand frame (S1–Ni1–S2, 79.29(2)°). The bond angles about the bridged Nip center (Ni2 in Figure 9) in 10 also deviate from the ideal value (P1–Ni2–P2, 86.18(2)°; P1–Ni2– S1, 99.13(2)°) due to steric constraints imposed by the four phenyl groups of the diphosphine ligand. The average Ni–S and Ni–P bond distances of the Nip site in 10 (2.2384(6) and 2.1799(6) Å, respectively) are similar to those found in other dinuclear Ni complexes with P2S2 coordination spheres.19,22
Figure 9.

ORTEP diagram of [Ni(dppe)Ni(PhPepS)] (10) (50 % probability) with the atom-labeling scheme (left). H atoms are omitted for the sake of clarity. A different view is shown on left showing the dihedral angle between the NiN2S2 and NiS2P2 plane.
The dinuclear structure of 10 is readily identified by the blue-shift of the νco to 1538 cm-1 from 1521 cm-1 in case of 2. The two square-planar Ni(II) centers of 10 give rise to a diamagnetic species that exhibits a clean 1H NMR spectrum in CDCl3.
Reactivity with 1,10-phenanthroline
In some of the earliest studies on ACS/CODH (before any structural analysis), Lindahl and coworkers showed that a portion of the Ni at the A-cluster site is labile and easily removed with the bidentate chelator 1,10-phenanthroline (phen).11b This removal of “labile Ni” by phen completely inhibits the ACS activity but leaves the CODH activity intact. The phen-treated enzyme also displays no NiFeC EPR signal. We were therefore curious to determine whether the sulfur-bridged Ni in the model complexes such as 6, 8 and 10 can be removed with phen. Previous studies on a trinuclear model complex reported by Darensbourg and coworkers, namely [{(BME-DACO)Ni}2Ni]Br2, have shown that the Ni(II) center coordinated by four metallosulfur moieties from the NiN2S2 units in the complex is indeed removed by phen.23 However, the absence of key carboxamido groups in the NiN2S2 fragments in these trinuclear species, as well as the lack of phen reactivity with a trinuclear complex similar to 6 (and 8) with carboxamido coordination synthesized by Hegg and coworkers21 prompted us to study the phen reactivity with our complexes.
Titration of solutions of 6 or 8 in DMF with phen results in immediate removal of the central Ni (NiC) ions in both these trinuclear species. The reaction can be followed by monitoring the electronic absorption spectrum. As shown in Figure 10, the spectrum completely stops changing only after addition of 3 mol equiv of phen to 6 suggesting formation of [Ni(phen)3]2+ and 2 mol equiv of the monomer 1.27b Similar results were obtained with complex 8. Since no further change is observed upon addition of a large excess (~ 100 mol equiv) of phen to these solutions, it is evident that the Ni(II) centers in the Nid mimics 1 and 2 are strongly coordinated to their dicarboxamido-dithiolato ligand frames and are resistant to removal by phen.
Figure 10.
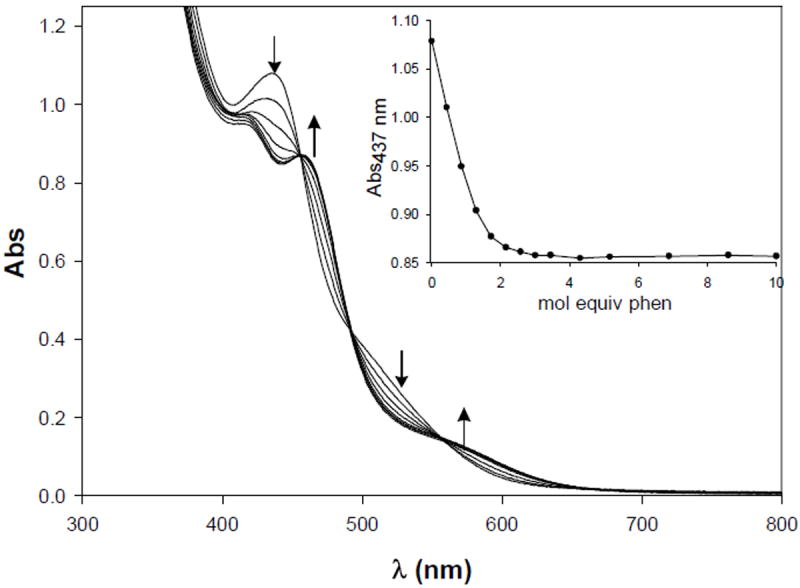
Changes in the electronic absorption spectrum of (Et4N)2[Ni(DMF)2{Ni(NpPepS)}2] (6, 0.087 mM) upon addition of 0.5 mol equiv aliquots of phen in DMF. Inset: change in Absorbance at 437 nm vs. mol equiv of phen.
The reactivity of phen with the dinuclear model 10 is slightly different from its trinuclear counterparts 6 and 8.27a Addition of phen to a solution of 10 in CH2Cl2 does remove the sulfur-bridged Nip model site in this complex resulting in the formation of 2, [Ni(phen)3]2+, and free dppe in solution. However, the removal of Nip in 10 requires excess phen (~ 25 mol equiv) and takes nearly 35 min to reach the end point (beyond which no further change occurs in the electronic absorption spectrum, Figure 11). The observed pseudo first order rate constant (kobs) for the phen removal of Nip in dimer 10 is 1.50 × 10-4 s-1. The slower phen reactivity of 10 compared to 6 and 8 presumably arises from the more stable square-planar low-spin nature of the Nip site in 10 as opposed to the high-spin octahedral NiC centers in 6 and 8. Interestingly, treatment of the Ni-Cu dimers 3 and 4 with phen does not result in any reaction. However, addition of the Cu(I) specific chelator neocuproine to 3 and 4 affords the respective monomers 1 and 2 and [Cu(neo)2]+. Neocuproine exhibits no reaction with the Ni centers of 6, 8, or 10. Collectively, the phen reactivity of the Ni complexes 6, 8, and 10 lends further support to the notion that the “labile Ni” in the enzyme originates from the Mp site and not the Nid site.11,12 The results also provide indirect evidence in favor of Ni at the Mp site of the Acluster of catalytically active ACS/CODH.
Figure 11.
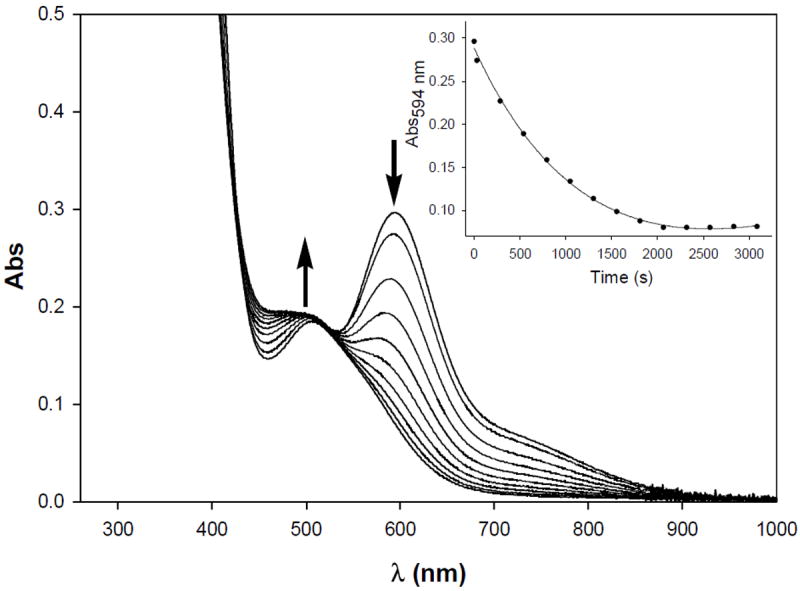
Changes in the electronic absorption spectrum of [Ni(dppe)Ni(PhPepS)] (10, 0.0974 mM) upon mixing with excess 1,10-phenanthroline (25 mol equiv) in CH2Cl2 at 298 K. Inset: change in Absorbance at 594 nm vs. Time (s).
Reactivity of Ni-Cu Models
The Cu(I) sites in all the synthetic models reported so far exhibit no affinity toward CO. For example, the dinuclear models synthesized by Riordan and coworkers undergo rupture of the (μ-SR)2 bridge and breakdown of the dinuclear structure upon reaction with CO.25 The Ni-Cu dinuclear complex, [{P(iPr)3Cu}{NiS2N’2}]-, synthesized by Rauchfuss and coworkers with a coordinatively unsaturated Cu(I) center, does not exhibit any CO binding.24 Although some of these models utilize Ni(II)-diamino-dithiolato complexes as the Nid synthon and hence lack crucial Ni(II)-carboxamido-N interaction(s),25 the consistent inertness of the Cu(I) centers in these model complexes indicates a non-catalytic role for Cu in ACS/CODH.
The Ni-Cu models of the present work namely, 3 and 4, consist of Nid synthons that include carboxamido ligation to the Ni(II) center. Both these models exhibit no CO binding to either metal center nor is any breakdown of the Cu-(μ-SR)2-Ni core observed upon passage of CO through the solutions of these complexes. No breakdown of the Cu- (μ-SR)2-Ni core is observed upon treatment with excess phen either. Clearly, the dinuclear structures are very stable and are in accord with results of previous experiments showing Cu(I) ion as a stronger binder to metallosulfur donors compared to Ni(II) and Zn(II) ions.23 Although the lack of reactivity of the Cu(I) centers of 3 and 4 toward CO could simply arise from coordinative saturation (both tetrahedrally coordinated), it is important to note that exposure to CO does not cause any break in the Cu-S bridge to provide a binding site for CO. This indicates that Cu(I) binds the thiolato sulfurs of the Nid site quite strongly and hence the presence of Cu at the Mp site in Drennan’s structure could be a consequence of binding of adventitious metal ions present in the crystallization buffers.11 Indeed, in a recent paper, Darensbourg and coworkers have examined the metal ion affinity of an analogous NiN2S2 thiolato complex, ([(BMEDACO) Ni]), and the results indicate that the affinity decreases in the order Cu(I)>Ni(II)>Zn(II).23
Reactivity of Ni-Ni Models
Since one of the steps of the proposed ACS mechanism invokes the formation of a Ni(I)-CO species, responsible for the NiFeC EPR signal, we have studied the redox properties and CO-affinities of the bridged Ni(II) centers in 6 and 8-10. The redox behaviors of the models of the Nid sites reported so far suggest that the Nid site of the A-cluster is unlikely to participate in any redox process during catalysis and in turn imply that the Nip site is a candidate for reduction and CO binding during the ACS catalyzed reaction.19-25 The NiC centers of the trinuclear species 6 and 8 are ligated to four metallosulfur ligands and hence resemble the Nip site to a great extent. Both these models can be chemically reduced with reductants like Na2S2O4 and NaBH4 at low temperature (−40 °C) to afford EPR-active species. The X-band EPR spectrum of the reduced species 6red in DMF glass (100 K) consists of an axial EPR signal (g = 2.33, 2.09) while that of 8red is more rhombic (g = 2.25, 2.12, 2.07). Since 1 and 2 cannot be reduced by Na2S2O4 or NaBH4 (no EPR signal), it is evident that the Ni(II)C centers in the trinuclear complexes undergo reduction. The similarity of the EPR spectra of 6red and 8red with those of other [Ni(I)L4] species leads us to propose that the coordination geometry about the Ni(I)C center in these reduced complexes is distorted tetrahedral.39
Passage of CO into DMF solutions of 6red and 8red results in the formation of their CO-adducts that exhibit intense νco bands in their solution IR spectra at 1960 and 1971 cm-1, respectively. These νco values are consistent with terminal coordination of CO to the Ni(I)C centers of 6red-CO and 8red-CO much like that proposed in ACS. The reduced Ni(I)S4 complex [Ni(tpttd)]− (tpttd = 2,2,11,11-tetraphenyl-1,5,8,12-tetrathiadodecane) with two alkyl thiolato-S donors also binds CO in a similar manner and exhibits its νco value at 1940 cm-1.40 The νco values for 6red-CO and 8red-CO are lower than that recorded for the enzyme (1996 cm-1). This red shift presumably arises from the enhanced electron density at the Ni(I) centers ligated by four metallosulfur donors (instead of the three observed in the enzyme) which in turn promotes more π back donation to the π* orbital of CO. The reduced species and their CO-adducts are moderately stable at low temperatures. All efforts to isolate solids from the reaction mixtures have so far met with little success.
The Ni-Ni model complexes 9 and 10 provided the opportunity to study the reactions of CO with Ni(I) centers at modeled Mp sites (with different coordination geometries) as more accurate mimics of the bimetallic A-cluster subsite.27a When complex 9 with N3S2 donor set at the Nip site is reduced with Na2S2O4 in DMF, the product exhibits an axial EPR spectrum with g = 2.23 and 2.12 (Figure 12, top panel). This spectrum is nearly identical to the EPR spectra of [NiI(terpy)(SR)2]− species with distorted trigonal bipyramidal geometry.41 It is therefore clear that 9red retains its dinuclear structure and that the Nip center in 9 is reduced to the Ni(I) state. Passage of CO through the solution of 9red in DMF affords 9red-CO that exhibits a strong νco band in its solution IR spectrum at 2044 cm-1.
Figure 12.
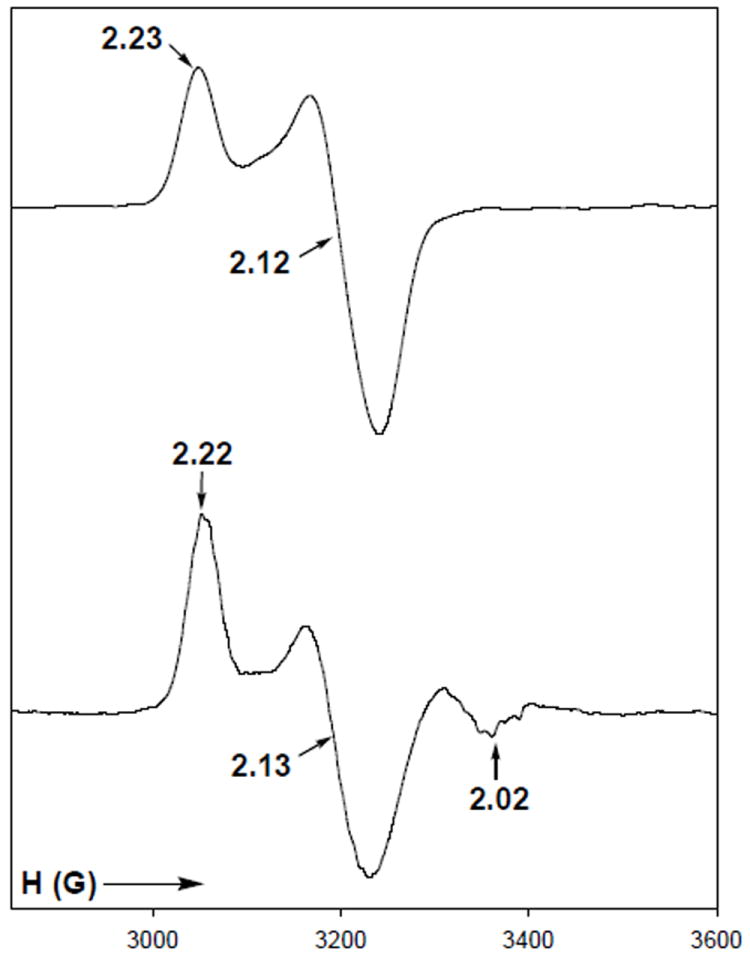
X-band EPR spectra of [NiI(terpy)NiII(NpPepS)]− (9red, Top) and [NiI(terpy)(CO)NiII(NpPepS)]− (9red-CO, Bottom) in DMF glass at 100 K. Selected g values are indicated. Spectrometer settings: microwave frequency, 9.50 GHz; microwave power, 20 mW; modulation frequency, 100 kHz; modulation amplitude, 5 G.
Since both 1 and 9 do not show any affinity toward CO, one can easily conclude that the Ni(I) center binds CO in 9red-CO. The EPR spectrum of 9red-CO is rhombic compared to 9red with g = 2.22, 2.13, and 2.02 (Figure 12, bottom panel) and resembles the EPR spectra of [NiI(terpy)(SR)2(CO)]– species reported by this group.41 For example, the Ni(I) complex, [NiI(terpy)(C6F5S)2(CO)]− exhibits a rhombic EPR signal in DMF glass with g = 2.20, 2.15, 2.02 and displays νco band at 2045 cm-1.41c These data clearly indicate that the Ni(I) center in 9red-CO exists in a distorted octahedral geometry and the CO is bound in a terminal fashion. The reduced complex, 9red, and its CO-adduct, 9red-CO, are moderately stable at low temperatures and decompose rapidly at room temperature. Prolonged passage of CO through DMF solutions of 9red also affords [NiI(terpy)(CO)2]+.
Since the Nip site of ACS exists in a planar geometry in the structurally characterized (as isolated) enzyme,6,7 the modeled Nip site of 10 was a more attractive target to study. It was expected that phosphine coordination to Nip in 10 would make the Ni(I) oxidation state accessible and result in a more stable CO-adduct. Both expectations have been met with 10. Complex 10 can be readily reduced by reductants such as NaBH4 and cobaltocene. The X-band EPR spectrum of 10red is consistent with a distorted tetrahedral Ni(I) center with P2S2 coordination and the resulting g values are close to 2.00 with significant hyperfine splitting originating from the two non-equivalent 31P nuclei of the dppe ligand frame.22,42 The reduced species is quite stable at room temperature. Under anaerobic condition, one can monitor the reduction of 10 to 10red in CH2Cl2 with the use of one equiv of cobaltocene. Reaction of one equiv of cobaltocene with 10 affords 10red, the species exhibiting the EPR spectrum. The violet color of 10 changes to light red-brown upon reduction. When the solution of 10red in CH2Cl2 is exposed to air, it is immediately reoxidized to 10 in nearly quantitative yield (Figure 13). This confirms that the (μ-SR)2 bridge remains intact during reduction.
Figure 13.
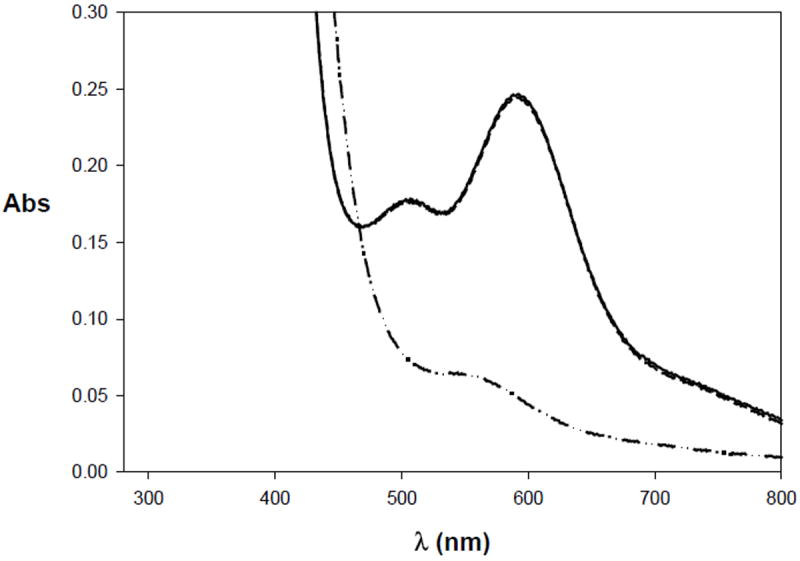
Electronic absorption spectrum of [[NiII(dppe)NiII(PhPepS)] (10) (solid line) and its reduced species [NiI(dppe)NiII(PhPepS)]- (10red) (dash-dotted line). Reoxidation of 10red to 10 upon exposure to air in CH2Cl2 is shown by the dashed line. The complex 10 was reduced with cobaltocene.
Passage of CO through a solution of 10red in CH2Cl2 (or DMF) gives rise to the CO-adduct 10red-CO that exhibits a strong νco band in its IR spectrum at 1997 cm-1, consistent with terminally bound CO to the Ni(I)p center. Complete removal of the solvent affords a dark red solid which also displays the same νco band in KBr matrix (supporting information).43 Since the νco value of 10red-CO is very close to the enzyme value of 1996 cm-1, it is reasonable to conclude that the Nip center in the enzyme binds CO in the +1 oxidation state. In [{Ni(CO)2}{NiS2N’2}]2− (Figure 2a), two molecules of CO are bound to a Ni(0) center in terminal fashion.24 This Nip mimic displays two νco bands at 1948 and 1866 cm-1. Holm and coworkers have reported a Ni(0) complex [Ni(bpy)(CO)2] (bpy = 2,2′-bipyridine) which exhibits νco bands at 1872 and 1973 cm-1 (KBr matrix).44 Since these νco values are all lower than that displayed by 10red-CO, it is evident that the latter species does contain Ni(I) and not Ni(0) at the Nip site. As mentioned above, the Nid site of 10 is quite resistant to reduction. Indeed, the νco values arising from the ligated carboxamido groups at the Nid sites of 10 and 10red-CO are very close to each other and further support the presence of Ni(II) at the Nid site in 10red-CO.
The EPR spectrum of 10red-CO in DMF glass is rhombic with g1 = 2.20, g2, = 2.12, g3 = 2.05; each line is split into a triplet from coupling to two non-equivalent 31P nuclei (Figure 14). This spectrum resembles the EPR spectrum of [Ni(psnet)]+, a structurally characterized five-coordinate complex with distorted square pyramidal P2S2N ligation to a Ni(I) center.42a The magnetic hyperfine interaction of 31P nuclei with an unpaired electron of Ni(I) has been shown to be very susceptible to the coordination geometry around the metal center.42b For example, the EPR spectrum of the tetrahedral Ni(I) complex [Ni(PMe3)4]+ (av. P-N-P angle = 109°) consists of an isotropic broad resonance with g = 2.12 with no 31P hyperfine coupling45 while the Ni(I) complex [Ni(psnet)]+ with distorted square pyramidal structure exhibits a rhombic EPR signal with g1 = 2.21, g2 = 2.13 and g3 = 2.01 and a1 = 45 G, a2 = 40 G and a3 = 44 G respectively.42a In the present case, three distinct 31P hyperfine couplings with a values in the range of 30-60 G are noted with 10red-CO (Figure 14). We therefore assign a {NiIP2S2(CO)}–(μ-SR)2–{NiII(N2S2)} formulation to 10red-CO.
Figure 14.
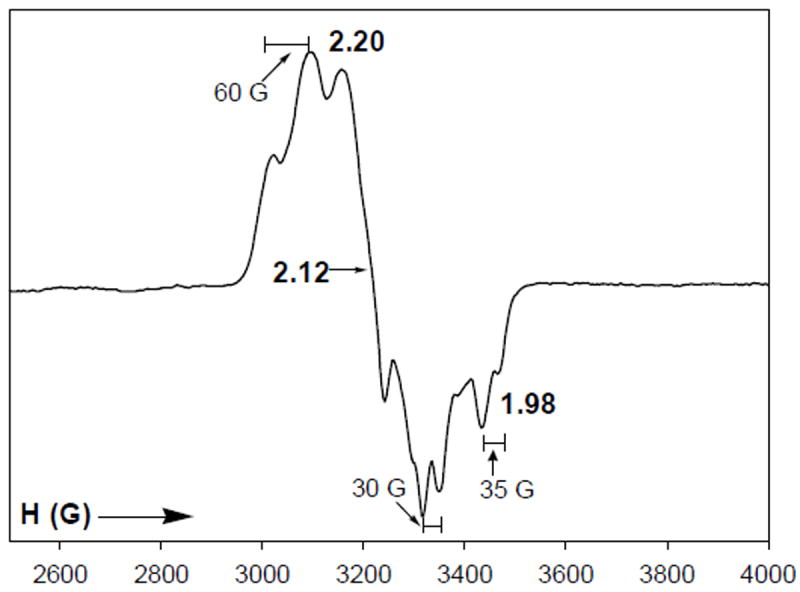
X-band EPR spectrum of [NiI(dppe)(CO)NiII(PhPepS)]- (10red-CO) in DMF glass at 100 K. Selected g and a values are indicated. Spectrometer settings: microwave frequency, 9.50 GHz; microwave power, 20 mW; modulation frequency, 100 kHz; modulation amplitude, 5 G.
Recently, three groups have reported sulfur-bridged dinuclear Ni complexes with P2S2 coordination19,20a,22 at the bridged Ni center (Nip mimic) that exhibit low Ni(I)/Ni(II) redox potentials.20,22 Unfortunately, very little spectroscopic data are currently available on the CO adducts of these complexes in the reduced state. Complex 10 deserves attention in this regard as the first structurally characterized Ni-Ni model that includes dicarboxamido-dithiolato ligation (Nid mimic) to a bridged Ni(II) center that (i) can be reduced to the Ni(I) state and (ii) binds CO in the reduced state (Nip mimic) much like that proposed for the A-cluster of ACS/CODH.
In the absence of the Fe4S4 portion of the A-cluster in the sulfur-bridged Ni-Ni models, it is impossible to assess the effect(s) of the iron-sulfur cluster on the chemical and redox properties of the Nip center. Although recent studies have indicated that electron flow to and from the Fe4S4 cluster is too slow compared to the rate of methyl transfer,46 the proposed mechanisms of ACS activity mostly involve redox changes at the Fe4S4 cluster during key steps of the catalytic cycle.5,6,47,48 Theoretical studies suggest that the Ni(0) state49 is only stabilized by either removal of the cluster15 or by the presence of a neighboring reduced iron-sulfur cluster [Fe4S4]1+ (S = ½).48 Clearly, the next generation models of the ACS A-cluster have to incorporate the Fe4S4 cluster in order to address these issues.47 Such attempts are in progress in this laboratory at this time.50
Conclusions
The results of the modeling work reported here reveal valuable insight into the novel A-cluster site involved in acetyl CoA synthesis by ACS/CODH. For example, it is quite evident that coordination of both carboxamido nitrogen and thiolato sulfur stabilizes the Ni(II) state to a great extent (like in 1 and 2) and hence the Nid site in the enzyme is not expected to participate in any redox chemistry. Binding of potential substrates such as CO at this site is also unlikely. Furthermore, Ni(II) centers bridged to metallosulfur moieties (Nip mimics) appear quite capable of binding substrates like CO when in the reduced Ni(I) state but not in the Ni(II) state. The IR spectra of such CO-adducts exhibit νco bands in the range 1960-2044 cm-1, indicating terminal Ni(I)-CO coordination similar to that proposed in the enzyme (νco = 1996 cm-1). Since the Ni-Ni models of this work (such as 9 and 10) are devoid of the strong dipolar coupling of the Nip site to the Fe4S4 cluster, the EPR spectra of the CO-adducts like 10red-CO do not resemble the NiFeC EPR signal. The removal of Nip sites from selected models by phen provides strong evidence in favor of Nip being the source of “labile Ni” in the enzyme. No reactivity toward CO has been noted with the dinuclear Ni-Cu models (3 and 4). Collectively, these observations lend support to the present notion that the Mp site in catalytically active ACS/CODH is occupied by Ni.
Supplementary Material
Acknowledgments
T. C. H. received financial support from the NIH IMSD grant GM58903.
Footnotes
Supporting Information Available: The FTIR spectra of 10 and 10red-CO in KBr matrix (Figure S1), machine parameters, crystal data, and data collection parameters for all the complexes (Table S1), selected bond distances and angles are reported (Table S2), and X-ray crystallographic data (in CIF format) and tables for the structure determination of complexes (Et4N)2[Ni(PhPepS)] (2), (Et4N)[Cu(neo)Ni(NpPepS)]•0.5Et2O•0.5H2O (3•0.5Et2O•0.5H2O), (Et4N)[Cu(neo)Ni(PhPepS)]•H2O (4•H2O), (Et4N)2[Ni{Ni(NpPepS)}2]•DMF (5•DMF), (Et4N)2[Ni(DMF)2{Ni(NpPepS)}2]•3DMF (6•3DMF), (Et4N)2[Ni(DMF)2{Ni(PhPepS)}2] (8), and [Ni(dppe)Ni(PhPepS)]•CH2Cl2 (10•CH2Cl2). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Ragsdale SW. Crit Rev Biochem Mol Biol. 2004;39:165–195. doi: 10.1080/10409230490496577. [DOI] [PubMed] [Google Scholar]; (b) Ragsdale SW, Kumar M. Chem Rev. 1996;96:2515–2539. doi: 10.1021/cr950058+. [DOI] [PubMed] [Google Scholar]
- 2.Lindahl PA. Biochemistry. 2002;41:2097–2105. doi: 10.1021/bi015932+. [DOI] [PubMed] [Google Scholar]
- 3.Wood HG, Ljungdahl LG. Variation in Autotrophic Life. Academic Press; New York, NY: 1991. [Google Scholar]
- 4.Huber C, Wächtershäuser G. Science. 1997;276:245–247. doi: 10.1126/science.276.5310.245. [DOI] [PubMed] [Google Scholar]
- 5.Doukov TI, Iverson TM, Seravalli J, Ragsdale SW, Drennan CL. Science. 2002;298:567–572. doi: 10.1126/science.1075843. [DOI] [PubMed] [Google Scholar]
- 6.Darnault C, Volbeda A, Kim EJ, Legrand P, Vernède X, Lindahl PA, Fontecilla-Camps JC. Nat Struct Biol. 2003;10:271–279. doi: 10.1038/nsb912. [DOI] [PubMed] [Google Scholar]
- 7.Svetlitchnyi V, Dobbek H, Meyer-Klaucke W, Meins T, Thiele B, Römer P, Huber R, Meyer O. Proc Natl Acad Sci USA. 2004;101:446–451. doi: 10.1073/pnas.0304262101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) George SJ, Seravalli J, Ragsdale SW. J Am Chem Soc. 2005;127:13500–13501. doi: 10.1021/ja0528329. [DOI] [PubMed] [Google Scholar]; (b) Seravalli J, Kumar M, Ragsdale SW. Biochemistry. 2002;41:1807–1819. doi: 10.1021/bi011687i. [DOI] [PubMed] [Google Scholar]; (c) Xia J, Hu Z, Popescu CV, Lindahl PA, Münck E. J Am Chem Soc. 1997;119:8301–8312. [Google Scholar]; (d) Ragsdale SW, Wood HG, Antholine WE. Proc Natl Acad Sci USA. 1985;82:6811. doi: 10.1073/pnas.82.20.6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Huang S, Seravalli J, Gutzman H, Jr, Swartz DJ, Ragsdale SW, Bagley KA. Biochemistry. 2003;42:14822–14830. doi: 10.1021/bi0349470. [DOI] [PubMed] [Google Scholar]
- 10.(a) Tan XS, Sewell C, Lindahl PA. J Am Chem Soc. 2002;124:6277–6284. doi: 10.1021/ja016676r. [DOI] [PubMed] [Google Scholar]; (b) Barondeau DP, Lindahl PA. J Am Chem Soc. 1997;119:3959–3970. [Google Scholar]
- 11.(a) Bramlett MR, Tan X, Lindahl PA. J Am Chem Soc. 2003;125:9316–9317. doi: 10.1021/ja0352855. [DOI] [PubMed] [Google Scholar]; (b) Shin W, Lindahl PA. J Am Chem Soc. 1992;114:9718–9719. [Google Scholar]
- 12.Seravalli J, Xiao Y, Gu W, Cramer SP, Antholine WE, Krymov V, Gerfen GJ, Ragsdale SW. Biochemistry. 2004;43:3944–3955. doi: 10.1021/bi036194n. [DOI] [PubMed] [Google Scholar]
- 13.(a) Funk T, Gu W, Friedrich S, Wang H, Gencic S, Grahame DA, Cramer SP. J Am Chem Soc. 2004;126:88–95. doi: 10.1021/ja0366033. [DOI] [PubMed] [Google Scholar]; (b) Gencic S, Grahame DA. J Biol Chem. 2003;278:6101–6110. doi: 10.1074/jbc.M210484200. [DOI] [PubMed] [Google Scholar]
- 14.Webster CE, Darensbourg MY, Lindahl PA, Hall MB. J Am Chem Soc. 2004;126:3410–3411. doi: 10.1021/ja038083h. [DOI] [PubMed] [Google Scholar]
- 15.(a) Brunold TC. J Biol Inorg Chem. 2004;9:533–541. doi: 10.1007/s00775-004-0566-8. [DOI] [PubMed] [Google Scholar]; (b) Schenker RP, Brunold TC. J Am Chem Soc. 2003;125:13962–13963. doi: 10.1021/ja037893q. [DOI] [PubMed] [Google Scholar]
- 16.Peters JW, Stowell MHB, Soltis M, Finnegan MG, Johnson MK, Rees DC. Biochemistry. 1997;36:1181–1187. doi: 10.1021/bi9626665. [DOI] [PubMed] [Google Scholar]
- 17.(a) Harrop TC, Mascharak PK. Acc Chem Res. 2004;37:253–260. doi: 10.1021/ar0301532. [DOI] [PubMed] [Google Scholar]; (b) Mascharak PK. Coord Chem Rev. 2002;225:201–214. [Google Scholar]; (c) Nagashima S, Nakasako M, Dohmae N, Tsujimura M, Takio K, Odaka M, Yohda M, Kamiya N, Endo I. Nat Struct Biol. 1998;5:347–351. doi: 10.1038/nsb0598-347. [DOI] [PubMed] [Google Scholar]; (d) Huang W, Jia J, Cummings J, Nelson M, Schneider G, Lindqvist Y. Structure. 1997;5:691–699. doi: 10.1016/s0969-2126(97)00223-2. [DOI] [PubMed] [Google Scholar]
- 18.(a) Barondeau DP, Kassmann CJ, Bruns CK, Tainer JA, Getzoff ED. Biochemistry. 2004;43:8038–8047. doi: 10.1021/bi0496081. [DOI] [PubMed] [Google Scholar]; (b) Wuerges J, Lee J-W, Yim Y-I, Yim H-S, Kang S-O, Carugo KD. Proc Natl Acad Sci USA. 2004;101:8569–8574. doi: 10.1073/pnas.0308514101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rao PV, Bhaduri S, Jiang J, Holm RH. Inorg Chem. 2004;43:5833–5849. doi: 10.1021/ic040055s. [DOI] [PubMed] [Google Scholar]
- 20.(a) Krishnan R, Riordan CG. J Am Chem Soc. 2004;126:4484–4485. doi: 10.1021/ja038086u. [DOI] [PubMed] [Google Scholar]; (b) Krishnan R, Voo JK, Riordan CG, Zahkarov L, Rheingold AL. J Am Chem Soc. 2003;125:4422–4423. doi: 10.1021/ja0346577. [DOI] [PubMed] [Google Scholar]
- 21.Hatlevik Ø, Blanksma MC, Mathrubootham V, Arif AM, Hegg EL. J Biol Inorg Chem. 2004;9:238–246. doi: 10.1007/s00775-003-0518-8. [DOI] [PubMed] [Google Scholar]
- 22.Wang Q, Blake AJ, Davies ES, McInnes EJL, Wilson C, Schröder M. Chem Commun. 2003:3012–3013. doi: 10.1039/b310183e. [DOI] [PubMed] [Google Scholar]
- 23.Golden ML, Rampersad MV, Reibenspies JH, Darensbourg MY. Chem Commun. 2003:1824–1825. doi: 10.1039/b304884p. [DOI] [PubMed] [Google Scholar]
- 24.Linck RC, Spahn CW, Rauchfuss TB, Wilson SR. J Am Chem Soc. 2003;125:8700–8701. doi: 10.1021/ja035606c. [DOI] [PubMed] [Google Scholar]
- 25.Harrop TC, Mascharak PK. Coord Chem Rev. 2005;249:3007–3024. [Google Scholar]
- 26.Harrop TC, Olmstead MM, Mascharak PK. Inorg, Chim Acta. 2002;338:189–195. [Google Scholar]
- 27.(a) Harrop TC, Olmstead MM, Mascharak PK. J Am Chem Soc. 2004;126:14714–14715. doi: 10.1021/ja045284s. [DOI] [PubMed] [Google Scholar]; (b) Harrop TC, Olmstead MM, Mascharak PK. Chem Commun. 2004:1744–1745. doi: 10.1039/b405572c. [DOI] [PubMed] [Google Scholar]
- 28.Gill NS, Taylor FB. Inorg Synth. 1967;9:136–143. [Google Scholar]
- 29.(a) Healy PC, Pakawatchai C, White AH. J Chem Soc Dalton Trans. 1985:2531–2539. [Google Scholar]; (b) Rao CP, Dorfman JR, Holm RH. Inorg Chem. 1986;25:428–439. [Google Scholar]; (c) Anderson OP, Brito KK, Laird SK. Acta Cryst. 1990;C46:1600–1603. doi: 10.1107/s0108270189011728. [DOI] [PubMed] [Google Scholar]
- 30.Judge JS, Reiff WM, Intille GM, Ballway P, Baker WA., Jr J Inorg Nucl Chem. 1967;29:1711–1716. [Google Scholar]
- 31.(a) Krüger H-J, Peng G, Holm RH. Inorg Chem. 1991;30:734–742. [Google Scholar]; (b) Krüger H-J, Holm RH. Inorg Chem. 1987;26:3645–3647. [Google Scholar]
- 32.Dutton JC, Fallon GD, Murray KS. Chem Lett. 1990:983–986. [Google Scholar]
- 33.Hanss J, Krüger H-J. Angew Chem Int Ed. 1998;37:360–363. doi: 10.1002/(SICI)1521-3773(19980216)37:3<360::AID-ANIE360>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 34.Marlin DS, Mascharak PK. Chem Soc Rev. 2000;29:69–74. [Google Scholar]
- 35.Smeets WJJ, Spek AL, Henderson RK, Bouwman E, Reedijk J. Acta Cryst Sect C. 1997;C53:1564–1566. [Google Scholar]
- 36.Chapman RL, Vagg RS. Inorg Chim Acta. 1979;33:227–234. [Google Scholar]
- 37.Pallenberg AJ, Koenig KS, Barnhart DM. Inorg Chem. 1995;34:2833–2840. [Google Scholar]
- 38.(a) Farmer PJ, Solouki T, Mills DK, Soma T, Russell DH, Reibenspies JH, Darensbourg MY. J Am Chem Soc. 1992;114:4601–4605. [Google Scholar]; (b) Turner MA, Driessen WL, Reedijk J. Inorg Chem. 1990;29:3331–3335. [Google Scholar]
- 39.(a) Craft JL, Mandimutsira BS, Fujita K, Riordan CG, Brunold TC. Inorg Chem. 2003;42:859–867. doi: 10.1021/ic020441e. [DOI] [PubMed] [Google Scholar]; (b) Musie G, Farmer PJ, Tuntulani T, Reibenspies JH, Darensbourg MY. Inorg Chem. 1996;35:2176–2183. doi: 10.1021/ic9515968. [DOI] [PubMed] [Google Scholar]
- 40.Yamamura T, Sakurai S, Arai H, Miyamae H. J Chem Soc Chem Commun. 1993:1656–1658. [Google Scholar]
- 41.(a) Marganian CA, Vazir H, Baidya N, Olmstead MM, Mascharak PK. J Am Chem Soc. 1995;117:1584–1594. [Google Scholar]; (b) Baidya N, Olmstead MM, Whitehead JP, Bagyinka C, Maroney MJ, Mascharak PK. Inorg Chem. 1992;31:3612–3619. [Google Scholar]; (c) Baidya N, Olmstead MM, Mascharak PK. Inorg Chem. 1991;30:929–937. [Google Scholar]
- 42.(a) James TL, Cai L, Muetterties MC, Holm RH. Inorg Chem. 1996;35:4148–4161. doi: 10.1021/ic960216v. [DOI] [PubMed] [Google Scholar]; (b) Kim JS, Reibenspies JH, Darensbourg MY. J Am Chem Soc. 1996;118:4115–4123. [Google Scholar]; (c) James TL, Smith DM, Holm RH. Inorg Chem. 1994;33:4869–4877. [Google Scholar]; (d) Bowmaker GA, Boyd PDW, Campbell GK. Inorg Chem. 1982;21:2403–2412. [Google Scholar]
- 43.The red solid is moderately stable under anaerobic conditions, however, it decomposes slowly in solution and to date, we have not been able to characterize it by structural studies. The EPR spectrum of a solution of the freshly isolated red solid in DMF (same as Figure 14) confirms that the solid is indeed 10red-CO.
- 44.Tucci GC, Holm RH. J Am Chem Soc. 1995;117:6489–6496. [Google Scholar]
- 45.Gleizes A, Dartiguenave M, Dartiguenave Y, Galy J, Klein HF. J Am Chem Soc. 1977;99:5187–5189. [Google Scholar]
- 46.Tan X, Sewell C, Yang Q, Lindahl PA. J Am Chem Soc. 2003;125:318–319. doi: 10.1021/ja028442t. [DOI] [PubMed] [Google Scholar]
- 47.Hegg EL. Acc Chem Res. 2004;37:775–783. doi: 10.1021/ar040002e. [DOI] [PubMed] [Google Scholar]
- 48.Amara P, Volbeda A, Fontecilla-Camps JC, Field MJ. J Am Chem Soc. 2005;127:2776–2784. doi: 10.1021/ja0439221. [DOI] [PubMed] [Google Scholar]
- 49.Lindahl PA. J Biol Inorg Chem. 2004;9:516–524. doi: 10.1007/s00775-004-0564-x. [DOI] [PubMed] [Google Scholar]
- 50.In a recent paper, Holm and coworkers have reported the results of such an attempt. However, only complexes of the thiolato-bridged [Fe4S4]-NiL type have been isolated. See: Rao PV, Bhaduri S, Jiang J, Hong D, Holm RH. J Am Chem Soc. 2005;127:1933–1945. doi: 10.1021/ja040222n.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.