

Abstract
Control of plasma glucose level is essential to organismal survival. Sustained inflammation has been implicated in control of glucose homeostasis in cases of infection, obesity, and type 2 diabetes; however, the precise role of inflammation in these complex disease states remains poorly understood. Here, we find that sustained inflammation results in elevated plasma glucose due to increased hepatic glucose production. We find that sustained inflammation suppresses CYP7A1, leading to accumulation of intermediate metabolites at the branch point of the mevalonate pathway. This results in prenylation of RHOC, which is concomitantly induced by inflammatory cytokines. Subsequent activation of RHO-associated protein kinase results in elevated plasma glucose. These findings uncover an unexpected mechanism by which sustained inflammation alters glucose homeostasis.
Introduction
The plasma glucose level is homeostatically maintained to ensure its continuous supply to target tissues while avoiding the toxic effects of hyperglycemia. Multiple physiologic states are associated with altered plasma glucose levels: pregnancy, infection, and persistent stress all are accompanied by hyperglycemia (Andrews and Walker, 1999; Barbour et al., 2007; Butte, 2000; McGuinness, 2005), while extended food restriction is associated with reduced plasma glucose level (Redman and Ravussin, 2009). The mechanism and rationale for these observed changes in plasma glucose level remain largely unknown, although the alteration in glucose concentration likely functions as a means to redistribute nutrients in an effort to adapt to changing physiologic priorities (Kotas and Medzhitov, 2015).
Inflammation is a protective response to infection and injury, but it operates at the expense of normal tissue function (Okin and Medzhitov, 2012). In particular, it is increasingly appreciated that inflammation can affect systemic glucose homeostasis. Critically ill patients in the intensive care unit are known to develop hyperglycemia (Mizock, 2001), and the degree of hyperglycemia correlates with increased morbidity and mortality (Falciglia et al., 2009). Additionally, patients with rheumatoid arthritis are known to have a 50% increased risk of developing type 2 diabetes mellitus (T2DM), and this risk is reduced by treatment with anti-inflammatory therapies (Solomon et al., 2010; 2011). Furthermore, work over the past two decades has indicated that low-grade inflammation plays an important role in the altered glucose homeostasis seen in obesity (Donath and Shoelson, 2011; Gregor and Hotamisligil, 2011; Olefsky and Glass, 2010).
Several models have been developed to examine the effects of inflammation on glucose homeostasis. Continuous intraperitoneal (i.p.) infusion of lipopolysaccharide (LPS) in mice results in fasting hyperglycemia due to increased hepatic glucose production (Cani et al., 2007). Additionally, chronic activation of IκB kinase (IKK) in the livers of mice results in a T2DM-like phenotype (Cai et al., 2005) while inactivation of hepatic IKK in obese mice prevents hepatic insulin resistance (Arkan et al., 2005). Although these studies suggest the liver is a target for inflammatory control of glucose homeostasis, the mechanisms involved remain incompletely understood.
There are many possible mechanisms whereby inflammation could affect glucose homeostasis. For example, inflammation could directly control glucose production in the liver. However, inflammatory signals rapidly suppress the rate-limiting enzymes of gluconeogenesis (Feingold et al., 2012; McGuinness, 2005). Alternatively, inflammatory signals, such as TNF, IL-1ß, and IL-6, could suppress glucose utilization by muscle, liver, and fat by suppressing insulin signaling in these organs, possibly through activation of inflammatory kinases, including IKK and JNK (Donath and Shoelson, 2011; Gregor and Hotamisligil, 2011; Johnson and Olefsky, 2013; Odegaard and Chawla, 2013). However, kinase activity is rapid and tightly controlled by multiple negative feedback mechanisms, suggesting that physiologic alterations of metabolism induced by acute inflammation are likely to be distinct from pathologic alterations associated with sustained inflammation. Thus, the mechanisms through which sustained inflammation regulates glucose homeostasis remain poorly understood, despite the well appreciated importance of sustained inflammation in a variety of pathological conditions (Nathan and Ding, 2010).
Here, we investigated the effect of sustained inflammation on systemic glucose homeostasis. We found that sustained inflammation resulted in fasting hyperglycemia and hyperinsulinemia due to increased hepatic glucose production. Sustained inflammation suppressed CYP7A1, the rate-limiting enzyme of the bile acid biosynthesis pathway, and this suppression was required for the development of hyperglycemia. Suppression of CYP7A1 led to accumulation of intermediate metabolites of the mevalonate pathway, resulting in prenylation and stabilization of RHOC, a small GTPase induced by inflammation in hepatocytes, with subsequent activation of RHO-associated protein kinase (ROCK), leading to fasting hyperglycemia. Finally, we demonstrate that inhibition of ROCK in obese mice can improve glucose homeostasis. Together, our study provides insight into mechanisms that control glucose homeostasis under conditions of sustained inflammation.
Results
Sustained Inflammation Alters Glucose Homeostasis
To study the impact of sustained inflammation on glucose homeostasis, we devised a model whereby mice were given daily i.p. injections of low-dose LPS for 7 days (7D-LPS), or PBS as a control. 7D-LPS treatment induced fasting hyperglycemia and hyperinsulinemia (Figure 1A) without any effect on body weight (Figure S1A). Glucose tolerance test (GTT) revealed that 7D-LPS mice displayed mild glucose intolerance (Figures 1B and S1B). The shapes of the GTT curves from 7D-LPS and PBS treated mice were similar, suggesting that the observed difference was likely due to the initial difference in fasting glucose levels. Indeed, 7D-LPS treated mice retained elevated plasma glucose levels 120 minutes after glucose injection (Figure 1B) despite similar glucose-induced insulin secretion (Figure S1C). Additionally, insulin tolerance test (ITT) of 7D-LPS mice demonstrated mildly impaired insulin tolerance (Figures 1C and S1D). 7D-LPS mice had a similar initial response to insulin injection, with a nadir identical to PBS mice; however, serum glucose levels of 7D-LPS mice returned to an elevated level over the course of the ITT and remained elevated for up to 6 hours after insulin injection (Figure 1C). These data suggest that the main effect of 7D-LPS treatment is the establishment of an elevated fasting glucose level that is stable in the face of perturbation.
Figure 1. Sustained inflammation induces fasting hyperglycemia.

(A) Fasting plasma glucose and insulin levels in mice given LPS for 7 days (7D-LPS) or mice receiving PBS as control (n=10/condition).
(B) Glucose tolerance test (GTT) in PBS or 7D-LPS treated mice (n=10/condition).
(C) Insulin tolerance test (ITT) in PBS or 7D-LPS treated mice (n=5/condition).
(D) Hyperinsulinemic-euglycemic clamp was performed on PBS or 7D-LPS treated mice and endogenous glucose production and glucose uptake were measured (n=4-7/condition).
(E) Insulin stimulated phosphorylated-AKT on serine 473 (pS473-AKT) was evaluated in tissues from overnight fasted PBS or 7D-LPS mice. Bar plots represent densitometry analysis of pS473-AKT/AKT (n=3-4/condition). Data is presented as mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 by Student’s T-Test. # p < 0.05, ## p < 0.01 by two-way analysis of variance (2-ANOVA). See also Figure S1.
Plasma glucose levels are regulated primarily by hepatic glucose production (HGP) and glucose uptake into skeletal muscle and adipose tissue. Two possibilities exist to explain the fasting hyperglycemia seen in 7D-LPS mice: increased glucose production or decreased glucose uptake by peripheral tissues. To evaluate these possibilities, we performed a hyperinsulinemic-euglycemic clamp, and found that 7D-LPS mice demonstrated increased endogenous glucose production without any difference in peripheral glucose uptake (Figure 1D). Alongside elevated glucose production, we found that 7D-LPS mice had elevated ‘homeostatic model assessment’ (HOMA) (Figure S1E), consistent with prior work (Berglund et al., 2008). Studies of insulin signaling in muscle, adipose tissue, and liver revealed that 7D-LPS treated mice had reduced insulin-stimulated AKT phosphorylation in the liver and adipose tissue while the muscle was largely unaffected (Figure 1E). The preservation of muscle insulin signaling is consistent with our hyperinsulinemic-euglycemic clamp data indicating no difference in glucose uptake during clamp conditions (Figure 1D), as skeletal muscle is the primary initial consumer of glucose in response to an insulin bolus (Shulman, 2014). Taken together, these data suggest that the primary effects of 7D-LPS are on the liver, resulting in increased HGP and fasting hyperglycemia, and reduced hepatic insulin signaling.
Sustained Inflammation Alters Liver Size and Reduces Triglyceride Content
We next sought to characterize the effects of 7D-LPS on the liver. 7D-LPS treated mice had increased liver weight as a percentage of body weight (Figure S2A). Furthermore, liver weight was highly correlated with fasting glucose levels (Figure S2B); we did not observe alteration in hepatic tissue architecture (Figure S2C). Prior work suggested that elevated liver triglycerides can be one cause of fasting hyperglycemia and reduced hepatic insulin signaling (Perry et al., 2014). Surprisingly, livers from 7D-LPS mice had reduced triglyceride content without alteration in non-esterified fatty acids (Figure S2D). Additionally, 7D-LPS treatment resulted in a small but significant increase in liver cholesterol levels (Figure S2D). Increased expression of gluconeogenesis genes could explain the fasting hyperglycemia seen with 7D-LPS treatment; however, 7D-LPS suppressed PEPCK expression without altering other genes involved in HGP (Figure S2E). These data indicate that previously described mechanisms cannot fully explain the increased HGP seen after 7D-LPS treatment.
Establishment of Ex Vivo Model System Using Primary Hepatocytes
To investigate the mechanism driving increased HGP, we employed primary hepatocytes as an ex vivo model system, using reduced insulin signaling as a readout for the effects of sustained inflammation on hepatocyte function. The parenchymal effects of LPS are mediated largely through tissue resident macrophage production of inflammatory cytokines, such as IL-1β and TNF. 7D-LPS treatment induced the expression of IL1B and TNF in the liver (Figure S2F); we selected TNF to model sustained inflammation ex vivo. Prolonged stimulation with TNF for 2 days (2D-TNF) was required to reduce insulin-stimulated AKT phosphorylation (Figures 2A & S2G). AKT phosphorylates multiple downstream proteins that are responsible for carrying out the cellular effects of insulin signaling (Pearce et al., 2010). 2D-TNF treatment suppressed phosphorylation of two AKT target proteins, forkhead box O1 (FOXO1) and glycogen synthase kinase 3ß (GSK3ß) (Figure 2B). These results indicate that 2D-TNF treatment of primary hepatocytes ex vivo recapitulates the reduced insulin signaling seen after 7D-LPS treatment.
Figure 2. Sustained inflammation suppresses primary hepatocyte insulin signaling.

(A) Insulin-stimulated pS473-AKT in primary hepatocytes after 2 days of 2ng/mL TNF (2D-TNF) treatment. Bar plots represent densitometry analysis of pS473-AKT/AKT (n=3/condition).
(B) Insulin-stimulated phosphorylation of targets downstream of AKT after 2D-TNF treatment.
(C) Phosphorylation of insulin receptor substrate 1 (IRS-1) on serine 307 after 1 hour or 2 days TNF treatment.
(D) Insulin-stimulated pS473-AKT in hepatocytes isolated from mice with targeted replacement of IRS-1 serine 307 with alanine (A/A) or control (S/S) after 2D-TNF treatment. Data is presented as mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 by Student’s T-Test. See also Figure S2.
Prior work has indicated that TNF alters insulin signaling in adipocytes through JNK mediated phosphorylation of insulin receptor substrate 1 (IRS-1) on serine 307 (S307) (Gregor and Hotamisligil, 2011). The initial work demonstrating importance of IRS-1 serine phosphorylation showed that this only explained altered insulin signaling in the adipose tissue and skeletal muscle, but had no effect in the liver (Hotamisligil et al., 1996). In agreement with these earlier studies, treatment with TNF did not result in increased IRS-1 S307 phosphorylation (IRS-1 pS307), even during times where JNK was activated (Figures 2C & S2H). Furthermore, in primary hepatocytes isolated from mice carrying a point mutation in IRS-1 where S307 is mutated to alanine (Copps et al., 2010), we observed no difference in the 2D-TNF suppression of insulin-induced AKT phosphorylation (Figure 2D). These data indicate that consistent with prior work, IRS-1 pS307 does not explain the effect of 2D-TNF on hepatocyte insulin signaling.
Suppression of CYP7A1 is Required for the Effects of Sustained Inflammation on Fasting Glucose Level
The requirement for sustained inflammation suggested that a transcriptional change might drive the observed effects on the liver. TNF regulates transcription through the NF-κB and cJun pathways (Chen, 2002). Inhibition of NF-κB transcriptional activity using adenoviral over-expression of the NF-κB super repressor (dominant negative IκBα, with serine 32 and 36 mutated to alanine) (Ghosh et al., 1998) prevented 2D-TNF suppression of insulin signaling (Figure 3A). These data indicate that TNF suppresses insulin signaling through NF-κB-mediated transcription.
Figure 3. Suppression of CYP7A1 is required for induction of fasting hyperglycemia by sustained inflammation.

(A) Insulin-stimulated pS473-AKT in primary hepatocytes transduced with control adenovirus [Ad-CMV] or adenovirus expressing dominant negative IκBα [Ad-IκBα(DN)] after treatment with 2D-TNF. All cells were cultured in the presence of 10µM zVAD-FMK, which did not change the effect of 2D-TNF treatment on insulin-stimulated pS473-AKT in the absence of adenoviral transduction (data not shown).
(B) Heat map displaying the fold change in mRNA expression, as measured by microarray, after either 2 hours or 2 days of TNF treatment. Expression levels were normalized to untreated controls (not shown) and displayed as relative values (log2).
(C) Pathway analysis on the subset of genes suppressed >1.5-fold after 2D-TNF treatment, but unchanged after 2 hours of TNF treatment.
(D) CYP7A1 expression in primary hepatocytes after 2D-TNF treatment (n=3/condition).
(E) CYP7A1 expression in primary hepatocytes treated as described in Figure 3A (n=3/condition).
(F) CYP7A1 expression in primary hepatocytes treated with 2D-TNF +/− 10µM TO901317 (n=3/condition).
(G) Insulin-stimulated pS473-AKT and CYP7A1 levels in primary hepatocytes treated with 2D-TNF and 10µM TO901317 or DMSO control.
(H) Insulin-stimulated pS473-AKT in primary hepatocytes isolated from mice with a liver specific transgenic expression of CYP7A1 (CYP7A1-TG) or wild type (WT) mice after 2D-TNF treatment.
(I) Fasting glucose levels in WT and CYP7A1-TG animals after treatment with 7D-LPS or PBS (n=3-5/condition). Data is presented as mean ± SEM. * p < 0.05,
** p < 0.01, *** p < 0.001, **** p < 0.0001 by Student’s T-Test. See also Figure S3 and Table S1.
Based upon these results, we performed whole-genome gene expression analysis on primary hepatocytes after short (2 hour) and long (2 day) treatment with TNF. Use of these two time points allowed us to isolate changes in transcription that were unique to 2D-TNF suppression of insulin signaling. 2D-TNF altered expression of 870 genes by > 1.5 fold (Figure 3B, full data available in Table S1). To identify patterns of gene expression changes, we subjected the genes induced or suppressed exclusively after 2D-TNF treatment to pathway analysis using the NIH’s DAVID software (Huang et al., 2009a; 2009b). We found numerous pathways enriched in genes suppressed after 2D-TNF treatment (Figure 3C). No pathways were enrichment in genes induced after 2D-TNF treatment (Figure S3A). We were particularly interested in the observed enrichment of the bile acid (BA) biosynthetic pathway given the multitude of research indicating that BAs play a role in glucose homeostasis (Calkin and Tontonoz, 2012; de Aguiar Vallim et al., 2013). BA synthesis is controlled by the rate limiting enzyme, CYP7A1 (Thomas et al., 2008). 2D-TNF treatment suppressed expression of CYP7A1 and this suppression depended upon NF-κB transcriptional activity (Figures 3D-E). 2D-TNF treatment of primary hepatocytes suppressed CYP7A1 protein levels while 7D-LPS treatment suppressed liver CYP7A1 protein levels (Figures S3B-C). These data indicate that sustained inflammation suppresses CYP7A1.
To test whether suppression of CYP7A1 was important for the effects of sustained inflammation on glucose homeostasis, we utilized two methods to augment CYP7A1 expression. First, Liver X Receptor (LXR) is known to induce CYP7A1 transcription (Calkin and Tontonoz, 2012). Treatment of primary hepatocytes with LXR agonist TO901317 blunted the 2D-TNF effect on CYP7A1 expression and improved insulin signaling (Figures 3F-G). Second, we used mice with hepatocyte specific transgenic over-expression of CYP7A1 (CYP7A1-TG) (Miyake et al., 2001). Primary hepatocytes isolated from CYP7A1-TG animals were refractory to effects of 2D-TNF on insulin signaling (Figure 3H). Additionally, CYP7A1-TG animals were resistant to 7D-LPS induction of hyperglycemia, hyperinsulinemia, and HOMA (Figures 3I & S3D). Collectively, these data indicate that suppression of CYP7A1 is required for 7D-LPS induction of fasting hyperglycemia.
Suppression of CYP7A1 Does Not Regulate Hepatic Glucose Production Through Altered Transcription
The observation that suppression of CYP7A1 was required for 7D-LPS induction of fasting hyperglycemia prompted us to further investigate the mechanism by which this occurred. Given the importance of CYP7A1 in regulation of BA synthesis, and BA ability to regulate glucose metabolism, we envisioned three possible mechanisms that could explain how suppression of CYP7A1 is required for 7D-LPS induction of fasting hyperglycemia and how CYP7A1-TG mice are protected. First, suppression of CYP7A1 could increase plasma BAs. Second, CYP7A1 suppression could alter interstitial BA concentrations. Third, it is possible that overexpression of CYP7A1 alters the effects of 7D-LPS or preferentially suppresses machinery responsible for HGP.
We began by measuring plasma BA concentrations, and found no difference between PBS and 7D-LPS treated animals (Figure 4A). To address options 2 and 3, we performed RNA sequencing (RNA-seq) on livers of wild type (WT) and CYP7A1-TG animals treated with 7D-LPS or PBS. RNA-seq revealed that over 3900 genes were differentially expressed in at least one condition (Figures 4B & S4A, full data available in Table S2). BAs mediate transcriptional effects through FXR, and evaluation of FXR target genes revealed generalized suppression by 7D-LPS regardless of genotype (Figures 4C-D). Furthermore, there did not appear to be substantial induction of FXR target genes in CYP7A1-TG livers (Figures 4C-D). These data suggest that local BA concentrations are likely unaffected by 7D-LPS treatment or CYP7A1 overexpression.
Figure 4. Regulation of CYP7A1 does not have transcriptional effects on FXR target genes or 7D-LPS responsiveness.

(A) Serum total bile acids in mice treated with 7D-LPS or PBS (n=5/condition).
(B) Heat map displaying fold change of mRNA expression, as measured using RNA sequencing, in whole livers from WT and CYP7A1-TG animals treated with 7D-LPS or PBS. Expression levels were normalized to WT, Untreated livers (not shown), and fold change plotted as log2 values.
(C) Heatmap of FXR target genes from RNA sequencing experiment. Values represent log10 of FPKM+1.
(D-E) Expression analysis of genes in the FXR pathway (SHP, APOE, ABCB11) and TNF in WT and CYP7A1-TG animals treated with 7D-LPS or PBS (n=4-6/condition).
(F) Heatmap representing sample relatedness as calculated by Jensen-Shannon divergence. Lower numbers mean samples are similar, or more related. Data is presented as mean ± SEM. * p < 0.05, ** p < 0.01 by Student’s T-Test. See also Figure S4 and Table S2.
To evaluate whether CYP7A1 overexpression altered response to 7D-LPS, we first asked whether cytokine expression was altered in CYP7A1-TG animals, and found no difference in induction of TNF (Figure 4E). Bioinformatics analysis demonstrated that genes in 7D-LPS treated animals clustered together, suggesting that this was primary driver of gene expression change (Figures 4F & S4A). Further analysis revealed that CYP7A1 overexpression affected transcript levels of genes involved in sterol and cholesterol metabolism, and that this effect was driven by a core group of 123 genes that were similarly regulated when comparing between genotypes in either the PBS or 7D-LPS treated conditions (Figures S4B-D). Additionally, evaluation of 7D-LPS gene induction demonstrated altered transcript levels of genes involved in the immune response, and this was driven by a group of 1505 genes with shared regulation regardless of genotype (Figure S4E). We did not observe enrichment for genes involved in gluconeogenesis (Figures S4B-E).
To further evaluate whether CYP7A1-TG expression altered 7D-LPS responsiveness, we performed weighted gene correlation network analysis (WGCNA) of the differentially expressed genes (Langfelder and Horvath, 2008). WGCNA identified 7 distinct gene modules whose expression is highly correlated across conditions (Figure S4F). The yellow and green modules were highly correlated with CYP7A1-TG animals treated with PBS, and gene ontology (GO) analysis revealed that these genes clustered in the same pathways as the 123 differentially expressed genes above (Figure S4G). The brown module was enriched in 7D-LPS treated CYP7A1-TG animals, however GO analysis of these genes did not reveal significant enrichment in any pathway (Figure S4G). Thus, these data suggest that transcriptional effects are insufficient to explain the mechanism by which altering CYP7A1 expression regulates fasting glycemia.
Sustained Inflammation Alters Hepatic Mevalonate Pathway Flux
The absence of a transcriptional explanation for the effects of CYP7A1 prompted us to explore alternative mechanisms. CYP7A1 acts to convert cholesterol into 7-α-hydroxycholesterol (Figure 5A). This first step is also at the end of the cholesterol biosynthetic pathway, a branch of the mevalonate pathway (Goldstein and Brown, 1990). Therefore, we examined whether 2D-TNF suppression of CYP7A1 led to accumulation of cellular cholesterol. However, 2D-TNF treatment had no effect on cellular cholesterol levels (Figure S5A), likely due to compensatory regulatory mechanisms that prevent cholesterol elevation to avoid toxicity (Tabas, 2002).
Figure 5. Accumulation of intermediate metabolites in the mevalonate pathway mediate inflammatory control of glucose homeostasis.

(A) Diagram of the mevalonate pathway with molecules (black) and enzymes (green).
(B) Farnesyl pyrophosphate, FPP, and geranylgeranyl pyrophosphate, GGPP, levels in primary hepatocytes treated with 2D-TNF (n=6/condition).
(C) FPP and GGPP levels in WT or CYP7A1-TG primary hepatocytes treated with 2D-TNF (n=6/condition).
(D) FPP and GGPP levels in primary hepatocytes treated with 2D-TNF and 10µM atorvastatin hemicalcium or DMSO (n=6/condition).
(E) Insulin stimulated pS473-AKT in primary hepatocytes treated with 2D-TNF and 10µM atorvastatin or DMSO.
(F) Fasting glucose levels in mice treated with PBS or 7D-LPS and 10mg/kg/day atorvastatin or vehicle control (n=8-10/condition). Data is presented as mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 by Student’s T-Test. See also Figure S5.
Cholesterol is one of many products of the mevalonate pathway, along with ubiquinone, sterols, and isoprenoids (Goldstein and Brown, 1990). Thus, the absence of cholesterol accumulation suggested that suppression of CYP7A1 led to accumulation of other metabolites. The last common metabolite in the synthesis of all products of the mevalonate pathway is farnesyl pyrophosphate (FPP). We reasoned that the suppression of cholesterol conversion into BA caused a general reduction in flux through that arm of the pathway, and that substrate was being redistributed to other branches of the mevalonate pathway. Given that cholesterol is the major product of the mevalonate pathway in the liver, we thus reasoned that FPP would accumulate in response to 2D-TNF treatment. To test this hypothesis, we developed a liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay to analyze isoprenoid composition of primary hepatocyte lipid extracts. LC-MS/MS analysis of lipid extracts from primary hepatocytes demonstrated that 2D-TNF treatment led to a 40% increase in FPP levels (Figure 5B). This was not due to increased synthesis, as we did not detect accumulation of the upstream metabolite dimethylallyl pyrophosphate, nor did we detect changes in expression of HMG-CoA reductase or HMG-CoA synthase 1, two proximal enzymes responsible for initiating sterol synthesis (Figures S5B-C). The accumulation of FPP prompted us to investigate what other metabolites accumulated under inflammatory conditions. FPP can undergo a single enzymatic modification by geranylgeranyl diphosphate synthase to form geranylgeranyl pyrophosphate (GGPP), another lipid species used for isoprenylation (Liao and Laufs, 2005). We found that 2D-TNF treatment substantially increased GGPP levels (Figure 5B). To determine whether accumulation of FPP and GGPP depended upon suppression of CYP7A1, we analyzed lipid extracts from WT and CYP7A1-TG primary hepatocytes. Overexpression of CYP7A1 prevented 2D-TNF induction of FPP and GGPP concentrations (Figure 5C). These data indicate that 2D-TNF treatment induces accumulation of FPP and GGPP and that this depends on suppression of CYP7A1.
Based upon these observations, we set out to determine whether accumulation of FPP and GGPP are required for the effects of sustained inflammation on glucose homeostasis. Both FPP and GGPP are biosynthetic products of the mevalonate pathway, which can be inhibited using the statin drugs. Co-treatment with atorvastatin hemicalcium prevented the 2D-TNF mediated accumulation of FPP and GGPP (Figure 5D). Furthermore, statin treatment was sufficient to prevent 2D-TNF mediated suppression of insulin signaling (Figure 5E). Finally, co-administration of atorvastatin alongside 7D-LPS prevented the development of fasting hyperglycemia, hyperinsulinemia and elevation in HOMA, without effecting LPS induction of TNF (Figures 5F & S5D-E). Taken together, these data suggest that accumulation of FPP and GGPP mediate the effects of 7D-LPS on fasting glucose levels.
Inflammation-Induced Stabilization of RHOC and Activation of RHO-Associated Protein Kinase Regulate Glucose Homeostasis
Although FPP is a common precursor for all biosynthetic products of the mevalonate pathway, the accumulation of GGPP suggested that increased substrate availability could lead to increased isoprenylation, and that this may be one way by which accumulation of FPP and GGPP regulated glucose metabolism. Isoprenylation is a post-translational modification of proteins whereby either FPP or GGPP are covalently added to the C-terminus of a protein containing a −CaaX consensus sequence by either farnesyltransferase (FTase) or geranylgeranyltransferase (GGTase) (Magee and Seabra, 2005; Zhang and Casey, 1996). We found that inhibition of both enzymes with prenyltransferase inhibitors (PTIs; LB42708, FTase, and GGTI-2133, GGTase) blunted the 2D-TNF mediated reduction of insulin signaling (Figure 6A).
Figure 6. RHO-associated protein kinase as a mediator of sustained inflammatory control of glucose homeostasis.

(A) Insulin-stimulated pS473-AKT in primary hepatocytes treated with 2D-TNF and prenyltransferase inhibitors, PTIs, (1µM LB42708 and 10µM GGTI-2133).
(B) Expression of genes with known isoprenylation motifs in primary hepatocytes after 2D-TNF treatment (n=3/condition).
(C) RHOC expression in primary hepatocytes treated as described in Figure 3A (n=3/condition).
(D) RHOC and GTP-bound RHOC protein levels in primary hepatocytes treated with 2D-TNF and PTIs or DMSO.
(E) Myosin light chain 2 (MLC2) phosphorylation on serine 19 (pS19-MLC2) in primary hepatocytes treated with 2D-TNF and PTIs or DMSO.
(F) Insulin-stimulated pS473-AKT in primary hepatocytes treated with 2D-TNF and one of two Rho-associated protein kinase (ROCK) inhibitors, H-1152 (1µM) or Y-27632 (40µM).
(G) Fasting glucose levels in mice treated with PBS or 7D-LPS and 30mg/kg/day Y-27632 or vehicle control (n=4-5/condition).
(H-I) GTT and AUC of mice fed a HFD for 12 weeks followed by daily injections with 30mg/kg/day Y-27632 or PBS for 2 weeks. During the 2 weeks of injections, mice were maintained on HFD (n=5/condition).
(J-K) ITT and AUC of mice treated as in (H-I), (n=6/condition). Data is presented as mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 by Student’s T-Test. # p < 0.05, ## p < 0.01 by 2-ANOVA. See also Figures S6 & S7.
Isoprenylation of target proteins typically confers two properties: protein stability and subcellular localization (Zhang and Casey, 1996). At baseline, most constitutively expressed isoprenylated proteins exist exclusively in the prenylated form, suggesting that the increase in prenylation precursors alone would not be sufficient to regulate insulin signaling, unless the target protein is either inducible or has a short half-life. Because TNF-induced NF-κB was required to alter insulin signaling in hepatocytes, we hypothesized that 2D-TNF induces expression of a protein that is a target for isoprenylation and thus functions to sense the increased FPP and GGPP abundance. Such protein should have three properties: (1) it must be induced by 2D-TNF treatment, (2) its function and/or stability must depend upon isoprenylation, and (3) it is likely to be a relatively poor substrate for FPP and GGPP transferases, so that its prenylation only occurred when the abundance of FPP and GGPP is increased. Interrogation of 2D-TNF-induced gene expression data revealed 17 differentially expressed proteins containing –CaaX motifs (Figure S6A). RT-qPCR confirmation of genes whose expression changed > 1.5 fold identified RHOC as the only target with reproducible induction by 2D-TNF treatment (Figure 6B). Furthermore, induction of RHOC required NF-κB (Figure 6C), suggesting that it was part of the transcriptional program important for inhibiting insulin signaling (requirement 1 above). Treatment of primary hepatocytes with 2D-TNF and PTIs prevented induction of RHOC without altering RHOC transcript levels (Figures 6D & S6B), indicating that isoprenylation is required for stability of RHOC during inflammation (requirement 2 above). The activity of RHOC is controlled through GTP loading, with RHOC-GTP active and RHOC-GDP inactive (Etienne-Manneville and Hall, 2002). 2D-TNF treatment increased the active, RHOC-GTP bound form, and this increase was prevented by co-treatment with PTIs (Figure 6D). Finally, the −CaaX motif of RHOC (−CPIL) has proline at position +2, which is conserved among RHOC in mammals and birds, and unusual for −CaaX motifs in that small hydrophobic amino acids are preferred at that position (Maurer-Stroh and Eisenhaber, 2005). The presence of proline at position +2 of the −CaaX motif, deviating from the consensus sequence, might make RHOC a suboptimal substrate for FTase and GGTase, and therefore it may be only isoprenylated when FPP and GGPP are abundant, as would occur when CYP7A1 is suppressed (requirement 3 above). Collectively, these data suggest that isoprenylation may be one mechanism through which sustained inflammation regulates hepatic insulin signaling and that this effect may be mediated through stabilization of RHOC.
RHO proteins are known to activate RHO-associated protein kinase (ROCK), and one unique aspect of RHOC, compared to RHOA and RHOB, is its high affinity for activating ROCK (Huang et al., 2013). ROCK coordinates multiple cellular functions and has been implicated in the regulation of insulin signaling, although its role in the liver has not been explored (Huang et al., 2013). 2D-TNF treatment increased ROCK activity in an isoprenylation dependent manner, as measured by phosphorylation of ROCK target myosin light chain 2 (MLC2) (Figure 6E). To test whether inhibition of ROCK would prevent 2D-TNF effects on hepatocyte insulin signaling, we co-treated cells with two separate ROCK inhibitors, H-1152 and Y-27632. Inhibition of ROCK prevented 2D-TNF suppression of insulin signaling (Figure 6F), suggesting that this could be one pathway through which inflammation regulates insulin signaling. 7D-LPS treatment of mice had similar effects, increasing RHOC levels and ROCK activity in the liver (Figure S6C-E). Co-treatment of mice with 7D-LPS and Y-27632 prevented the elevation in fasting glucose (Figure 6G), indicating that this pathway may be important in the inflammatory control of glucose homeostasis.
Inhibition of ROCK Ameliorated Glucose Homeostasis in High Fat Fed Mice
The observation that ROCK plays a role in the regulation of glucose metabolism by 7D-LPS suggested to us that this could represent a pharmacologic target to treat the dysglycemia seen in obesity. To test the role of ROCK in regulation of metabolic function during obesity, we fed mice a high fat diet (HFD) for 12 weeks, after which we treated them with ROCK inhibitor Y-27632 for 2 weeks (Figure S7A). Treatment with Y-27632 did not change body weight, fasting glucose, fasting insulin, or HOMA (Figures S7B-C). However, Y-27632 treatment resulted in improved glucose and insulin tolerance (Figures 6H-K) in HFD mice without any effect on mice fed normal chow (Figures S7D-G). These data suggest that inhibition of ROCK can improve glucose homeostasis in already obese mice.
Discussion
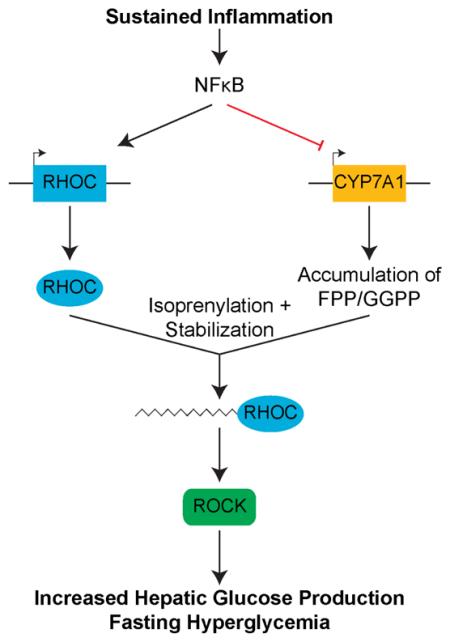
Although it is widely appreciated that inflammation plays an important role in the control of plasma glucose levels, direct insight into the rationale for such control, especially in the context of chronic or sustained inflammation, has remained unclear. Inflammation’s unique ability to alter target tissue function as a means to restore homeostasis suggested that this capability could explain inflammation’s effect on glucose metabolism. Here, we provide evidence that suppression of a tissue function unique to the liver is responsible for inflammation’s ability to regulate fasting glycemia (Figure 7A). Specifically, our data indicate that sustained inflammation suppresses the rate-limiting enzyme of bile acid (BA) biosynthesis, CYP7A1, and that this suppression results in elevated fasting glucose through regulation of the hepatic mevalonate pathway.
Figure 7. Model of how sustained inflammation affects glucose homeostasis.

(A) Model depicting the effects of TNF treatment on hepatocyte function. TNF activates NFκB, suppressing CYP7A1, inducing accumulation of FPP and GGPP. Accumulated FPP and GGPP stabilize TNF induced RHOC, which then goes on to activate ROCK, inducing fasting hyperglycemia in mice.
(B) The model above can be represented as a coherent feed-forward loop whereby both induction of RHOC and accumulation of FPP and GGPP work together to regulate fasting glucose levels.
Previous work has indicated that regulation of BAs plays a role in control of glucose homeostasis (Thomas et al., 2008). Studies suggesting an important role for FXR (Prawitt et al., 2011) and serum BA signaling through TGR5 (Thomas et al., 2009) have demonstrated how BAs can function as signaling molecules to achieve cell extrinsic regulation of glucose homeostasis. Our finding that suppression of CYP7A1 is required for hyperglycemia seen with 7D-LPS treatment (Figure 3I) supports existing data by demonstrating that direct control of the BA biosynthetic machinery effects glucose homeostasis.
The requirement of suppression of CYP7A1 led us to ask whether it functioned in a cell extrinsic or cell intrinsic fashion. The primary cell extrinsic effect of CYP7A1 suppression is on BA production. 7D-LPS treatment did not alter circulating BA concentration (Figure 4A). Circulating BAs could remain stable secondary to alterations in enterohepatic recirculation, and it is possible that hepatic interstitial BA concentration is increased by 7D-LPS treatment, resulting in activation of FXR. However, we did not find an effect of 7D-LPS treatment on FXR target genes (Figures 4C-D), suggesting that interstitial BA concentrations were unchanged. The cell intrinsic effect of CYP7A1 suppression would be on flux through the mevalonate and bile acid pathway (Figure 5A). Indeed, we found that suppression of CYP7A1 led to accumulation of FPP and GGPP, two metabolites in the mevalonate pathway (Figures 5B-C). Using a statin inhibited accumulation of FPP and GGPP, prevented 2D-TNF suppression of primary hepatocyte insulin signaling, and prevented 7D-LPS induction of hyperglycemia (Figures 5D-F). These data indicate that inflammation regulates fasting glucose homeostasis through cell intrinsic effects on metabolite flux through the mevalonate pathway, consistent with research indicating that the primary mechanism for control of glucose homeostasis is posttranslational (Lin and Accili, 2011). Furthermore, it is known that BA sequestrants, drugs that induce BA biosynthesis and increase flux through the mevalonate pathway, improve glucose tolerance in diabetic patients (Goldberg et al., 2008; Staels and Fonseca, 2009). Our data suggest a mechanism for how BA sequestrants improve glucose homeostasis in diabetic individuals.
The observation that statin use could prevent the effects of inflammation on glucose metabolism was surprising. Recent work has identified an association between long-term statin use and an increased incidence of T2DM in humans (Ridker et al., 2012; Sattar et al., 2010). It is likely that the discordance between our data and the observations in humans is can be explained by the combination of at least two differences. First, the studies linking long-term statin use in humans evaluated the effects of the drug on T2DM secondary to obesity, while our study looked at the effect of statins in the context of the effect of sustained inflammation on fasting glucose levels. Obesity is a complex disease where multiple mechanisms are involved in the regulation of glucose homeostasis. Thus, statin treatment could positively influence the effects of inflammation while negatively influence a different mechanism. Second, statin use is known to be pleiotropic (Liao and Laufs, 2005) and influenced by duration of treatment. The studies which link statin use to human development of T2DM are conducted over years, compared to the week-long treatment we used in mice. Indeed, short term treatment of rats on a HFD with statins has been shown to improve glucose homeostasis (Lalli et al., 2008) while prolonged statin treatment of mice worsens glucose tolerance (Nakata et al., 2006). Although the association between statin use and development of T2DM has been well documented, the mechanism behind this association remains unknown. We have provided a mechanism explaining how statin treatment can improve dysregulated glucose homeostasis involved with inflammation, but given the pleiotropic effects of obesity, more research will be needed to tease apart the differences in the effects of statin treatment on glucose metabolism.
Previous work has implicated RHO-associated protein kinase (ROCK) in the regulation of glucose homeostasis (Huang et al., 2013). Much of this work has focused on the role of ROCK in adipose tissue (Chun et al., 2012; Lee et al., 2014) and muscle (Lee et al., 2009), while its role in the liver remains unexplored. Interestingly, the role of ROCK differs based upon the tissue evaluated, with adipose tissue ROCK activity associated with impaired insulin signaling and muscle ROCK activity associated with improved insulin signaling (Huang et al., 2013) Furthermore, the upstream activators of ROCK have yet to be identified (Huang et al., 2013). Here, we provide evidence that sustained inflammation induces hepatic RHOC, which is subsequently stabilized by isoprenylation (Figures 6B-D). RHOC is known to be a strong inducer of ROCK (Wheeler and Ridley, 2004), and we find that induction of RHOC results in activation of ROCK, which is required for the hyperglycemia and hepatic insulin resistance seen in sustained inflammation (Figures 6E-G). These data provide evidence that hepatic ROCK can play a role in the regulation of glucose metabolism.
The identification of ROCK as a downstream modulator of glucose homeostasis in response to sustained inflammation suggests it could be an attractive target in conditions of obesity. Sustained inflammation is one of many mechanisms known to alter glucose homeostasis during obesity, with others including ER stress, lipid deposition, and dysbiosis (Gregor and Hotamisligil, 2011; Johnson and Olefsky, 2013; Shulman, 2014). Applying our finding that ROCK is induced by sustained inflammation, we found that inhibition of ROCK could restore glucose homeostasis in already obese mice (Figures 6H-K).
In summary, our data suggest a two-component model for how sustained inflammation regulates glucose metabolism. Two branches of inflammation-induced effects in hepatocytes (induction of RHOC expression and accumulation of FPP and GGPP) combine to increase fasting glucose. Inflammatory signaling in this context operates through a coherent feed-forward loop with an AND gate (Figure 7B) (Alon, 2007). When one branch of the pathway is delayed compared to the other, this signaling network motif can detect signal persistence. In this case, the time required for accumulation of FPP and GGPP may explain why sustained inflammation is required to alter glucose homeostasis. These data suggest that sustained inflammation likely regulates homeostasis through persistent suppression of tissue metabolic function in combination with specific programs induced by inflammatory signaling.
Limitations, Caveats and Open Questions
Our data suggest that sustained inflammation controls glucose homeostasis through regulation of the hepatic mevalonate pathway. As with most studies, there are several limitations of this work that we would like to point out:
The difference between statin use in our model and the observed effects of statins on increasing development of T2DM remains unclear.
While we implicate ROCK in regulation of insulin signaling and fasting glucose levels, the direct target of ROCK kinase activity remains unknown.
Because the effect of ROCK inhibitors in vivo likely affects this enzyme in many tissues, the critical role of ROCK in the liver is suggested but not formally proven. It is possible that ROCK activity in other tissues may contribute to the in vivo phenotype.
The distinction between ROCK inhibitors affecting fasting glucose levels during conditions of sustained inflammation while altering glucose excursion in obesity is unknown.
Pathological states are often due to trade-offs between different physiological functions. In this case, it remains unclear as to why sustained inflammation impacts glucose homeostasis through such an indirect and complex mechanism.
Experimental Procedures
Mouse Experiments
Wild type mice were obtained from Jackson Laboratories (Bar Harbor, ME), ApoE-Cyp7a1 transgenic mice have been described previously (Miyake et al., 2001) and were obtained from the University California Davis Mutant Mouse Regional Resource Center. IRS-1 serine 307 to alanine mutant s were a kind gift of Morris White and Kyle Copps (Copps et al., 2010) Mice were injected intraperitoneally with PBS or LPS for 7 days (7D-LPS), fasted overnight, and then glucose tolerance tests, insulin tolerance tests, or hyperinsulinemic-euglycemic clamp experiments were performed, or serum and organs were harvested for further analysis. For insulin signaling studies, animals were treated with PBS or 7D-LPS, fasted overnight, and then injected i.p. with recombinant human insulin at 1U/kg; 15 minutes later, tissues were harvested and snap frozen on liquid N2. Obesity was modeled by feeding mice a high fat diet (60% kCal from fat) for 12-weeks, after which they were given daily injections of Y-27632 at 30mg/kg or PBS as control for 2 weeks, after which glucose and insulin tolerance tests were performed.
Ex Vivo Primary Hepatocytes
Primary hepatocytes were isolated using in situ collagenase digestion, plated on collagen coated plates, and then treated with TNF after which gene expression, insulin signaling, or other biochemical processes were evaluated.
Western Blotting Analysis
Tissue that was snap frozen on liquid N2 was pulverized and protein extracted and then subjected to western blotting. Primary hepatocyte protein was lysed and solubilized in 1X SDS buffer. Equal amounts of protein were loaded onto mini-gels (Invitrogen) and transferred on to PVDF membrane using established protocols. Samples were probed for pS473-AKT, AKT, ACTIN, CYP7A1, RHOC, pS19-MLC2, and MLC2.
Gene Expression Analysis
Whole RNA was extracted from either pulverized liver or primary hepatocytes using RNA Bee. RNA was then subjected to a cleanup step using Qiagen RNeasy Mini kit with on-column DNase treatment. Poly-A selected RNA was then subjected to microarray analysis or RNA sequencing using Illumina kits and according to standard protocols. Microarray and RNA sequencing data have been deposited at the NCBI Gene Expression Omnibus under accession numbers GSE67422 and GSE75477, respectively.
Lipid Analysis
Lipids were extracted from livers of PBS or 7D-LPS mice using the method of Bligh & Dyer or from primary hepatocytes using hexane/isopropanol (3:2) and then solvent was evaporated under a nitrogen stream. Samples were then analyzed for triglyceride, cholesterol, or non-esterified fatty acid levels using commercially available kits, or samples were resuspended in 50% methanol/50% H2O before being subjected to LC-MS/MS analysis.
Online Experimental Procedures
More details are provided in the Extended Experimental Procedures section available online as part of the supplementary information associated with this article.
Supplementary Material
Highlights.
Inflammation induces hepatic glucose production, leading to fasting hyperglycemia
Inflammatory suppression of CYP7A1 is required to induce fasting hyperglycemia
Reduced CYP7A1 leads to accumulation of metabolites in mevalonate pathway (MP)
MP metabolites stabilize RHOC to activate ROCK and regulate serum glucose levels
Acknowledgements
The authors would like to thank members of the Medzhitov lab for discussion and advice, Maya Kotas for experimental advice and technical expertise, Scott Pope for assistance and advice on RNA sequencing experiments and analysis, Sophie Cronin and Chuck Annicelli for technical assistance, Gerald Shulman for experimental advice and discussions, and Morris White for kindly providing IRS-1 S307A mice. Hyperinsulinemic-euglycemic clamp experiments were performed by the Yale Mouse Metabolic Phenotyping Center (supported by NIH DK059635). LC-MS/MS analysis was performed by the Yale Keck facility. DO is supported by NIH training grant (2T32GM7205-36) and a NIDDK fellowship (1F30DK094480). RM is supported by the HHMI, The Blavatnik Family Foundation, Else Kröner Fresenius Foundation, and grants from the NIH (AI046688, AI089771, CA157461).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: D.O. and R.M. designed the study; D.O. performed all the experiments; D.O. and R.M. wrote the paper.
Supplementary Information:
Extended Experimental Procedures
Figures S1-S7
Tables S1-S2
References
- Alon U. Network motifs: theory and experimental approaches. Nat Rev Genet. 2007;8:450–461. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- Andrews RC, Walker BR. Glucocorticoids and insulin resistance: old hormones, new targets. Clin Sci. 1999;96:513–523. doi: 10.1042/cs0960513. [DOI] [PubMed] [Google Scholar]
- Arkan MC, Hevener AL, Greten FR, Maeda S, Li Z-W, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- Barbour LA, McCurdy CE, Hernandez TL, Kirwan JP, Catalano PM, Friedman JE. Cellular Mechanisms for Insulin Resistance in Normal Pregnancy and Gestational Diabetes. Diabetes Care. 2007;30:S112–S119. doi: 10.2337/dc07-s202. [DOI] [PubMed] [Google Scholar]
- Berglund ED, Li CY, Poffenberger G, Ayala JE, Fueger PT, Willis SE, Jewell MM, Powers AC, Wasserman DH. Glucose metabolism in vivo in four commonly used inbred mouse strains. Diabetes. 2008;57:1790–1799. doi: 10.2337/db07-1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butte NF. Carbohydrate and lipid metabolism in pregnancy: normal compared with gestational diabetes mellitus. Am. J. Clin. Nutr. 2000;71:1256S–61S. doi: 10.1093/ajcn/71.5.1256s. [DOI] [PubMed] [Google Scholar]
- Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkin AC, Tontonoz P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat Rev Mol Cell Biol. 2012;13:213–224. doi: 10.1038/nrm3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- Chen G. TNF-R1 signaling: A beautiful pathway. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- Chun KH, Araki K, Jee Y, Lee DH, Oh B-C, Huang H, Park KS, Lee SW, Zabolotny JM, Kim Y-B. Regulation of glucose transport by ROCK1 differs from that of ROCK2 and is controlled by actin polymerization. Endocrinology. 2012;153:1649–1662. doi: 10.1210/en.2011-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AW, Razani B, Wang XB, Combs TP, Williams TM, Scherer PE, Lisanti MP. Caveolin-1-deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue. Am. J. Physiol., Cell Physiol. 2003;285:C222–C235. doi: 10.1152/ajpcell.00006.2003. [DOI] [PubMed] [Google Scholar]
- Copps KD, Hancer NJ, Opare-Ado L, Qiu W, Walsh C, White MF. Irs1 Serine 307 Promotes Insulin Sensitivity in Mice. Cell Metab. 2010;11:84–92. doi: 10.1016/j.cmet.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic Roles of Bile Acids in Metabolism. Cell Metab. 2013 doi: 10.1016/j.cmet.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Falciglia M, Freyberg RW, Almenoff PL, D'Alessio DA, Render ML. Hyperglycemia-related mortality in critically ill patients varies with admission diagnosis. Crit. Care Med. 2009;37:3001–3009. doi: 10.1097/CCM.0b013e3181b083f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold KR, Moser A, Shigenaga JK, Grunfeld C. Inflammation inhibits the expression of phosphoenolpyruvate carboxykinase in liver and adipose tissue. Innate Immun. 2012;18:231–240. doi: 10.1177/1753425911398678. [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB. NF-κB AND REL PROTEINS: Evolutionarily Conserved Mediators of Immune Responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- Goldberg RB, Fonseca VA, Truitt KE, Jones MR. Efficacy and safety of colesevelam in patients with type 2 diabetes mellitus and inadequate glycemic control receiving insulin-based therapy. Arch. Intern. Med. 2008;168:1531–1540. doi: 10.1001/archinte.168.14.1531. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271:665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research. 2009a;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009b;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang H, Lee DH, Zabolotny JM, Kim Y-B. Metabolic actions of Rho-kinase in periphery and brain. Trends Endocrinol Metab. 2013;24:506–514. doi: 10.1016/j.tem.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AMF, Olefsky JM. The origins and drivers of insulin resistance. Cell. 2013;152:673–684. doi: 10.1016/j.cell.2013.01.041. [DOI] [PubMed] [Google Scholar]
- Kotas ME, Medzhitov R. Homeostasis, Inflammation, and Disease Susceptibility. Cell. 2015;160:827. doi: 10.1016/j.cell.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalli CA, Pauli JR, Prada PO, Cintra DE, Ropelle ER, Velloso LA, Saad MJA. Statin modulates insulin signaling and insulin resistance in liver and muscle of rats fed a high-fat diet. Metab Clin Exp. 2008;57:57–65. doi: 10.1016/j.metabol.2007.07.021. [DOI] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Shi J, Jeoung NH, Kim MS, Zabolotny JM, Lee SW, White MF, Wei L, Kim Y-B. Targeted Disruption of ROCK1 Causes Insulin Resistance in Vivo. J Biol Chem. 2009;284:11776–11780. doi: 10.1074/jbc.C900014200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-H, Huang H, Choi K, Lee DH, Shi J, Liu T, Chun KH, Seo JA, Lima IS, Zabolotny JM, et al. ROCK1 isoform-specific deletion reveals a role for diet-induced insulin resistance. AJP: Endocrinology and Metabolism. 2014;306:E332–E343. doi: 10.1152/ajpendo.00619.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao JK, Laufs U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HV, Accili D. Hormonal Regulation of Hepatic Glucose Production in Health and Disease. Cell Metab. 2011;14:9–19. doi: 10.1016/j.cmet.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee T, Seabra MC. Fatty acylation and prenylation of proteins: what's hot in fat. Curr Opin Cell Biol. 2005;17:190–196. doi: 10.1016/j.ceb.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Maurer-Stroh S, Eisenhaber F. Refinement and prediction of protein prenylation motifs. Genome Biol. 2005;6:R55. doi: 10.1186/gb-2005-6-6-r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuinness OP. Defective glucose homeostasis during infection. Annu Rev Nutr. 2005;25:9–35. doi: 10.1146/annurev.nutr.24.012003.132159. [DOI] [PubMed] [Google Scholar]
- Miyake JH, Doung XD, Strauss W, Moore GL, Castellani LW, Curtiss LK, Taylor JM, Davis RA. Increased production of apolipoprotein B-containing lipoproteins in the absence of hyperlipidemia in transgenic mice expressing cholesterol 7alpha-hydroxylase. J Biol Chem. 2001;276:23304–23311. doi: 10.1074/jbc.M101853200. [DOI] [PubMed] [Google Scholar]
- Mizock BA. Alterations in fuel metabolism in critical illness: hyperglycaemia. Best Practice & Research Clinical Endocrinology & Metabolism. 2001;15:533–551. doi: 10.1053/beem.2001.0168. [DOI] [PubMed] [Google Scholar]
- Nakata M, Nagasaka S, Kusaka I, Matsuoka H, Ishibashi S, Yada T. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): implications in glycaemic control. Diabetologia. 2006;49:1881–1892. doi: 10.1007/s00125-006-0269-5. [DOI] [PubMed] [Google Scholar]
- Nathan C, Ding A. Nonresolving Inflammation. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- Odegaard JI, Chawla A. Pleiotropic Actions of Insulin Resistance and Inflammation in Metabolic Homeostasis. Science. 2013;339:172–177. doi: 10.1126/science.1230721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okin D, Medzhitov R. Evolution of inflammatory diseases. Curr Biol. 2012;22:R733–R740. doi: 10.1016/j.cub.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky JM, Glass CK. Macrophages, Inflammation, and Insulin Resistance. Annu Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. 2014;510:84–91. doi: 10.1038/nature13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prawitt J, Abdelkarim M, Stroeve JHM, Popescu I, Duez H, Velagapudi VR, Dumont J, Bouchaert E, van Dijk TH, Lucas A, et al. Farnesoid X receptor deficiency improves glucose homeostasis in mouse models of obesity. Diabetes. 2011;60:1861–1871. doi: 10.2337/db11-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman LM, Ravussin E. Endocrine alterations in response to calorie restriction in humans. Mol Cell Endocrinol. 2009;299:129–136. doi: 10.1016/j.mce.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Pradhan A, MacFadyen JG, Libby P, Glynn RJ. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet. 2012;380:565–571. doi: 10.1016/S0140-6736(12)61190-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, De Craen AJM, Seshasai SRK, McMurray JJ, Freeman DJ, Jukema JW, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375:735–742. doi: 10.1016/S0140-6736(09)61965-6. [DOI] [PubMed] [Google Scholar]
- Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med. 2014;371:1131–1141. doi: 10.1056/NEJMra1011035. [DOI] [PubMed] [Google Scholar]
- Solomon DH, Love TJ, Canning C, Schneeweiss S. Risk of diabetes among patients with rheumatoid arthritis, psoriatic arthritis and psoriasis. Annals of the Rheumatic Diseases. 2010;69:2114–2117. doi: 10.1136/ard.2009.125476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon DH, Massarotti E, Garg R, Liu J, Canning C, Schneeweiss S. Association Between Disease-Modifying Antirheumatic Drugs and Diabetes Risk in Patients With Rheumatoid Arthritis and Psoriasis. Jama. 2011;305:2525–2531. doi: 10.1001/jama.2011.878. [DOI] [PubMed] [Google Scholar]
- Staels B, Fonseca VA. Bile acids and metabolic regulation: mechanisms and clinical responses to bile acid sequestration. Diabetes Care. 2009;32(Suppl 2):S237–S245. doi: 10.2337/dc09-S355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I. Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. Journal of Clinical Investigation. 2002;110:905–911. doi: 10.1172/JCI16452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, et al. TGR5-Mediated Bile Acid Sensing Controls Glucose Homeostasis. Cell Metab. 2009;10:167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7:678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- Wheeler AP, Ridley AJ. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp Cell Res. 2004;301:43–49. doi: 10.1016/j.yexcr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu. Rev. Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.