

Summary
Mutations in DMD disrupt the reading frame, prevent dystrophin translation, and cause Duchenne muscular dystrophy (DMD). Here we describe a CRISPR/Cas9 platform applicable to 60% of DMD patient mutations. We applied the platform to DMD-derived hiPSCs where successful deletion and non-homologous end joining of up to 725kb reframed the DMD gene. This is the largest CRISPR/Cas9-mediated deletion shown to date in DMD. Use of hiPSCs allowed for evaluation of dystrophin in disease relevant cell types. Cardiomyocytes and skeletal muscle myotubes derived from reframed hiPSC clonal lines had restored dystrophin protein. The internally deleted dystrophin was functional as demonstrated by improved membrane integrity and restoration of the dystrophin glycoprotein complex in vitro and in vivo. Furthermore, miR31 was reduced upon reframing, similar to observations in Becker muscular dystrophy. This work demonstrates the feasibility of using a single CRISPR pair to correct the reading frame for the majority of DMD patients.
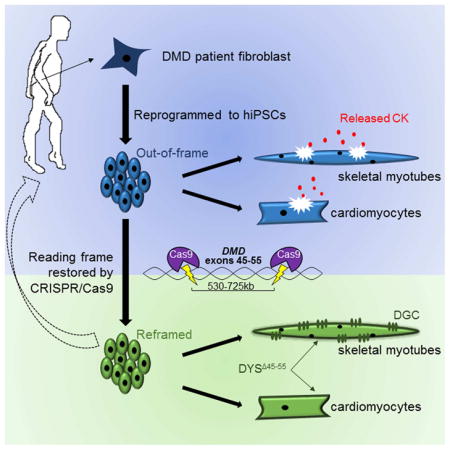
Graphical abstract
Introduction
Duchenne muscular dystrophy (DMD) is the most common fatal genetic disease of childhood, affecting ~1 in 3500–5000 boys. In DMD, progressive muscle degeneration generally leads to death in the twenties, and there are currently no highly effective therapies. DMD is often caused by frameshifting exonic deletions in DMD, which encodes dystrophin. Dystrophin stabilizes the dystrophin glycoprotein complex (DGC) at the sarcolemma; loss of functional dystrophin leads to the degradation of DGC components which results in muscle membrane fragility and leakage of creatine kinase (CK) (Pearce et al., 1964). Approximately 60% of mutations causing Duchenne occur between DMD exons 45–55 (Beroud et al., 2007). Multiple independent clinical reports in patients and dystrophic mice have revealed that in-frame deletions of exons 45–55 produce an internally deleted dystrophin protein and are associated with a very mild Becker muscular dystrophy (BMD) disease course, with some patients still asymptomatic in their 60s (Beroud et al., 2007; Echigoya et al., 2015; Nakamura et al., 2008; Taglia et al., 2015). Thus, genetic manipulation to create a large deletion of exons 45–55 is a therapeutic strategy to restore the reading frame for 60% of DMD patients with mutations in this region.
One promising approach to induce genetic correction of DMD is through the use of the bacterially acquired immune surveillance system known as clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated nuclease (Cas) 9. In this system a short guide RNA (gRNA), which is complimentary to a specific site in the genome, is used to target the Cas9 nuclease and induce double stranded breaks (DSBs). The DSB can be repaired through non-homologous end joining (NHEJ) or homology directed repair.
Previous work has shown that CRISPR/Cas9 components can modify the DMD gene (Li et al., 2015; Long et al., 2015, 2014; Nelson et al., 2015; Ousterout et al., 2015; Tabebordbar et al., 2015; Wojtal et al., 2016; Xu et al., 2015). In this investigation, we describe a therapeutically relevant CRISPR/Cas9 platform that we designed to modify DMD. Our platform involves excision of exons 45–55 and NHEJ to reframe dystrophin through creation of an internally deleted protein that is stable and functional. The internally deleted protein mimics the naturally occurring exon 45–55 deletion observed in mild BMD patients and encompasses 60% of DMD patient mutations.
For the first time, we demonstrate CRISPR/Cas9-mediated deletion and NHEJ of up to 725kb of the DMD gene in human induced pluripotent stem cell (hiPSC) lines. We show that CRISPR/Cas9 reframed hiPSC-derived skeletal and cardiac muscle cells express stable dystrophin that improves membrane stability and restores a DGC member, β-dystroglycan. We also demonstrate reduced microRNA 31 (miR31) levels after the reading frame is restored, consistent with the observations made in BMD patients (Cacchiarelli et al., 2011). Furthermore, we show restoration of dystrophin and β-dystroglycan in vivo after engraftment of reframed hiPSC-derived skeletal muscle cells into a mouse model of DMD. This work sets the stage for use of reframed DMD hiPSC-derived cells or in vivo correction strategies using CRISPR/Cas9 for direct translation to patients with DMD.
Results
DMD hiPSC lines are pluripotent and genetically stable
We have developed several xenobiotic-free hiPSC lines derived from wild type and DMD patient fibroblasts using current good manufacturing practice protocols. Each DMD hiPSC line harbors a unique frame-shifting DMD mutation within the exon 45–55 hotspot region. All hiPSC lines (Center for Duchenne Muscular Dystrophy (CDMD) 1003, 1006 and 1008) express pluripotency markers (NANOG and SOX2) and are karyotypically normal (Figures 1A and 1B). CDMD hiPSCs maintain pluripotency, as they form teratomas in vivo that represent all three germ layers (Figure 1C), and each harbor unique mutations (Figure 1D).
Figure 1. CDMD hiPSCs are pluripotent and genetically stable.

A. CDMD hiPSCs were generated from DMD fibroblasts. Brightfield images depict fibroblasts before and after reprogramming to hiPSCs. Immunocytochemical staining reveals that cells express pluripotency markers NANOG (green) and SOX2 (red). Scale bar 100μm.
B. Karyotyping of all lines is shown.
C. CDMD hiPSCs were injected into mice to test teratoma formation in vivo. Representative hematoxylin and eosin stainings of the three germ layers (endoderm, mesoderm, and ectoderm) are shown.
D. Patient mutations for each CDMD hiPSC line are shown. In addition, the number of exons and the approximate distance necessary for successful NHEJ is indicated, based on comparative genomic hybridization data for the patient’s underlying mutation size.
CRISPR/Cas9-mediated deletion and NHEJ of up to 725kb in the DMD gene
In order to delete exons 45–55 of DMD, gRNAs were designed to target introns 44 and 55. gRNA sites were chosen to only retain ~500bp of the intron next to each of the flanking exons (44 and 56). The rationale for this design is to develop gRNAs applicable to as many patient mutations as possible and to ensure a small functional chimeric intron is generated. During NHEJ, the 3′ end of intron 44 and the 5′ end of intron 55 join to create a ~1kb chimeric intron (Figure 2A). We expect introns generated in this manner are functional and splice correctly to create an in-frame transcript, with exon 44 joined with exon 56.
Figure 2. Generation of stable, pluripotent CDMD hiPSC lines with an exon 45–55 deletion.

A. Shown is a cartoon (not to scale) of the region of DMD targeted for CRISPR/Cas9-mediated deletion using gRNAs specific to introns 44 and 55 (lightning bolts). Successful NHEJ deletes exons 45–55 and restores the reading frame for mutations within this region. Different deletion sizes are required depending on the patient’s underlying mutation (black arrow heads).
B. PCR genotyping of 117 and 109 single cell clones from parental lines CDMD 1006 and 1003, respectively, was carried out on cells nucleofected with gRNAs 44C4 and 55C3. One clone from CDMD 1006 (CDMD 1006-1) and three from CDMD 1003 (CDMD 1003-49, 1003-57, 1003-81) were identified as stably deleted. Deletion PCR genotyping results for 6 hiPSC clonal lines is shown. One pair of primers (red arrows in A) was located internal to the deletion and only produced a 1201bp band in the undeleted clones CDMD 1003-13 and 1003-51. Another primer set (purple arrows in A) flanked the deletion region and produced a 788bp band only when the deletion and NHEJ occurred successfully, as in the reframed clones CDMD 1006-1, 1003-49, 1003-57, 1003-81.
C. Each clonal line maintained normal morphology (brightfield) and expressed NANOG (green) and SOX2 (red) by immunocytochemistry. Scale bar 100μm. Shown to the right is the sequence of the gDNA at the rejoining site between introns 44 (I44) and 55 (I55). Sequencing revealed a 16bp deletion in CDMD 1006-1, a 2bp insertion in CDMD 1003-49, and 1bp insertions in CDMD 1003-57 and CDMD 1003-81.
See also Figure S1, S2, S3, and S4 A–B.
Since hiPSCs are challenging to genetically manipulate, human embryonic kidney (HEK) 293FT cells were used to screen five gRNAs at each intronic region. All gRNAs demonstrated individual cutting activity on Surveyor assay up to 34% (Figures S1A and S1B). Using multiplex PCR, gRNAs transfected in pairs were shown to effectively delete the entire 708kb region encompassing exons 45–55 (Figures S1C and S1D).
In order to assess the feasibility of an exon 45–55 deletion across different patient mutations, we applied our gRNAs to three DMD hiPSC lines. The lines (CDMD 1003, 1006, 1008) require ~530kb, 670kb, or 725kb for successful deletion and NHEJ of DMD respectively. The gRNAs used were shown to be active in all three lines and effectively deleted exons 45–55 (Figures S2 and S3). Transient puromycin selection of cells nucleofected with the CRISPR plasmids improved the efficiency of deletion in CDMD 1003 and 1006 hiPSCs (Figure S3D).
Clonal reframed DMD hiPSC lines contain no off target activity at candidate sites
Stably deleted DMD hiPSC lines were generated from CDMD 1003 and 1006 by clonal selection after nucleofection with the gRNA pair 44C4 and 55C3 (Figure 2B and 2C) and are pluripotent (Figures 2C and S4B). All reframed lines were karyotypically normal except for one clone (CDMD 1003-81), which was found to contain a 1q32 amplification confirmed via FISH analysis (Figure S4A), also observed in the original parental line and in all daughter clones after post-hoc analysis. The 1q32 amplification is common in hPSCs after extended propagation in culture (Dekel-Naftali et al., 2012), and thus was not a result of CRISPR-mediated off target activity. To determine off target activity of our gRNAs, the top 10 homologous sites per guide were determined by COSMID (Cradick et al., 2014) and sequenced in all clonal and parental lines. No off target mutations were observed at any site (Table S2). All variants, besides a heterozygous SNP in chromosome 11, were detected in less than 1% of reads which is consistent with error in the sequencing method.
Dystrophin (DYSΔ45–55) expression is restored in reframed DMD hiPSC-derived cardiomyocytes and skeletal myotubes
CRISPR/Cas9-mediated deletion of DMD should result in an internally deleted dystrophin protein lacking exons 45–55 (hereafter referred to as DYSΔ45–55). As hiPSCs do not express dystrophin, we differentiated the reframed DMD hiPSC clonal lines to two disease-relevant cell types, cardiomyocytes and skeletal muscle myotubes, using directed differentiation or overexpression of MyoD to evaluate rescue of DYSΔ45–55. PCR and sequencing of the exon 44/56 boundary in cDNA from the reframed cardiomyocyte clones demonstrated correct splicing of the dystrophin transcript (Figures S4C and S4D). Additionally, both the reframed cardiac and skeletal muscle cell lines restored dystrophin expression as assayed by immunocytochemistry and Western blot (Figures 3A–3C). Compared to wild type CDMD 1002 or human skeletal muscle myotubes (HSMM), the band was truncated by ~66kDa as expected.
Figure 3. Reframed CDMD hiPSC-derived skeletal muscle and cardiomyocytes restore dystrophin expression.

A. Immunocytochemical staining of human myosin heavy chain (MyHC, red) and dystrophin (green) of wild type (CDMD 1002), out-of-frame (CDMD 1003, 1006) or reframed (CDMD 1003-49, 1006-1) cardiomyocytes derived from hiPSCs by directed differentiation. Inset depicts zoomed in region defined by the white box. Scale bar 50μm.
B. Immunocytochemical staining of MyHC (red) and dystrophin (green) of wild type (CDMD 1002), out-of-frame (CDMD 1006) or reframed (CDMD 1006-1, 1003-49) skeletal muscle myotubes derived from hiPSCs. Myotubes were fused after MyoD overexpression (OE) or from sorted NCAM+ cells after an adapted directed differentiation 50-day protocol. Inset depicts zoomed in region defined by the white box. Scale bar 100μm.
C. Western blots of cell extracts probed with anti-dystrophin. Extracts were from out-of-frame and reframed cardiomyocytes (left) and skeletal muscle myotubes (right), derived from CDMD hiPSCs. Wild type (wt) hiPSCs (CDMD 1002) or human skeletal muscle myotubes (HSMM) were used as a control for dystrophin. The molecular weight shift caused by the exon 45–55 deletion (1779bp, ~66kDa) is evident in reframed vs. wild type dystrophin (arrows). A non-specific band around 220kDa was seen in some samples. Samples were also probed with anti-MyHC as a loading control (bottom panels).
See also Figure S4C–D.
DYSΔ45–55 protein restores membrane functionality to cardiomyocytes and skeletal myotubes in vitro
Cardiomyocytes or skeletal myotubes lacking dystrophin demonstrate membrane fragility in vitro and respond to osmotic stress by releasing elevated levels of CK (Guan et al., 2014; Menke and Jockusch, 1995), as is seen in human patients (Pearce et al., 1964). To determine whether DYSΔ45–55 could restore stability to dystrophic plasma membranes, we subjected differentiated cardiomyocytes and skeletal muscle myotubes derived from reframed and out-of-frame hiPSCs to hypoosmotic conditions. Cells were stressed by incubation in hypoosmolar solutions (66–240mosmol) and CK release into the supernatant was measured to show functional improvement after dystrophin restoration. Both the reframed CDMD 1003-49 cardiomyocytes and skeletal muscle cells demonstrated reduced CK release, similar to wild type (CDMD 1002), versus the out-of-frame CDMD 1003 cells, indicating that DYSΔ45–55 was capable of reducing membrane fragility (Figure 4A). The same trend was also observed with CDMD 1006/1006-1 cardiomyocytes (Figure S4E). After normalizing and pooling all experiments, we observed that significantly less CK was released at 93, 135, and 240mosmol in the reframed and wild type cells compared to out-of-frame (Figure S4F).
Figure 4. Reframed hiPSC-derived cardiomyocytes and skeletal muscle cells demonstrate restored function in vitro and in vivo.

A. Representative graphs of CK release assays from cells exposed to hypoosmotic conditions. Cardiomyocytes and skeletal muscle myotubes derived from hiPSCs were subjected to a range of osmolarities below 240mosmol and CK release to the supernatant was measured as an indication of membrane fragility. Data are presented as average ± standard error.
B. Fold change in expression of miR31 measured by ddPCR in myotubes derived from out-of-frame or reframed hiPSCs by MyoD OE, normalized to wild type (CDMD 1002). Data are presented as average ± standard deviation.
C. Western blots of cell extracts probed with anti-β-dystroglycan. Extracts were from out-of-frame and reframed skeletal muscle myotubes derived by MyoD OE. HSMM was used as a positive control. Samples were also probed with anti-MyHC as a loading control (bottom panel).
D. Immunocytochemical staining of MyHC (red) and β-dystroglycan (green), a component of the DGC, in wild type (CDMD 1002), out-of-frame (CDMD 1006) or reframed (CDMD 1006-1) skeletal muscle myotubes. Inset depicts zoomed in region defined by the white box. Scale bar 50μm.
E. Assessment of human dystrophin restoration in wild type (CDMD 1002), out-of-frame (CDMD 1003), and reframed (CDMD 1003-49) MyoD OE cells engrafted into the TA of NSG-mdx mice. Engrafted human cells were identified by co-immunostaining for human spectrin and lamin A/C (shown in red). Positive staining for human dystrophin is shown in green and all fibers are shown using laminin (grey). All sections were stained with DAPI (blue) to identify nuclei. Scale bar 100μm.
F. Assessment of β-dystroglycan restoration in human fibers from wild type (CDMD 1002), out-of-frame (CDMD 1003), and reframed (CDMD 1003-49) MyoD OE cells engrafted into the TA of NSG-mdx mice. Engrafted human cells were identified by co-immunostaining for human spectrin and lamin A/C (shown in red). Positive staining for dystrophin is shown in grey and β-dystroglycan is shown in green. All sections were stained with DAPI (blue) to identify nuclei. Cell order is same as noted in E. Scale bar 20μm.
See also Figure S4E–F.
CRISPR/Cas9 reframing correlates with miR31 levels in skeletal myotubes in vitro
Elevated levels of miR31 have been observed in DMD patient biopsies compared to wild type or BMD (Cacchiarelli et al., 2011). We measured levels of miR31 using droplet digital PCR (ddPCR) after differentiation of out-of-frame and reframed CDMD hiPSCs to skeletal myotubes. Reframing DMD reduced levels of miR31 (similar to wild type cells), compared to out-of-frame DMD, as is observed in human dystrophinopathies (Figure 4B). Thus, reframing the DMD gene normalizes miR31 levels similar to BMD, demonstrating functional rescue of the dystrophic phenotype to a BMD phenotype.
DYSΔ45–55 protein restores the DGC in vitro and in vivo
As a third assay of DYSΔ45–55 functionality, we evaluated its ability to restore the DGC in vitro and in vivo. The DGC member, β-dystroglycan, was restored and detected at the membrane of reframed hiPSCs, but not out-of-frame hiPSCs, after directed differentiation to skeletal muscle in vitro by immunostaining and Western blot (Figure 4C and 4D). Additionally, skeletal muscle cells derived from a wild type (CDMD 1002), out-of-frame (CDMD 1003), or reframed (CDMD 1003-49) hiPSC line were injected into the tibialis anterior of NSG-mdx mice. Correctly localized dystrophin and β-dystroglycan was only observed in engrafted human cells (demarked by human lamin A/C and spectrin) from the reframed or wild type lines (Figure 4E and 4F). These studies taken together with the hypoosmotic stress assays demonstrate the ability of DYSΔ45–55 to functionally reassemble the DGC and restore membrane stability in vitro and in vivo.
Discussion
Using CRISPR/Cas9 gene editing, we have induced the largest deletion accomplished to date in DMD hiPSCs and restored a functional dystrophin protein. Deletion of DMD exons 45–55 has the potential to be therapeutically relevant to 60% of Duchenne patients. Since this internal deletion has been associated with a very mild disease course in multiple independent patients, a therapy utilizing this approach should create a highly functional dystrophin. We showed successful deletion of exons 45–55 using a single gRNA pair and did not identify any off target activity at the top 10 homologous sites, however a more comprehensive and unbiased approach should be undertaken such as whole genome sequencing. Importantly, removal of exons 45–55 resulted in stable dystrophin protein (DYSΔ45–55) in both cardiomyocytes and skeletal myotubes in vitro. Functionality of DYSΔ45–55 was tested in cardiomyocytes and skeletal muscle derived from reframed DMD hiPSCs and demonstrated improved membrane stability by a physiologically relevant measure of CK release, similar to wild type. The ability to evaluate cardiomyocyte functionality is an advantage of using hiPSCs, as some current preclinical and clinical studies for DMD therapies do not efficiently target the heart (e.g. exon skipping; Arechavala-Gomeza et al., 2012). Additionally, we demonstrated a normalization in miR31 levels, a microRNA that inhibits dystrophin, after reading frame restoration similar to what is observed in human BMD patients (Cacchiarelli et al., 2011). Finally, we show restored DGC localization in vitro and in vivo, which further validates the functionality of DYSΔ45–55.
Previous work by Ousterout et al. (2015) demonstrated that multiplexed gRNAs can restore the DMD reading frame in primary myoblasts. However, myoblasts do not provide a renewable source of stem cells, which is a requirement for long-term therapeutic efficacy (Partridge, 2002). In contrast, we used hiPSCs which offer the opportunity to evaluate the internally deleted dystrophin protein in multiple cell types that are affected in DMD, and in future studies, may provide a renewal source of corrected progenitor cells. Our work is further distinguished from previous studies as we are the only group to show restoration of dystrophin function on membrane integrity, miR31 expression, and the DGC in cardiac and skeletal muscle cells following CRISPR-mediated gene editing.
An advantage of our CRISPR platform is the therapeutic potential of a single pair of gRNAs to treat the majority of DMD patients. By designing gRNAs that accomplish a deletion that encompasses the majority of DMD mutations, this approach is optimized for future clinical studies. It would be unreasonable to design, validate, and evaluate off targets for every new CRISPR pair tailored for each individual patient. Additionally, CRISPR/Cas9 is advantageous over exon skipping, as it results in permanent restoration of reading frame as opposed to transient effects on RNA splicing. Previously, Li et al. (2015) used CRISPR/Cas9 to induce exon skipping, frameshifting, or exon knockin to restore dystrophin in a DMD hiPSC line with an exon 44 deletion; however, their platform is only applicable to 3–9% of DMD patients (Bladen et al., 2015), and two of their strategies relied on creation of indels which would be difficult to apply consistently to each patient. While Ousterout et al. deleted exons 45–55, they removed significantly less of the intervening region (336kb) and thus their approach would cover fewer patient mutations within the hotspot region. This is because many mutations extend into the intronic region, thus by designing gRNAs that encompass more of the intron, our platform is applicable to more patients.
Another benefit of using this platform to delete a large portion of DMD, as opposed to single exons, is the known correlation of DYSΔ45–55 with a mild BMD phenotype. Large deletions in the rod domain of dystrophin often produce a more functional (more like wild type) protein, than even very small deletions (Harper et al., 2002). Larger deletions, which remove hinge III (exons 50–51), are believed to lead to a milder BMD phenotype than smaller deletions, or those which retain hinge III (Carsana et al., 2005). Thus, in many cases larger deletions are more therapeutically beneficial than smaller ones, due to the way they affect the secondary structure of the protein.
In summary, we have developed a potentially therapeutic gene editing platform for DMD to permanently restore dystrophin reading frame in multiple patient-derived hiPSCs. Our approach using CRISPR/Cas9 and NHEJ deletes up to 725kb of DMD encompassing exons 45–55 and restores dystrophin protein function in both cardiomyocytes and skeletal muscle cells derived from corrected hiPSCs. A current limitation of this platform is that clinical protocols still need to be developed that allow for rapid clonal line derivation, and the utilization of hiPSC-derived cardiac and skeletal muscle progenitors combined with gene correction. Alternatively, CRISPR/Cas9 to restore the reading frame in DMD mouse models has been delivered directly in vivo (Long et al., 2015; Nelson et al., 2015; Tabebordbar et al., 2015). Thus, applications of this platform in the future will allow for the development of an in situ gene correction strategy or ex vivo gene correction followed by autologous cell transplantation, either of which offers tremendous potential for DMD.
Experimental Procedures
Differentiation of hiPSCs to skeletal muscle cells and cardiomyocytes
Skeletal muscle differentiation from hiPSCs was induced using overexpression of a tamoxifen inducible MyoD-ERT lentivirus or an adapted 50 day directed differentiation protocol where NCAM+ HNK1− cells were fluorescently activated cell sorted at day 50. Cardiomyocytes were derived through aggregates over 30 days. See Supplemental Experimental Procedures.
Engraftment into immunodeficient mice
NOD scid IL2Rgamma (NSG) immunodeficient mice (Jackson Laboratory) were crossed to mdx scid mice (Jackson Laboratory) to generate NSG-mdx mice, see Supplemental Experimental Procedures. 5–7 week-old NSG-mdx mice were pretreated with 50μl of 10μM cardiotoxin (Sigma-Aldrich) injected into the right tibialis anterior (TA) 24hrs prior to engraftment. For MyoD OE cells, 100μl of 5mg/ml tamoxifen (Sigma-Aldrich) was IP injected for 5 days beginning the day prior to engraftment. 1×106 cells in HBSS were injected intramuscularly and the TA was harvested after 30 days. See Supplemental Experimental Procedures.
Hypoosmotic stress CK release assay
Terminally differentiated skeletal muscle cells and cardiomyocytes plated in duplicate were stressed by incubation in hypoosomolar solutions ranging from 66–240mosmol, see Supplemental Experimental Procedures, for 20mins at 37°C. CK was measured in triplicate from the supernatant and cell lysate with the Creatine Kinase-SL kit (Sekisui Diagnostics) according to the manufacturer’s instructions.
Supplementary Material
Highlights.
Largest CRISPR/Cas9-mediated deletion of 725kb of DMD
Reframed DMD hiPSCs differentiated to cardiac & skeletal muscle express dystrophin
Internally deleted dystrophin demonstrates functionality in vitro and in vivo
This single gRNA pair is therapeutically relevant to 60% of DMD mutations
Acknowledgments
We would like to thank Doug Black, Richard Wang, and Haibin Xi for suggestions and Diana Becerra and Jane Wen for technical assistance. The following cores were utilized: CDMD Muscle Phenotyping and Imaging Core, HTS and Cell Repository Core, and Bioinformatics and Genomics Core (supported by NIAMS-P30 AR057230), the UCLA Broad Stem Cell Research Center (BSCRC) Flow Cytometry Core Resource, the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research (CFAR) Flow Cytometry Core Facility (supported by NIH P30 CA016042, 5P30 AI028697), the UCLA Integrated Molecular Technologies Core (supported by CURE/P30 DK041301), the UCLA Humanized Mouse Core (supported by the JCCC, BSCRC, CFAR, NIH/NIAID AI028697), and the UCLA GenoSeq core. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1144087 (CSY). Funding was provided by NIAMS of the NIH (P30 AR057230-MJS, R01AR064327-ADP) and the Eli & Edythe Broad Center of Regenerative Medicine and Stem Cell Research at UCLA (MJS, ADP) and Rose Hills Foundation Research Award (ADP).
Footnotes
Author contributions
Conceptualization, Methodology, Writing-Original Draft, Visualization, Project Administration - CSY, MJS, ADP; Validation- CSY, MRH, NVE; Formal Analysis- CSY; Investigation- CSY, MRH, NVE, HN, MJ, SY, SK CK-C, DW; Resources- AN, SFN, MCM, MJS, ADP; Writing- Review and Editing- CSY, MRH, NEV, SK, CK-C, DBK, AN, SFN, MCM, MJS, ADP; Supervision- JAZ, DBK, AN, MCM, MJS, ADP; Funding Acquisition- MJS, ADP.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arechavala-Gomeza V, Anthony K, Morgan J, Muntoni F. Antisense Oligonucleotide-Mediated Exon Skipping for Duchenne Muscular Dystrophy: Progress and Challenges. Curr Gene Ther. 2012;12:152–160. doi: 10.2174/156652312800840621. [DOI] [PubMed] [Google Scholar]
- Beroud C, Tuffery-Giraud S, Matsuo M, Hamroun D, Calemard LM, Boisseau P, Blayau M, Philippe C, Cosse M, Page M, et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum Mutat. 2007;28:196–202. doi: 10.1002/humu.20428. [DOI] [PubMed] [Google Scholar]
- Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, Dawkins H, Lamont L, Roy AJ, Chamova T, et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36:395–402. doi: 10.1002/humu.22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacchiarelli D, Incitti T, Martone J, Cesana M, Cazzella V, Santini T, Sthandier O, Bozzoni I. miR-31 modulates dystrophin expression: new implications for Duchenne muscular dystrophy therapy. EMBO Rep. 2011;12:136–141. doi: 10.1038/embor.2010.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carsana A, Frisso G, Tremolaterra M, Lanzillo R, Vitale D, Santoro L, Salvatore F. Analysis of dystrophin gene deletions indicates that the hinge III region of the protein correlates with disease severity. Ann Hum Genet. 2005;69:253–9. doi: 10.1046/J.1469-1809.2005.00160.x. [DOI] [PubMed] [Google Scholar]
- Cradick TJ, Qiu P, Lee CM, Fine EJ, Bao G. COSMID: A web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol Ther Nucleic Acids. 2014;3:e214. doi: 10.1038/mtna.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekel-Naftali M, Aviram-Goldring A, Litmanovitch T, Shamash J, Reznik-Wolf H, Laevsky I, Amit M, Itskovitz-Eldor J, Yung Y, Hourvitz A, et al. Screening of human pluripotent stem cells using CGH and FISH reveals low-grade mosaic aneuploidy and a recurrent amplification of chromosome 1q. Eur J Hum Genet. 2012;20:1248–55. doi: 10.1038/ejhg.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echigoya Y, Aoki Y, Miskew B, Panesar D, Touznik A, Nagata T, Tanihata J, Nakamura A, Nagaraju K, Yokota T. Long-term efficacy of systemic multiexon skipping targeting dystrophin exons 45–55 with a cocktail of vivo-morpholinos in mdx52 mice. Mol Ther Nucleic Acids. 2015;4:e225. doi: 10.1038/mtna.2014.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan X, Mack DL, Moreno CM, Strande JL, Mathieu J, Shi Y, Markert CD, Wang Z, Liu G, Lawlor MW, et al. Dystrophin-deficient cardiomyocytes derived from human urine: new biologic reagents for drug discovery. Stem Cell Res. 2014;12:467–80. doi: 10.1016/j.scr.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, Harper HA, Robinson AS, Engelhardt JF, Brooks SV, et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002;8:253–61. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- Li HL, Fujimoto N, Sasakawa N, Shirai S, Ohkame T, Sakuma T, Tanaka M, Amano N, Watanabe A, Sakurai H, et al. Precise correction of the dystrophin gene in Duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Reports. 2015;4:143–54. doi: 10.1016/j.stemcr.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, Olson EN. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2015 doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN. Prevention of muscular dystrophy in mice by CRISPR/Cas9 – mediated editing of germline DNA. Science. 2014;345:1184–1188. doi: 10.1126/science.1254445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke A, Jockusch H. Extent of shock-induced membrane leakage in human and mouse myotubes depends on dystrophin. J Cell Sci. 1995;108:727–733. doi: 10.1242/jcs.108.2.727. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Yoshida K, Fukushima K, Ueda H, Urasawa N, Koyama J, Yazaki Y, Yazaki M, Sakai T, Haruta S, et al. Follow-up of three patients with a large in-frame deletion of exons 45–55 in the Duchenne muscular dystrophy (DMD) gene. J Clin Neurosci. 2008;15:757–63. doi: 10.1016/j.jocn.2006.12.012. [DOI] [PubMed] [Google Scholar]
- Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Rivera RMC, Madhavan S, Pan X, Ran FA, Yan WX, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2015 doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousterout DG, Kabadi AM, Thakore PI, Majoros WH, Reddy TE, Gersbach CA. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun. 2015;6:6244. doi: 10.1038/ncomms7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge T. Myoblast transplantation. Neuromuscul Disord. 2002;12:S3–6. doi: 10.1016/s0960-8966(02)00076-7. [DOI] [PubMed] [Google Scholar]
- Pearce JMS, Pennington RJT, Walton JN. Serum enzyme studies in muscle disease: Part III Serum creatine kinase activity in relatives of patients with the Duchenne type of muscular dystrophy. J Neurol Neurosurg Psychiatry. 1964;27:181–185. doi: 10.1136/jnnp.27.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2015 doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglia A, Petillo R, Ambrosio PD, Picillo E, Torella A, Orsini C, Ergoli M, Scutifero M, Passamano L, Palladino A, et al. Clinical features of patients with dystrophinopathy sharing the 45–55 exon deletion of DMD gene. Acta Myol. 2015;34:9–13. [PMC free article] [PubMed] [Google Scholar]
- Wojtal D, Kemaladewi DU, Malam Z, Abdullah S, Wong TWY, Hyatt E, Baghestani Z, Pereira S, Stavropoulos J, Mouly V, et al. Spell Checking Nature: Versatility of CRISPR/Cas9 for Developing Treatments for Inherited Disorders. Am J Hum Genet. 2016;98:90–101. doi: 10.1016/j.ajhg.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Park KH, Zhao L, Xu J, El Refaey M, Gao Y, Zhu H, Ma J, Han R. CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol Ther. 2015 doi: 10.1038/mt.2015.192. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.