

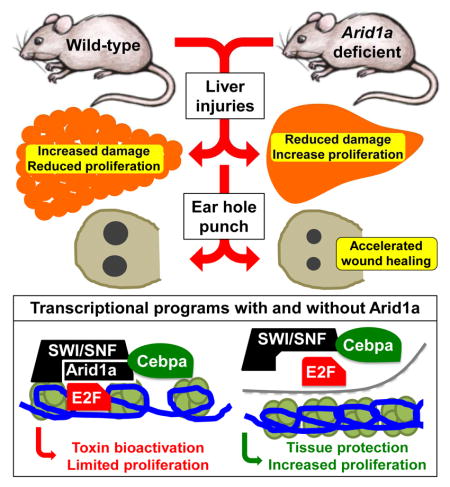
SUMMARY
Mammals have partially lost the extensive regenerative capabilities of some vertebrates, possibly as a result of chromatin-remodeling mechanisms that enforce terminal differentiation. Here, we show that deleting the SWI/SNF component Arid1a substantially improves mammalian regeneration. Arid1a expression is suppressed in regenerating tissues, and genetic deletion of Arid1a increases tissue repair following an array of injuries. Arid1a deficiency in the liver increases proliferation, reduces tissue damage and fibrosis, and improves organ function following surgical resection and chemical injuries. Hepatocyte-specific deletion is also sufficient to increase proliferation and regeneration without excessive overgrowth, and global Arid1a disruption potentiates soft tissue healing in the ear. We show Arid1a loss reprograms chromatin to restrict promoter access by transcription factors such as C/ebpα, which enforces differentiation, and E2F4, which suppresses cell cycle reentry. Thus, epigenetic reprogramming mediated by deletion of a single gene improves mammalian regeneration and suggests strategies to promote tissue repair after injury.
Graphical abstract
INTRODUCTION
While many species exhibit robust organ regeneration, this capacity has been partially lost in mammals (Poss, 2010; Tanaka and Reddien, 2011). Urodele amphibians and teleost fish regenerate in part through de-differentiation, by which regenerating cells revert to developmental states that are both plastic and proliferative in order to replenish lost body parts. It is unclear if epigenetic mechanisms enforcing tissue differentiation limit mammalian regeneration. Epigenetic reprograming has a role in induced pluripotent stem cell reprogramming (Onder et al., 2012; Stadtfeld et al., 2010), but whether altering the chromatin state might enhance regenerative capacity in vivo has not been addressed.
We hypothesized that the SWI/SNF ATP-dependent chromatin-remodeling complex, known to support terminal differentiation in multiple contexts (Hargreaves and Crabtree, 2011), might suppress regenerative capacity. SWI/SNF complexes contain a core ATPase (Brg1 or Brm), plus noncatalytic subunits with various DNA-binding and protein-binding domains that influence targeting and activity of complexes. The loss of Arid1a, which directly interacts with DNA, disrupts SWI/SNF targeting and nucleosome remodeling and results in aberrant gene regulation (Chandler et al., 2013; Dykhuizen et al., 2013; Kadoch et al., 2013). Arid1a is also thought to be a tumor suppressor, but this hypothesis awaits further functional interrogation (Hargreaves and Crabtree, 2011). Intriguingly, an Arid1a paralog is one of the most suppressed genes in the regenerative tissues of Brittle stars, close relatives of the famously regenerative Starfish (Purushothaman et al., 2015). Here, we also demonstrate that Arid1a is suppressed during physiologic regeneration and wound healing in mammals, and when deleted, potentiates greater regenerative capacity.
We focused on the liver because it is a regenerative tissue that permits extensive epigenetic analysis. The ability for mammalian livers to regenerate by compensatory cell proliferation and hypertrophy is distinct from epimorphic regeneration in fish and amphibians, but is still exceptional for its astounding potency (Michalopoulos, 2010; Miyaoka et al., 2012). Nevertheless, severe chronic insults can outstrip these healing abilities and lead to fibrosis and ultimately cirrhosis, the end-stage dysfunctional state of the liver that afflicts up to 1% of the population (Schuppan and Afdhal, 2008). Few therapies exist for cirrhosis in part because there is poor understanding of how tissue damage and liver regeneration can be modulated. We found that Arid1a loss lead to increased proliferation and cellular survival after injury, but also modestly reduced tissue maturation—a combination that improved regenerative capacity after and conferred resistance to diverse forms of tissue damage.
This work predicts that loss of SWI/SNF chromatin-remodeling components may represent adaptive events during tissue injury. Previously, Pajcini et al. showed that the transient inactivation of Rb and ARF was sufficient to coax post-mitotic myocytes into proliferation (Pajcini et al., 2010). Our study also implies that transient inactivation of a single gene would promote regeneration in a therapeutic context. We demonstrate that Arid1a is a potent regeneration suppressor in multiple tissues and that chromatin remodeling by SWI/SNF is a mechanism that limits regenerative capacity in vivo.
RESULTS
Arid1a loss results in improved liver regeneration after surgical and chemical injuries
Consistent with the idea that SWI/SNF is involved in the maintenance of the differentiated state, Arid1a was absent in neonatal liver but was expressed by ten days of age and into adulthood (Figure 1A). Within regenerating liver tissues, Arid1a mRNA and protein levels were suppressed (Figure 1B). To determine if further ablating Arid1a expression might enhance regeneration, liver-specific Arid1a knockout (LKO) mice were generated. Albumin-Cre excision of exon 8 of Arid1a causes a frameshift mutation and leads to nonsense-mediated decay (first reported in Gao et al., 2008) in bile duct epithelia and hepatocytes (Figure 1C). LKO mice were viable and healthy (Figure 1D) and possessed normal liver function (Figure S1A).
Figure 1. Arid1a loss results in improved liver regeneration after surgical resection.

(A) Arid1a expression in whole liver at different times measured by western blot.
(B) Arid1a mRNA and protein expression after partial hepatectomy, as measured by qPCR (n = 5 males per timepoint) and western blot.
(C) Arid1a protein levels in 4 week-old males Arid1aFl/Fl, Alb-Cre; Arid1a+/Fl and Alb-Cre; Arid1aFl/Fl livers, as measured by western blot.
(D) Alb-Cre; Arid1aFl/Fl mice were viable and healthy as compared to Arid1aFl/Fl mice.
(E) Liver/body weight ratios (%) at the time of hepatectomy (n = 5 WT and 5 LKO) and 40 hours after hepatectomy (n = 9 WT and 11 LKO).
(F) Hepatocyte proliferation prior to and 40 hours after partial hepatectomy in WT and LKO mice, as measured by Ki-67 immunostaining. Scale bar = 50μm.
(G) Ki-67 quantification of proliferation prior to and 40 hours after partial hepatectomy in WT and LKO livers (n = 3 WT and 6 LKO).
All data in this figure are represented as mean +/− SEM, * P<0.05, ** P< 0.01. “n” refers to biological replicates. See also Figures S1.
To assess Arid1a’s role in regeneration, we performed the partial hepatectomy, which involves surgically resecting two-thirds of the liver. WT and LKO mice had the same amount of residual liver mass after partial hepatectomy (Figure 1E). Forty hours after hepatectomy, LKO mice exhibited modestly reduced steatosis, a stress response seen after injury, but other histological differences were not observed (Figure 1F and Figure S1B). At this time point, LKO livers had more than two-fold greater Ki-67+ proliferating cells compared to WT (Figure 1F,G), resulting in greater regenerated liver mass by 40 hours after resection (Figure 1E). By 11 days after hepatectomy, both WT and LKO livers recovered nearly 100%, but not in excess of their original liver masses (Figure S1C). Outside of the immediate post-resection period, when active regeneration is occurring, there were very low levels of basal proliferation (Figure 1F and Figure S1D). Altogether, this data demonstrates that Arid1a loss is permissive of enhanced growth after injury but does not constitutively drive excessive growth.
We also modeled acute and chronic tissue damage, as seen in clinical liver failure, using chemicals. Centrilobular hepatocyte necrosis was induced with either carbon tetrachloride (CCl4) or acetaminophen. After one injection of CCl4, LKO livers appeared healthy, while WT livers were pale, suggesting widespread damage (Figure 2A). Twenty-four to 72 hours after CCl4 injection, LKO livers showed reduced apoptosis (Figure S2A), necrosis, and inflammatory infiltration (Figure 2B). LKO livers also had two-fold more proliferation 48 hours after injury (Figure 2C), indicating an enhanced regenerative response. Even after chronic CCl4 injury for 12 weeks, LKO mice continued to have reduced necrosis and inflammation (Figure S2B), less fibrosis (Figure S2C and Figure 2D), and improved liver function (Figure 2E). Acetaminophen overdose, the most common cause of acute liver failure in the U.S., showed similar results (Figure S2D,E). In addition, chronic exposure to the biliary toxin 5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) also revealed a regeneration suppressing role for Arid1a. DDC fed WT mice appeared ill, became jaundiced (Figure 2F), and lost >50% body weight (Figure 2G) while Arid1a LKO mice were healthy and maintained their body weight. LKO mice had reduced serum bilirubin levels (Figure 2H) and biliary inflammation (Figure S2F) while on DDC food. The absolute weights of WT liver remained unchanged, while LKO livers increased significantly (Figure 2I), reflecting a robust proliferative/regenerative response (Figure S2G) associated with extended survival (Figure 2J, p = 0.0325).
Figure 2. Arid1a knockout livers show greater regeneration after various chemical injuries.

(A) The gross appearance of livers before and after a single CCL4 injection.
(B) Liver histology 72 hours after one dose of CCL4 (H+E, Scale bar = 100μm).
(C) Ki-67 proliferation 48 hours after one dose of CCL4 in WT and LKO mice (Scale bar = 50μm).
(D) In mice treated with 12 weeks of chronic biweekly CCl4 injections, fibrosis was measured and quantified after trichrome staining (n = 5 and 5 images).
(E) Aspartate aminotransferase (AST) measured in chronic CCl4 treated mice (n = 7 WT and 8 LKO mice).
(F) Mice given six weeks of DDC food. Skin is jaundiced in WT but not in LKO mice.
(G) Body weight over a 120-day period (n = 7 WT and 9 LKO).
(H) Total bilirubin levels after two and six weeks of DDC feeding (n = 7 WT and 9 LKO).
(I) Representative livers from mice given six weeks of DDC food.
(J) Kaplan–Meier of WT and LKO mice on chronic DDC diet (p = 0.0325, n = 7 WT and 9 LKO).
All data in this figure are represented as mean +/− SEM, * P<0.05, ** P< 0.01. “n” refers to biological replicates. See also Figures S2.
Conditional Arid1a deletion promotes regeneration and overexpression impairs regeneration
To determine if post-natal, rather than developmental, loss of Arid1a after injury had an impact on tissue repair, and to determine the cell type responsible for enhanced tissue repair, we examined regeneration after conditional deletion. We first subjected mice to surgical and chemical injuries, deleted Arid1a with virally-delivered Cre recombinase, and finally examined tissue regeneration in each case. Fourteen days prior to partial hepatectomy, Arid1a was conditionally deleted in hepatocytes only using intravenously injected AAV8.TBG.PI.GFP.rBG or AAV8.TBG.PI.Cre.rBG (hereafter called AAV-GFP or AAV-Cre, Figure 3A and Figure S3A,B). Experimental and control groups underwent equivalent resections but Arid1a deficient livers recovered significantly more mass and displayed greater proliferation 40 hours later (Figure S3C,D and Figure 3B,C). After 12 weeks of CCl4 injury, mice were randomized to Adenovirus-GFP or -Cre to conditionally delete Arid1a (Figure 3D). Cre treated mice showed reduced fibrosis, showing that Arid1a loss after chronic injury was sufficient to enhance liver health (Figure 3E,F). The same experiment was performed with AAV-Cre, demonstrating that the anti-fibrotic effect is mediated by hepatocytes (Figure S3E,F). Similarly, Arid1aFl/Fl and Arid1a+l+ mice were fed DDC for 4 weeks, then DDC was stopped and all mice were injected with AAV-Cre to delete Arid1a specifically in hepatocytes (Figure 3G and Figure S3G). Arid1a deficient mice regained body and liver weights faster via greater proliferation, indicating greater recovery after liver injury (Figure 3H–K). These experiments separate degree of initial injury from amount of regeneration, and show that post-injury Arid1a inhibition in hepatocytes is sufficient to promote regeneration.
Figure 3. Conditional deletion of Arid1a leads to improved liver regeneration after surgical and chemical injuries.

(A) Schema for conditional knockout experiment prior to partial hepatectomy. Arid1aFl/Fl mice were randomized to AAV-GFP or AAV-Cre to delete Arid1a in liver tissues. Fourteen days later, partial hepatectomy was performed.
(B) Resected liver/body weight ratio and regenerated liver mass 40 hours after hepatectomy (n = 4 and 4).
(C) Hepatocyte proliferation as measured by Brdu.
(D) Schema for post-CCL4 injury conditional knockout experiment. Arid1aFl/Fl mice were injected with biweekly CCL4 for 12 weeks, then CCL4 was stopped and mice were randomized to either 1 × 109 PFU of Adenovirus-GFP or Adenovirus-Cre to delete Arid1a in liver tissues.
(E) Livers were examined for fibrosis using Sirius Red after 20 days (n = 3 Ad-GFP and 5 Ad-Cre treated females, Scale bar = 200μm).
(F) Quantification of Sirius Red staining area (8 images for each mice were averaged, n = 3 and 5 mice were analyzed).
(G) Schema for post-DDC injury conditional knockout experiment. Arid1a+/+ and Arid1aFl/Fl mice were fed with 4 weeks of DDC food. Once severe injury was achieved, DDC was stopped and all mice were injected with 5 × 1010 particles of AAV-Cre to conditionally delete Arid1a in hepatocytes. Mice were allowed to recover for 10 days, then livers were examined (in all following experiments, n = 4 Arid1a+/+ and 6 Arid1aFl/Fl males).
(H) % of original body weight gained over 10 day recovery period.
(I) Liver/body weight ratio (%) after 10 day recovery period.
(J) Hepatocyte proliferation as measured by BrdU positive cells per High-Power Field (HPF) after 10-day recovery period.
(K) BrdU immunostaining (Scale bar = 100μm). Green arrowheads point to BrdU positive hepatocytes, other spots are non-specific.
(L) WT and Arid1a LKO mice were injected with Adenovirus carrying either GFP (Ad-GFP) or ARID1A (Ad-ARID1A) under the control of a CMV promoter. IHC detecting both human ARID1A and mouse Arid1a shown here (Black arrowheads point to hepatocyte nuclei, Scale bar = 25μm).
(M) Serum AST measured 24 hours after one dose of CCL4 (n = 10, 9, 7, 8 mice).
(N) Proliferation per HPF as quantified by Ki-67 in liver sections 48 hours after CCL4 (5 HPFs averaged per mouse, n = 5 mice for each condition).
All data in this figure are represented as mean +/− SEM, * P<0.05, ** P< 0.01. “n” refers to biological replicates. All experiments were performed in triplicates. See also Figures S3.
To test if conditional Arid1a overexpression is sufficient to disrupt liver regeneration after chemical injury, we injected WT and LKO mice with Adenovirus carrying either GFP or human ARID1A under the control of a CMV promoter (Figure 3L and Figure S3H). 24 hours after one dose of CCl4, ALT trended up in ARID1A overexpressing cohorts, indicating small non-significant increases in hepatocyte injury (Figure 3M). After 48 hours, ARID1A overexpression suppressed proliferation in WT mice, and could completely rescue the proliferative phenotype in LKO mice (Figure S3I and Figure 3N), demonstrating that ARID1A is sufficient to block liver regeneration after chemical damage.
Arid1a loss also improves regeneration in ear hole wounds
Next, we asked if Arid1a regulates regeneration in tissues other than the liver. We examined outer ear wound healing, a complex tissue that fails to regenerate completely upon injury (Heber-Katz, 1999; McBrearty et al., 1998). Arid1a, Arid1b, and Arid2 mRNA expression decreased in wound tissues after ear hole punch (Figure 4A). To determine the functional relevance of Arid1a suppression in this context, we developed an inducible whole-body Arid1a knockout mouse based on the Ubiquitin-CreER system (hereafter designated Arid1a KO). Importantly, mice with tamoxifen-induced global Arid1a deletion were healthy, maintained their weight, and did not develop cancer even after 1 year (Figure S4A–D). Two weeks after successful Arid1a deletion (Figure 4B), 2mm full-thickness ear holes were punched in the outer ear. Arid1a KO mice showed improved wound healing over WT and heterozygous mice, indicated by smaller wound sizes at 14 days (Figure 4C,D). After five days of repair, increased numbers of proliferating cells were found in the wounded edge of KO ear holes (Figure 4E). These data support more general roles for Arid1a in tissue repair.
Figure 4. Arid1a loss improved regeneration in multiple tissues.

(A) Arid1a, Arid1b, and Arid2 mRNA levels before and after 2mm full-thickness ear hole punch, as measured by qPCR (n = 3 non-injured and 6 injured male ears).
(B) In 4 week-old male Arid1aFl/Fl Ubiquitin-CreER; Arid1a+/Fl, and Ubiquitin-CreER; Arid1aFl/Fl mice, we induced Arid1a deletion with Tamoxifen, then examined Arid1a expression in the ear by western blot 14 days later.
(C) 14 days after Tamoxifen, we performed 2mm ear hole punches (2 holes per ear, 4 per mouse). Relative ear hole wound size 14 days after hole punching is shown (n = 6 WT, 4 Heterozygous, and 3 KO mice).
(D) Ear holes 14 days after injury.
(E) Ki-67 immunostaining at the ear hole wound edge five days after hole punch (Scale bar = 200μm), quantification on the right (n = 5 and 5).
All data in this figure are represented as mean +/− SEM, * P<0.05, ** P< 0.01. See also Figures S4.
Arid1a-SWI/SNF complexes regulate tissue injury through chromatin remodeling
To examine the molecular mechanisms underlying the improved regenerative capacity of Arid1a deficient mice, we focused on the liver because it is one of the only tissues present in sufficient quantities to permit extensive epigenetic and biochemical analysis in the context of regeneration. We first asked if SWI/SNF complexes were still intact in the absence of Arid1a. Co-immunoprecipitation (Co-IP) experiments from a mouse carrying a knock-in V5 tag within the Arid1a locus (Arid1afV5/+ mice) confirmed that liver-specific SWI/SNF complexes contained Arid1a along with Brg1 or Brm (Figure S5A). Co-IP with a Brm antibody in either WT or LKO livers also identified other SWI/SNF components (Figure S5B). Also, size-exclusion chromatography showed that complexes remained intact in the absence of Arid1a (Figure S5C), but was replaced by Arid1b, and suggests altered SWI/SNF function or targeting rather than a structural loss of complexes.
To determine how Arid1a containing SWI/SNF complexes establish an epigenetic state that is repressive of regeneration, we obtained a genome-wide view of Arid1a targeting in the context of normal, uninjured liver. Chromatin Immunoprecipitation-sequencing (ChIP-seq) showed that V5-Arid1a binding was enriched at promoters near transcriptional start sites (TSSs) and the top three de novo identified Arid1a DNA binding motifs overlap with regulatory motifs recognized by C/ebpα, Hnf4a, and Foxa2, critical transcription factors that repress proliferation and maintain liver differentiation, activities that antagonize regeneration (Figure 5A). As expected, Gene Set Enrichment Analysis (GSEA) on RNA-sequencing (RNA-seq) data revealed suppressed C/ebpα, Hnf4a, and Forkhead transcriptional signatures in Arid1a deficient livers (Figure 5B). This correlated with an increase in proliferation/growth genes in the LKO setting (Figure 5C) and an increase in liver differentiation/function gene signatures in WT livers (Figure 5C and Figure S5D).
Figure 5. Arid1a-SWI/SNF complexes regulate tissue injury through chromatin remodeling.

(A) ChIP-seq in Arid1afV5/+ livers showed that V5-Arid1a binding is enriched at TSSs (top). Binding sites are associated with DNA sequence-specific motifs that overlap with C/ebpα, Hnf4a, and Foxa2 motifs (bottom).
(B) GSEA shows that Hnf4a, C/ebpα, Forkhead transcriptional target signatures are suppressed in LKO livers.
(C) Heat map of 1145 differentially expressed genes from RNA-seq (n = 2 and 2 livers, red is higher and blue is lower expression).
(D) 91% and 86% of V5-Arid1a binding peaks were also bound by Hnf4a and C/ebpα, respectively.
(E) Amongst the C/ebpα peaks identified in both WT and Arid1a LKO livers, 3952 showed significantly altered levels of C/ebpα binding and are depicted in this heat map (FDR <0.001, peaks are listed in Table S1). 95% of these sites were bound with less affinity in the LKO setting. Associated V5-Arid1a peaks are shown in the rightmost lane.
(F) ChIP-seq shows all H3K4me2 marks near peaks differentially bound by C/ebpα in WT and LKO livers.
(G) Cyp450 mRNA levels at baseline prior to injury in WT and LKO liver (n = 3 WT and 6 KO). This was performed in triplicates.
(H) Cyp2e1 immunostaining prior to and 24 hours after CCL4 injection. Scale bar = 100μm.
ImageJ quantification of Cyp2e1 positive area in an uninjured liver on the right. See also Figures S5 and S6 and Table S1.
Chromatin structure is a determinant of lineage-specific gene expression. We reasoned that Arid1a loss altered transcription factor access by remodeling nucleosome occupancy or position. Indeed, it is known that embryonic stem cells without Arid1a have altered nucleosome occupancy and phase shifting at TSSs (Lei et al., 2015). Moreover, we observed suppression of H3K4me2 (Figure S5E,F) marks in livers lacking Arid1a, evidence of a chromatin state associated with reduced transcriptional activation. In addition, GSEA showed that 28 transcription factor signatures were enriched in WT while only three were enriched in LKO livers, also suggesting a general restriction of transcriptional access after Arid1a loss. Because Hnf4a and C/ebpα are enforcers of terminal differentiation, a chromatin state that hinders their activity might be more permissive of proliferation when regeneration ensues.
We further interrogated Hnf4a and C/ebpα as examples of how transcriptional access might be altered. ChIP-seq experiments showed that Hnf4a (data from Davidson, 2014) bound to 91% and C/ebpα bound to 86% of V5-Arid1a binding peaks (Figure 5D, Table S1), showing that these factors and SWI/SNF co-occupy a common set of loci. We then performed C/ebpα ChIP-seq in Arid1a LKO livers to understand how C/ebpα binding changes when Arid1a is lost. 95% of peaks with altered binding between WT and LKO showed reductions in C/ebpα binding affinity in the context of Arid1a loss (FDR <0.001, Figure 5E, Table S1, and examples shown in Figure S6A,B). ChIP-qPCR confirmed that both Brm and C/ebpα bind known targets with reduced efficiency before and after injury in Arid1a deficient contexts (Figure S6C–H), suggesting that Arid1a loss establishes a chromatin state less permissive of transcription factor access in a Brm and SWI/SNF specific manner. Especially in differentially bound C/ebpα targets, this restricted access correlates with reduced H3K4me2 marks, indicating reduced transcriptional activation (Figure 5F). Since C/ebpα and SWI/SNF directly interact in the liver (Figure S5A,C), it is possible that faithful targeting is dependent on this interaction. Overall, this suggests that Arid1a loss, through modulation of transcription, results in a more plastic state that is primed for regeneration.
Next, we assessed some direct targets of Hnf4a and C/ebpα that are particularly relevant to regeneration and injury. For example, Cytochrome P450 genes (Cyp450s) comprise a superfamily of monooxygenase enzymes that bioactivate liver toxins such as ethanol, CCl4, and acetaminophen to promote hepatotoxicity. Arid1a binds to multiple Cyp450 loci, and in Arid1a LKO livers, C/ebpα binding to Cyp450 loci was quantitatively reduced (Figure S6B) and 8/19 Cyp450 mRNAs were downregulated (Figure 5G). Twenty-four hours after CCl4 injection, peri-central vein hepatocytes with high Cyp2e1 underwent necrosis in WT but not in LKO liver lobules, where Cyp2e1 levels are lower (Figure 5H).
Loss of Arid1a increases post-injury proliferation by restraining repressive E2Fs
C/ebpα and Hnf4a not only maintain differentiation through transcriptional activation, but are also known to transcriptionally repress proliferation under normal, non-injury conditions (Harris et al., 2001; Iakova et al., 2003; Wang et al., 2002; Wang et al., 2001). To assess additional mechanisms that could account for increases in proliferation after injury, we performed qPCR on cell cycle genes before and after partial hepatectomy. We found that Ccna2, Ccnb1, Ccnb2, and Ccnd1 were elevated in LKO livers, especially during regenerative timepoints (Figure 6A). RNA-seq and subsequent GSEA were then performed on regenerating liver tissues 40 hours after partial hepatectomy. This confirmed increases in cell cycle and mitosis gene signatures, but also identified E2F transcriptional targets, which are known to regulate the cell cycle and DNA synthesis (Figure 6B). Of the 195 curated E2F target genes (GSEA HALLMARK_E2F_TARGETS), 56 of 57 differentially expressed genes are upregulated in regenerating LKO livers (Figure 6C). Since Arid1a and E2F4 directly interact in SWI/SNF complexes (Figure S7) (Nagl et al., 2007), we further examined the DNA-binding activities of E2f4, a repressive E2f factor, in a genome-wide manner. E2f4 ChIP-seq experiments performed after partial hepatectomy revealed that this repressive factor bound target gene promoters less efficiently in the absence of Arid1a (Figure 6D). Reduced E2f4 binding on the promoters of the 57 differentially expressed E2F target genes reflect an overall impairment of binding when Arid1a is absent (Figure 6E and Table S1). Correspondingly, these promoters showed increased H3k4me2 and H3k27ac marks (Figure 6F), indicating de-repression of these E2f target genes.
Figure 6. Loss of Arid1a increases post-injury proliferation by restraining repressive E2Fs.

(A) Cell cycle gene expression before and after partial hepatectomy (n = 3, 6).
(B) GSEA shows that cell cycle, mitosis, and E2F target gene expression signatures are increased in LKO livers 40 hours after partial hepatectomy. Nominal enrichment score (NES), nominal p-value, and False discovery rate (FDR) q-value are shown in each GSEA plot.
(C) 57 E2F target genes that are differentially expressed in Arid1a WT and LKO livers 40 hours after partial hepatectomy (n = 3 WT and 2 LKO livers, red is higher and blue is lower expression). Data is from RNA-seq.
(D) Amongst the E2f4 ChIP-seq peaks identified in both WT and Arid1a LKO livers post-hepatectomy, 200 peaks with the most significantly altered levels of E2f4 binding based on FDR are depicted in this heat map (peaks are listed in Table S1). 162/200 (82%) of these sites were bound with less affinity in the LKO setting.
(E) E2f4 ChIP-seq peaks near the TSSs (+/−500bp) of the 57 differentially expressed E2F target genes in WT and Arid1a LKO livers post-hepatectomy (peaks are listed in Table S1).
(F) Representative upregulated E2F target genes whose promoters show reduced E2f4 binding, increased H3K27ac, and increased H3K4me2 marks. Y-axis scales are the same between WT and KO.
All data in this figure are represented as mean +/− SEM, * P<0.05, ** P< 0.01. See also Figures S7 and Table S1.
These data support a model in which Arid1a-SWI/SNF normally remodel chromatin to promote transcriptional access by a cohort of transcription factors that support terminal differentiation and antagonize the proliferative state (Figure 7). In support of this, mice with a mutant C/ebpα protein that have reduced binding to p300 transcriptional complexes possess enhanced regenerative phenotypes in the liver, reminiscent of Arid1a deficiency (Jin et al., 2014).
Figure 7. Mechanistic model of Arid1a’s role in liver regeneration.

The Arid1a-SWI/SNF complex remodels chromatin to promote transcriptional access by a cohort of transcription factors that support terminal differentiation and antagonize the proliferative state. Arid1a loss results in a more plastic state that is primed for regeneration through modulation of transcription.
DISCUSSION
It is remarkable that regenerative capacity in mammals is genetically malleable. Some mus musculus strains and closely related species have abilities reminiscent of newts and zebrafish (McBrearty et al., 1998; Seifert et al., 2012). Also, age related changes in regenerative capacity suggest that functional declines could be reversed (Bucher et al., 1964; Shyh-Chang et al., 2013; Sinha et al., 2014). Interestingly, aged livers in mice (24 month old) are known to accumulate a multi-protein C/ebpα-Brm-HDAC1 complex that epigenetically suppresses genes required for regeneration (Iakova et al., 2003; Inayoshi et al., 2006). Furthermore, systemic factors in young mice can enhance liver regeneration in old mice, a phenomenon that correlates with quantitative suppression of C/ebpα-Brm complexes in old livers (Conboy et al., 2005). Though these biochemical studies implicated SWI/SNF in regeneration, none until now have thoroughly demonstrated the importance of SWI/SNF complex members in animal models of regeneration.
Our work shows that Arid1a is physiologically suppressed during liver regeneration and wound healing, and that complete ablation leads to improved regeneration. Our mechanistic model shows that without Arid1a, altered SWI/SNF nucleosome remodeling reshapes the chromatin landscape. This quantitatively restricts transcriptional access for factors such as C/ebpα before injury and E2f4 after injury, likely through direct interactions with SWI/SNF (Figure 7), but potentially also involves restriction of other transcription factors such as Hnf4a, which ordinarily maintains liver maturation and represses cell cycle re-entry. The loss of Arid1a establishes a chromatin state that does not abolish, but instead globally attenuates the lineage-enforcing transcriptional activities of C/ebpα and the anti-proliferative activities of E2f4. These modest alterations result in increased cellular plasticity that allows for improved regeneration upon injury, but does not impair organ function under normal conditions (Figure S1A,B). A form of this SWI/SNF suppression and global chromatin remodeling probably also occurs during physiological regeneration, where Arid1a is transiently repressed (Figure 1B).
There are likely to be functional differences or even defects in Arid1a deficient livers that we have not yet detected. Many CYPs other than Cyp2e1 are suppressed in these livers, and some of these CYPs are needed to detoxify endogenous or exogenous metabolites. If the ultimate trade-off for increased regenerative capacity was compromised tumor suppression, then evolutionary or therapeutic strategies to reduce Arid1a might not be beneficial. Since ARID1A loss-of-function mutations are commonly found in human cancer (Chan-On et al., 2013; Fujimoto et al., 2012; Guichard et al., 2012; Huang et al., 2012; Jiao et al., 2013), we tested if Arid1a is always a bona fide tumor suppressor. Neither global nor liver-specific Arid1a deficient mice spontaneously developed tumors by one year of age (Figure S4D,E), consistent with some reports showing that Arid1a loss alone is not tumorigenic (Chandler et al., 2015; Guan et al., 2014), and in some cases, are even protective against cancer (Helming et al., 2014; Zhai et al., 2015). Much additional investigation will be needed to evaluate impact of Arid1a loss in tumorigenesis. The roles of Arid1a are likely heavily dependent on oncogenomic and environmental contexts. It should be noted that Arid1a suppression is a transient event during normal regeneration, and any future therapeutic interventions would likely involve transient suppression of Arid1a rather than permanent, chronic suppression.
It has long been theorized that epigenetic conformation could be permissive of greater regenerative capacity, but this has mainly been explored in the context of stem cell reprogramming and plants. We contribute to this body of knowledge by demonstrating that chromatin plasticity is a mechanism by which mammalian regenerative capacity can be controlled in vivo. It is striking that deletion of a single chromatin-remodeling gene can appreciably improve regeneration. This work suggests that transient epigenetic reprogramming via therapeutic Arid1a inhibition during severe injuries could enhance regeneration without compromising tissue function.
EXPERIMENTAL PROCEDURES
Animals
All mice were handled in accordance with the guidelines of the Institutional Animal Care and Use Committee at UTSW. In Arid1a floxed mice, induced deletions between the two loxP sites produce cells lacking exon 8 of Arid1a (see Supplemental Information for a list of primers), which will create a frameshift mutation and induce nonsense-mediated decay in the resulting transcript. For a full description, please see Gao et al., 2008. These mice were a mix of C57/B6 and 129. Baf250afV5/+ or Arid1afV5/+ mice were generated by inserting a DNA fragment encoding V5 tag in-frame with the endogenous coding region of Baf250a. Homozygous Arid1afV5/fV5 mice were viable, demonstrating that the V5 fusion protein functions normally. Please see Wu et al., 2014 for more details. All experiments were done in an age and sex controlled fashion unless otherwise noted in the figure legends.
Partial hepatectomy
Two thirds of the liver was surgically excised as previously described (Mitchell and Willenbring, 2008).
Chemical injury experiments
CCl4 is diluted 1:10 in corn oil (Sigma), and administered IP at a dose of 0.5 ml/kg of mouse as described previously (Beer et al., 2008). Mice were injected and monitored twice per week. For the DDC experiments, mice were fed a diet (TestDiet) containing 0.1% DDC (Sigma).
Virus experiments
5 × 1010 genomic particles of AAV8.TBG.PI.Cre.rBG or AAV-Cre (University of Pennsylvania Vector Core) in 100μL was retro-orbitally injected to mediate Cre excision. AAV8.TBG.PI.GFP.rBG or AAV-GFP was used as a control. 1 × 109 PFU of Adenovirus-Cre or GFP (Ad5CMVCre or Ad5CMVeGFP, University of Iowa Viral Vector Core) in 100μL was retro-orbitally injected to mediate Cre excision. For overexpression experiments, full-length human full length ARID1A (NM_006015.4) was cloned into an Adenovirus serotype 5 construct containing a CMV promoter. Vector BioLabs generated Ad-GFP and Ad-ARID1A, and these viruses were injected at a dose 1×109 viral PFU/mouse.
RNA Extraction and RT-qPCR
Total RNA was isolated using Trizol reagent (Invitrogen). For RT-qPCR of mRNAs, cDNA synthesis was performed with 1ug of total RNA using miScript II Reverse Transcription Kit (Qiagen). See Supplemental Information for primers used in these experiments. Expression was measured using the ΔΔCt method as previously described (Zhu et al., 2010).
Western Blot Assay
Mouse liver tissues were ground with a pestle and lysed in T-PER Tissue Protein Extraction Reagent (Thermo Scientific Pierce). Western blots were performed in the standard fashion. The following antibodies were used: Anti-mouse Arid1a (Santa Cruz, sc-32761), Anti-mouse Arid1b (Santa Cruz, sc32762x), β-Actin (Cell signaling, #4970), Anti-rabbit Brm (Abcam, ab15597), Anti-mouse Arid1b (Abcam, ab57461), Anti-tubulin (Cell signaling #3873), Anti-rabbit IgG, HRP-linked Antibody (Cell signaling, #7074) and Anti-mouse IgG, HRP-linked Antibody (Cell signaling, #7076).
Co-immunoprecipitation
Co-IP was performed as described in Wu et. al. 2014 (Wu et al., 2014). Antibodies against Brm (Abcam, ab15597), Brg1 (Santa Cruz, sc10768), V5 (Abcam ab15828), Arid1a (Sigma HPA005456), Arid1b (Santa Cruz, sc32762x), Baf155 (Santa Cruz, sc9746), C/ebpα (Santa Cruz, sc166258), and E2f4 (Milipore 05-312) were used for western blot analysis and/or Co-IP.
Size-exclusion chromatography
Liver nuclear extracts were isolated using CelLytic NuCLEAR Extraction Kit (Sigma) following the manufacturer’s instructions. 1mg of nuclear extract was fractionated by a Superdex 200 10/300 GL (GE Healthcare Life Sciences) size-exclusion column in gel filtration buffer (50mM Tris-HCl, 100mM NaCl, 5mM β-mercaptoethanol, pH 7.5). Fractions of 500μl were collected and 30μl of each fraction was analyzed by western blot with corresponding antibodies.
RNA-sequencing
RNA from 2 WT and 2 LKO non-injured livers, and 3 and 3 post-partial hepatectomy livers were purified with the Qiagen miRNeasy Mini kit. Illumina or Nugen libraries were made with these RNAs. These indexed libraries were multiplexed in a single flow cell lane and received 75 base single-end sequencing on an Illumina HiSeq2500 using 50SR SBS v3 reagents at the UTSW McDermott Center Sequencing Core, or a NextSeq500 using the High Output kit v2 (75 cycles) at the CRI Sequencing Facility. Sequence reads were aligned to mouse genome version mm9 using tophat and differential expression analysis was performed using edgeR. For the heatmap in Figure 6C, one of the LKO samples was excluded for this particular analysis due to quality control problems in the sequencing.
Gene set enrichment analysis
GSEA was used to identify gene sets and pathways associated with gene expression data. The input for GSEA was a list of significantly differentially expressed genes between two biological states as determined by edgeR. Each gene in the list was weighted by its log fold change in expression. Please see http://software.broadinstitute.org/gsea/index.jsp. Gene Set Enrichment Analysis (version 2.2.0) is an analytic tool for relating gene expression data to gene sets to identify unifying biological themes (Mootha et al., 2003; Subramanian et al., 2005).
Chromatin immunoprecipitation (ChIP-qPCR and ChIP-seq)
Hnf4a ChIP-seq data from p12 livers was obtained from the GEO database (GSM1390711). Arid1a, C/ebpα, H3K4me2, H3K27ac, and E2f4 Chip-seq data can be accessed at the GEO database (GSE65167 and GSE76935). ChIP was performed as described (Wu et al., 2014). Antibodies against Brm (Abcam, ab15597), C/ebpα (Santa Cruz, sc-61) and V5 (Abcam ab15828), H3K4me2 (Millipore, 07-030). H3K27ac (Abcam, ab4729), and E2F4 C-20 (Santa Cruz, sc-866x) were used. See Supplemental Information for primers used in ChIP-qPCR. ChIP-seq libraries were prepared using KAPA HTP Library Preparation Kit or NEBNext® ChIP-Seq Library Prep Master Mix Set for Illumina kit, and sequenced on either an Illumina HiSeq2500 or NextSeq500. Single-end reads of 51 bp were generated. After mapping reads to mouse genome (mm10) by bowtie2 (version 2.2.3 (Langmead and Salzberg, 2012)) with parameter “--sensitive”, we perform filtering by (1) removing alignments with mapping quality less than 10, (2) removing duplicate reads identified by Picard Mark Duplicates (version 1.92, http://broadinstitute.github.io/picard). Then enriched regions (peaks) were identified using MACS2 (Zhang et al., 2008) (version 2.0.10.20131216), with a q-value cut-off of 0.05. For the occupancy-based analysis (Venn diagram results), common peaks of replicates were used. Peak regions were annotated by HOMER (Heinz et al., 2010). For motif discovery, we first ranked peak regions by q-value for each sample, and then supply the top 40% ones to HOMER. To compare C/ebpα binding affinity difference between WT and LKO, We also focused on the top 40% peak regions from the two. Differentially bound regions were identified using DiffBind package (Ross-Innes et al., 2012) (http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf).
Histology, Immunohistochemistry, and Immunofluorescence
Tissue samples were fixed in 10% neutral buffered formalin or 4% paraformaldehyde (PFA) and embedded in paraffin. In some cases, frozen sections were made. Immunohistochemistry was performed as previously described (Zhu et al., 2010). Two hours before euthanasia, 50 mg/kg of BrdU (Sigma) was administered intraperitoneally. Primary antibodies used: Arid1a (Cell signaling, #12354s), Ki-67 (Abcam, ab15580), Cyp2e1 (Abcam, ab28146), and BrdU (BD 555627). Fibrosis detection was performed with Sirius red staining kit from Thermo Fisher Scientific (#NC0249910) or trichrome (UTSW Pathology Core). Apoptosis detection was performed with a Tunel kit from Roche (#11684817910). Detection was performed with the Elite ABC Kit and DAB Substrate (Vector Laboratories), followed by Hematoxylin Solution counterstaining (Sigma).
Liver function tests
Blood samples (~ 50 ul) were taken retro-orbitally in heparinized tubes and allowed to clot at 4°C. Tests were run by the UTSW Molecular Genetics Core.
Tamoxifen
Mice were given 500uL of 20mg/mL Tamoxifen by oral gavage for two consecutive days to induce whole body Cre excision mediated by Ubc-CreERT.
Ear hole punch assay
Two 2-mm-diameter holes were punched near the center of each outer ear by using a clinical biopsy punch (Roboz, Gaithersburg, MD).
Statistical analysis
The data in most figure panels reflect multiple experiments performed on different days using mice derived from different litters. Variation is always indicated using standard error presented as mean ± SEM. Two-tailed Student’s t-tests (two-sample equal variance) were used to test the significance of differences between two groups. Statistical significance is displayed as p < 0.05 (*) or p < 0.01 (**) unless specified otherwise. In all experiments, no mice were excluded form analysis after the experiment was initiated. In the conditional Arid1a deletion experiments from Figure 3, mice were randomized between control and Cre viral treatments so as to ensure equal initial injury levels between groups. Image analysis for the quantification of cell proliferation and fibrosis were blinded.
Supplementary Material
Highlights.
Arid1a loss results in improved regeneration after diverse liver injuries
Arid1a overexpression impairs liver proliferation and regeneration
Arid1a loss also improves tissue repair after ear hole punch
Arid1a loss remodels chromatin, altering transcriptional access by C/EBPα and E2F4
Acknowledgments
We thank M. Buszczak and N. Shyh-Chang for critical input and advice; J. Shelton for histology. X.S. was supported by the Hamon Center for Regenerative Science and Medicine at UTSW. Y.Z. was supported by a SDG grant (AHA), a Basil O’Connor Starter Scholar Research Award (March of Dimes Foundation), and by NIH (1R01HL107376 & 1K02HL103597). W.Z. was supported by NIH 7R01HL109054. We thank the McDermott Center Sequencing Core, the UTSW Bioinformatics Core, and the Children’s Research Institute Sequencing Core for sequencing and analysis. C.X. is partially supported by NIH grant UL1TR001105. H.Z. was supported by the Pollack Foundation, a NIH K08 grant (1K08CA157727), a NIH/NCI R01 grant (1R01CA190525), a Burroughs Welcome Career Medical Award, a CPRIT New Investigator Grant (R1209), and a CPRIT Early Translation Grant (DP150077).
Footnotes
ACCESSION NUMBERS
The accession number for the RNA-seq data reported in this paper is GEO: GSE76926. The accession number for the ChIP-seq data reported in this paper is GEO: GSE65167 and GSE76935.
AUTHOR CONTRIBUTIONS
X.S., J.C.C., and H.Z. designed and performed the experiments and wrote the manuscript. M.K., H.L., C.X., T.M., M.S., X.L., and J.X. generated and analyzed the bioinformatic data. L.W. performed partial hepatectomies and mouse husbandry. C.C., L.L., S.Z., L.N., R.A.J.S., S.W., and I.N. assisted with experiments and mouse husbandry. M.W. and Y.Z. provided the Arid1afV5/+ mice. Y.C.K performed the size exclusion chromatography. Z.W. provided the Arid1a Floxed mice and helped write the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beer S, Komatsubara K, Bellovin DI, Kurobe M, Sylvester K, Felsher DW. Hepatotoxin-induced changes in the adult murine liver promote MYC-induced tumorigenesis. PloS one. 2008;3:e2493. doi: 10.1371/journal.pone.0002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucher NL, Swaffield MN, Ditroia JF. The Influence of Age Upon the Incorporation of Thymidine-2-C14 into the DNA of Regenerating Rat Liver. Cancer research. 1964;24:509–512. [PubMed] [Google Scholar]
- Chan-On W, Nairismagi ML, Ong CK, Lim WK, Dima S, Pairojkul C, Lim KH, McPherson JR, Cutcutache I, Heng HL, et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nature genetics. 2013;45:1474–1478. doi: 10.1038/ng.2806. [DOI] [PubMed] [Google Scholar]
- Chandler RL, Brennan J, Schisler JC, Serber D, Patterson C, Magnuson T. ARID1a-DNA interactions are required for promoter occupancy by SWI/SNF. Molecular and cellular biology. 2013;33:265–280. doi: 10.1128/MCB.01008-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RL, Damrauer JS, Raab JR, Schisler JC, Wilkerson MD, Didion JP, Starmer J, Serber D, Yee D, Xiong J, et al. Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nature communications. 2015;6:6118. doi: 10.1038/ncomms7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–764. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- Davidson I. Hnf4a.Taf4.wt.ChIP.Seq 2014 [Google Scholar]
- Dykhuizen EC, Hargreaves DC, Miller EL, Cui K, Korshunov A, Kool M, Pfister S, Cho YJ, Zhao K, Crabtree GR. BAF complexes facilitate decatenation of DNA by topoisomerase IIalpha. Nature. 2013;497:624–627. doi: 10.1038/nature12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nature genetics. 2012;44:760–764. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- Gao X, Tate P, Hu P, Tjian R, Skarnes WC, Wang Z. ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:6656–6661. doi: 10.1073/pnas.0801802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan B, Rahmanto YS, Wu RC, Wang Y, Wang Z, Wang TL, Shih Ie M. Roles of deletion of Arid1a, a tumor suppressor, in mouse ovarian tumorigenesis. Journal of the National Cancer Institute. 2014;106 doi: 10.1093/jnci/dju146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nature genetics. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell research. 2011;21:396–420. doi: 10.1038/cr.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TE, Albrecht JH, Nakanishi M, Darlington GJ. CCAAT/enhancer-binding protein-alpha cooperates with p21 to inhibit cyclin-dependent kinase-2 activity and induces growth arrest independent of DNA binding. The Journal of biological chemistry. 2001;276:29200–29209. doi: 10.1074/jbc.M011587200. [DOI] [PubMed] [Google Scholar]
- Heber-Katz E. The regenerating mouse ear. Seminars in cell & developmental biology. 1999;10:415–419. doi: 10.1006/scdb.1999.0328. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, Kim Y, Kryukov GV, Ghandi M, Aguirre AJ, et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nature medicine. 2014;20:251–254. doi: 10.1038/nm.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Deng Q, Wang Q, Li KY, Dai JH, Li N, Zhu ZD, Zhou B, Liu XY, Liu RF, et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nature genetics. 2012;44:1117–1121. doi: 10.1038/ng.2391. [DOI] [PubMed] [Google Scholar]
- Iakova P, Awad SS, Timchenko NA. Aging reduces proliferative capacities of liver by switching pathways of C/EBPalpha growth arrest. Cell. 2003;113:495–506. doi: 10.1016/s0092-8674(03)00318-0. [DOI] [PubMed] [Google Scholar]
- Inayoshi Y, Miyake K, Machida Y, Kaneoka H, Terajima M, Dohda T, Takahashi M, Iijima S. Mammalian chromatin remodeling complex SWI/SNF is essential for enhanced expression of the albumin gene during liver development. Journal of biochemistry. 2006;139:177–188. doi: 10.1093/jb/mvj015. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Pawlik TM, Anders RA, Selaru FM, Streppel MM, Lucas DJ, Niknafs N, Guthrie VB, Maitra A, Argani P, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nature genetics. 2013;45:1470–1473. doi: 10.1038/ng.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Hong IH, Lewis K, Iakova P, Breaux M, Jiang Y, Sullivan E, Jawanmardi N, Timchenko L, Timchenko NA. Cooperation of C/EBP family proteins and chromatin remodeling proteins is essential for termination of liver regeneration. Hepatology. 2014 doi: 10.1002/hep.27295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nature genetics. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei I, West J, Yan Z, Gao X, Fang P, Dennis JH, Gnatovskiy L, Wang W, Kingston RE, Wang Z. BAF250a regulates nucleosome occupancy and histone modifications in priming embryonic stem cell differentiation. The Journal of biological chemistry. 2015 doi: 10.1074/jbc.M115.637389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBrearty BA, Clark LD, Zhang XM, Blankenhorn EP, Heber-Katz E. Genetic analysis of a mammalian wound-healing trait. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:11792–11797. doi: 10.1073/pnas.95.20.11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalopoulos GK. Liver regeneration after partial hepatectomy: critical analysis of mechanistic dilemmas. The American journal of pathology. 2010;176:2–13. doi: 10.2353/ajpath.2010.090675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell C, Willenbring H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nature protocols. 2008;3:1167–1170. doi: 10.1038/nprot.2008.80. [DOI] [PubMed] [Google Scholar]
- Miyaoka Y, Ebato K, Kato H, Arakawa S, Shimizu S, Miyajima A. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Current biology: CB. 2012;22:1166–1175. doi: 10.1016/j.cub.2012.05.016. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature genetics. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Nagl NG, Jr, Wang X, Patsialou A, Van Scoy M, Moran E. Distinct mammalian SWI/SNF chromatin remodeling complexes with opposing roles in cell-cycle control. The EMBO journal. 2007;26:752–763. doi: 10.1038/sj.emboj.7601541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder TT, Kara N, Cherry A, Sinha AU, Zhu N, Bernt KM, Cahan P, Marcarci BO, Unternaehrer J, Gupta PB, et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature. 2012;483:598–602. doi: 10.1038/nature10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajcini KV, Corbel SY, Sage J, Pomerantz JH, Blau HM. Transient inactivation of Rb and ARF yields regenerative cells from postmitotic mammalian muscle. Cell stem cell. 2010;7:198–213. doi: 10.1016/j.stem.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss KD. Advances in understanding tissue regenerative capacity and mechanisms in animals. Nature reviews Genetics. 2010;11:710–722. doi: 10.1038/nrg2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushothaman S, Saxena S, Meghah V, Swamy CV, Ortega-Martinez O, Dupont S, Idris M. Transcriptomic and proteomic analyses of Amphiura filiformis arm tissue-undergoing regeneration. Journal of proteomics. 2015;112:113–124. doi: 10.1016/j.jprot.2014.08.011. [DOI] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389–393. doi: 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371:838–851. doi: 10.1016/S0140-6736(08)60383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert AW, Kiama SG, Seifert MG, Goheen JR, Palmer TM, Maden M. Skin shedding and tissue regeneration in African spiny mice (Acomys) Nature. 2012;489:561–565. doi: 10.1038/nature11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyh-Chang N, Zhu H, Yvanka de Soysa T, Shinoda G, Seligson MT, Tsanov KM, Nguyen L, Asara JM, Cantley LC, Daley GQ. Lin28 enhances tissue repair by reprogramming cellular metabolism. Cell. 2013;155:778–792. doi: 10.1016/j.cell.2013.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha M, Jang YC, Oh J, Khong D, Wu EY, Manohar R, Miller C, Regalado SG, Loffredo FS, Pancoast JR, et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. 2014;344:649–652. doi: 10.1126/science.1251152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Apostolou E, Akutsu H, Fukuda A, Follett P, Natesan S, Kono T, Shioda T, Hochedlinger K. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature. 2010;465:175–181. doi: 10.1038/nature09017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka EM, Reddien PW. The cellular basis for animal regeneration. Developmental cell. 2011;21:172–185. doi: 10.1016/j.devcel.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Goode T, Iakova P, Albrecht JH, Timchenko NA. C/EBPalpha triggers proteasome-dependent degradation of cdk4 during growth arrest. The EMBO journal. 2002;21:930–941. doi: 10.1093/emboj/21.5.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Iakova P, Wilde M, Welm A, Goode T, Roesler WJ, Timchenko NA. C/EBPalpha arrests cell proliferation through direct inhibition of Cdk2 and Cdk4. Molecular cell. 2001;8:817–828. doi: 10.1016/s1097-2765(01)00366-5. [DOI] [PubMed] [Google Scholar]
- Wu M, Peng S, Yang J, Tu Z, Cai X, Cai CL, Wang Z, Zhao Y. Baf250a orchestrates an epigenetic pathway to repress the Nkx2.5-directed contractile cardiomyocyte program in the sinoatrial node. Cell research. 2014;24:1201–1213. doi: 10.1038/cr.2014.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y, Kuick R, Tipton C, Wu R, Sessine M, Wang Z, Baker SJ, Fearon ER, Cho KR. Arid1a inactivation in an Apc and Pten-defective mouse ovarian cancer model enhances epithelial differentiation and prolongs survival. The Journal of pathology. 2015 doi: 10.1002/path.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome biology. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Shah S, Shyh-Chang N, Shinoda G, Einhorn WS, Viswanathan SR, Takeuchi A, Grasemann C, Rinn JL, Lopez MF, et al. Lin28a transgenic mice manifest size and puberty phenotypes identified in human genetic association studies. Nature genetics. 2010;42:626–630. doi: 10.1038/ng.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.